

Abstract
Chronic pain following spinal cord injury (SCI) is associated with electrical hyperactivity (spontaneous and evoked) in primary nociceptors. Cyclic adenosine monophosphate (cAMP) signaling is an important contributor to nociceptor excitability, and knockdown of the cAMP effector, exchange protein activated by cAMP (EPAC), has been shown to relieve pain-like responses in several chronic pain models. To examine potentially distinct roles of each EPAC isoform (EPAC1 and 2) in maintaining chronic pain, we used rat and mouse models of contusive spinal cord injury (SCI). Pharmacological inhibition of EPAC1 or 2 in a rat SCI model was sufficient to reverse SCI-induced nociceptor hyperactivity, indicating that EPAC1 and 2 signaling activity are complementary, with both required to maintain hyperactivity. However, EPAC activation was not sufficient to induce similar hyperactivity in nociceptors from naïve rats, and we observed no change in EPAC protein expression after SCI. In the mouse SCI model, inhibition of both EPAC isoforms through a combination of pharmacological inhibition and genetic deletion was required to reverse SCI-induced nociceptor hyperactivity. This was consistent with our finding that neither EPAC1−/− nor EPAC2−/− mice were protected against SCI-induced chronic pain as assessed with an operant mechanical conflict test. Thus, EPAC1 and 2 activity may play a redundant role in mouse nociceptors, although no corresponding change in EPAC protein expression levels was detected after SCI. Despite some differences between these species, our data demonstrate a fundamental role for both EPAC1 and EPAC2 in mechanisms maintaining nociceptor hyperactivity and chronic pain after SCI.
Keywords: cAMP signaling, Spontaneous activity, Hyperexcitability, Depolarizing spontaneous fluctuations, Neuropathic pain, Hyperalgesia
1. Introduction
Chronic pain afflicts at least 50 million American adults, yet the mechanisms promoting and maintaining it are not fully understood (Dahlhamer et al., 2018). Often resulting in a drastic drop in quality of life, chronic pain is associated with multiple inflammatory and neuropathic conditions (Finnerup and Baastrup, 2012, Finnerup, 2013), including spinal cord injury (SCI). More than 50% of SCI patients suffer from chronic neuropathic pain (Burke et al., 2017, Siddall et al., 2003), and for most of them none of the available treatments is adequate (Hatch et al., 2018), pointing to the need for new therapeutic targets.
Several studies indicate an unexpected contribution by somatic sensory neurons to the maintenance of at-level and below-level neuropathic pain after SCI; specifically, a switch of nociceptors within dorsal root ganglia (DRG) at and below the contusion level into a persistent hyperactive state (Bedi et al., 2010, Walters, 2012, Yang et al., 2014). Electrical hyperactivity can be observed months after injury, which arises from the combined effects of sustained depolarization of resting membrane potential (RMP), a decrease in action potential (AP) threshold, and an increase in amplitudes of depolarizing spontaneous fluctuations (DSFs) of membrane potential (Odem et al., 2018). These three intrinsic mechanisms promote and maintain spontaneous activity (SA) generated at resting membrane potential (RMP) and can also produce sustained ongoing activity (OA) if nociceptors are depolarized by prolonged extrinsic inputs after SCI (Odem et al., 2018).
Our previous study describing intracellular signaling mechanisms maintaining SCI- induced SA within small-diameter nociceptors showed that the hyperactive state depends on persistent cAMP-PKA signaling, as well as an alteration in adenylyl cyclase regulation (Bavencoffe et al., 2016). The change in adenylyl cyclase regulation suggests that other cAMP effectors might also be involved. Recent work has identified an additional cAMP effector involved in pain function, EPAC. EPAC activity was shown to be necessary for hyperalgesia induced by peripheral application of a cAMP analog (Hucho et al., 2005) and to increase sensory neuron excitability in vitro (Eijkelkamp et al., 2013, Gu et al., 2016, Shariati et al., 2016, Vasko et al., 2014, Wang et al., 2007), as well as to induce hyperalgesia in vivo (Gu et al., 2016, Hucho et al., 2005). Diverse models of injury- and inflammation-related pain have implicated divergent contributions from EPAC1 and 2 in nociceptors (Cao et al., 2016, Eijkelkamp et al., 2010, Gu et al., 2016, Hucho et al., 2005, Singhmar et al., 2016, Singhmar et al., 2018, Vasko et al., 2014) as well as differing alterations of EPAC1 and 2 expression (Cao et al., 2016, Gu et al., 2016, Vasko et al., 2014) in the DRG. Whether EPAC1 and EPAC2 play different roles in chronic pain signaling remain unclear.
Here we show that pharmacological inhibition of either EPAC1 or EPAC2 produces similar mitigation of electrical hyperactivity in putative nociceptors dissociated from SCI rats, indicating that enzymatic activity of both isoforms is necessary in this species. Pharmacological activation of both EPAC isoforms in uninjured rats increased excitability weakly, consistent with essential contributions from signals other than EPAC to produce hyperactivity. To further explore the contribution of each EPAC isoform to SCI-induced nociceptor hyperactivity, we employed mouse models. Genetic deletion of either EPAC1 or EPAC2 failed to attenuate pain-related behavior. However, examination of putative nociceptors isolated from EPAC1−/− and EPAC2−/− mice alone and in combination with pharmacological inhibitors of each EPAC isoform revealed functional redundancy between the two isoforms, in contrast to the necessity of both isoforms for hyperactivity in rat nociceptors. These results demonstrate that activity of both EPAC1 and EPAC2 contributes to nociceptor hyperactivity (albeit with some differences between rats and mice) and suggest that future translational studies should consider both isoforms of EPAC as potential therapeutic targets for chronic pain.
2. Materials and methods
2.1. Animals
All procedures followed the guidelines of the International Association for the Study of Pain and were approved by the McGovern Medical School at UT Health Animal Care and Use Committees. Male Sprague-Dawley rats (Envigo, USA) were used. After arrival at the McGovern Medical School, the rats (8–9 weeks old, 250–300 g, 2 per cage) were allowed to acclimate to a 12-hour reverse light/dark cycle for at least four days before beginning experiments.
Male and female C57BL/6 (Charles River, USA) wild-type, EPAC2−/−, and EPAC1−/− mice (Pereira et al., 2013) were generated within the McGovern Medical school animal facility and subsequently transferred at 6–8 weeks of age to a controlled environment where they were allowed to acclimate for at least 1 week before beginning experiments.
2.2. Spinal cord injury (SCI) procedures
Rat SCI surgeries were performed as previously described (Bavencoffe et al., 2016, Bedi et al., 2010, Wu et al., 2013, Yang et al., 2014). Rats were anesthetized with an intraperitoneal (i.p.) injection of ketamine (60 mg/kg, Henry Schein, Dublin, OH), xylazine (10 mg/kg, Henry Schein, Dublin, OH), and acepromazine (1 mg/kg, Henry Schein, Dublin, OH), or with isoflurane (induction 4–5%; maintenance 1–2%, Isothesia, Henry Schein, Dublin, OH). A T10 vertebral laminectomy was followed by a dorsal contusive spinal impact (150 kilodyne, 1-second dwell time) using an Infinite Horizon Spinal Cord Impactor (Precision Systems and Instrumentation, LLC, Fairfax Station, VA). Sham-operated rats received the same surgical treatment without the contusion. The analgesic buprenorphine hydrochloride (0.02 mg/kg in 0.9% saline 2 ml/kg; Buprenex, Reckitt Benckiser Healthcare Ltd., Hull, England, UK) and the antibiotic enrofloxacin (0.3 ml in 0.9% saline; Enroflox, Norbrook, Inc., Overland Park, KS) were injected i.p. twice daily for 5 days (buprenorphine) or 10 days (enrofloxacin). Manual bladder evacuations were performed twice daily until rats recovered neurogenic bladder voiding. Rats had free access to food and water. Rats included in this study received a score of 0 or 1 for both hind limbs the day after surgery, as measured on the Basso, Beattie, and Bresnahan (BBB) Locomotor Rating Scale (Basso et al., 1995).
Mouse SCI surgeries were conducted as previously described (Herrera et al., 2008, Herrera et al., 2010). Briefly, adult female and male mice were anesthetized by inhalation of 4.0% isoflurane and maintained by a mixture of 1.5% isoflurane, 30% oxygen, and air administered through a rodent ventilator (Harvard Apparatus, model 683, Holliston, MA) throughout the surgical procedure. Following laminectomy of the 9th thoracic (T9) vertebrae, a force-controlled contusion (60 kilodyne, 1-second dwell time) was performed using an Infinite Horizon Spinal Cord Impactor. Sham-operated animals received the same surgical treatment without the contusion injury. For 2–3 days post injury, mice received twice daily subcutaneous injections of 0.9% saline to ensure proper hydration (0.5 cc) and twice daily subcutaneous injections of buprenorphine (0.02–0.1 mg/kg) for pain management. For 10 days post injury, mice received twice daily subcutaneous injection of Baytril (0.2–0.5 mg/ml; Med-Vet International, Mettawa, IL) to prevent urinary tract infections. Manual bladder evacuations were performed twice daily for the duration of the study. Mice had free access to food and water. Mice included in this study received a score of 0 or 1 for both hind limbs the day after surgery, as measured by the Basso Mouse Scale for locomotion (BMS) (Basso et al., 2006). Animals that could not resume a minimum normal activity level after injury were euthanized.
2.3. Western blot
At 8–10 weeks post-surgery, mice (including naïve litter mates) were euthanized by inhalation of isoflurane followed by cervical dislocation and transcardial perfusion of ice cold PBS (Sigma-Aldrich, St Louis, MO). 3 months post-surgery, sham and SCI rats, as well as age matched naïves, were euthanized by i.p. injection of an overdose of pentobarbital/phenytoin (0.9 ml of Euthasol, Virbac AH, Inc., Fort Worth TX) followed by transcardial perfusion of ice cold PBS. DRGs from mice and rats were harvested below T9 and T10 levels, respectively, and subsequently frozen in liquid nitrogen. DRGs were homogenized in SDS lysis buffer (1% SDS, 320 mM sucrose, 5 mM Hepes pH 7.4, 1 mM NaF, 1 mM PMSF) with protease inhibitor cocktail P8340 (Sigma-Aldrich, St Louis, MO). The homogenates were centrifuged at 14,000 rpm for 5 min at room temperature and then heated to 100 °C for 5 min and stored at −80 °C. Protein concentrations were determined by the BCA method (Pierce BCA Protein Assay Kit, ThermoFisher Scientific, Waltham, MA). Equal amounts of cell lysates were separated on 9% SDS-PAGE gels and transferred to a PVDF membrane. Membranes were blocked in 5% (w/v) nonfat milk prior to incubation overnight (4 °C) in primary antibody. Primary antibodies included EPAC1 (1:1000; Cell Signaling; Cat# 4155), EPAC2 (1:1000; Cell Signaling; Cat# 4156), anti-actin (1:1000; Cytoskeleton; Cat# AAN01), and GAPDH (1:20,000; RDI; Cat#RDI-Trk5G4-6C5). Membranes were incubated with anti-mouse or anti-rabbit IgG for 1 h (room temperature) and developed using ECL or the Super Signal West Femto Kit (ThermoFisher Scientific, Waltham, MA). GAPDH, Actin, and Stain Free imaging were used as a loading controls. Protein expression was then quantified by optical density using Image Lab software (Bio-Rad Laboratories, Version 5.2.1 Build 11).
2.4. Dissociation and culture of DRG neurons
DRGs from mice and rats were harvested below vertebral levels T9 and T10, respectively, down to L6 in each case. Ganglia were surgically desheathed before being transferred in high glucose DMEM culture medium (Sigma-Aldrich, St Louis, MO) containing trypsin TRL (0.3 mg/ml, Worthington Biochemical Corporation, Lakewood, NJ) and collagenase D (1.4 mg/ml, Roche Life Science, Penzberg, Germany). After 40 min incubation under constant shaking at 34 °C, digested DRG fragments were washed by two successive centrifugations and triturated with a fire-polished glass Pasteur pipette. Cells were plated on 8 mm glass coverslips coated with poly-L-ornithine (Sigma-Aldrich, St Louis, MO) in DMEM without serum or growth factors, and incubated overnight at 37 °C, 5% CO2 and 95% humidity.
2.5. Recording from dissociated DRG neurons
Whole-cell patch clamp recordings were performed at room temperature 18–30 h after dissociation using a MultiClamp 700B (Molecular Devices, San Jose, CA) and an EPC10 USB (HEKA Elektronik, Lambrecht/Pfalz, Germany) amplifier. Patch pipettes were made of borosilicate glass capillaries (Sutter Instrument Co., Novato, CA) with a horizontal P-97 puller (Sutter Instrument Co., Novato, CA) and fire-polished with a MF-830 microforge (Narishige, Tokyo, Japan) to a final pipette resistance of 3–8 MΩ when filled with an intracellular solution composed as follows (in mM): 134 KCl, 1.6 MgCl2, 13.2 NaCl, 3 EGTA, 9 HEPES, 4 Mg-ATP, and 0.3Na-GTP, which was adjusted to pH 7.2 with KOH and 300 mOsM with sucrose. Isolated small neurons with a soma diameter ≤30 µM were observed at 20x magnification on IX-71 (Olympus, Tokyo, Japan) or 40x on TE2000-U (Nikon, Tokyo, Japan) and Axiovert 200 M (Zeiss, Oberkochen, Germany) inverted microscopes and recorded in a bath solution containing (in mM): 140 NaCl, 3 KCl, 1.8 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose, which was adjusted to pH 7.4 with NaOH and 320 mOsM with sucrose. After obtaining a tight seal (>3 GΩ), the plasma membrane was ruptured to achieve whole-cell configuration under voltage clamp at −60 mV. Recordings were acquired with Patchmaster v2x90.1 (HEKA Elektronik, Lambrecht/Pfalz, Germany) and Clampex v10.4 (Molecular Devices, San Jose, CA). The liquid junction potential was calculated to be ~4.3 mV and not corrected, meaning the actual potentials were ~4.3 mV more negative than indicated in the recordings and measurements presented herein.
2.6. Quantifying depolarizing spontaneous fluctuations of membrane potential (DSFs)
DSFs were analyzed as previously described (Odem et al., 2018). Briefly, we used a custom program (SFA.py) to quantify irregular DSFs in patch recordings, which we imported as time and voltage coordinate data for 30-second periods from recordings obtained with PatchMaster (HEKA Elektronik) sampled at 20 kHz and filtered with a 10 kHz Bessel filter. The program used a sliding median function to calculate resting membrane potential (RMP) at each point and returned coordinates, amplitudes, and durations of identified APs and DSFs (minimum amplitude and duration 1.5 mV and 5 ms), as well as a continuous color-coded plot of membrane potential generated using the matplotlib library (Python v3.6, Python Software Foundation, Beaverton,OR) (Odem et al., 2018). Manual inspection of each plot confirmed that each AP was generated by a suprathreshold DSF. Conservative estimates of the amplitude of the suprathreshold DSFs were obtained by taking the larger of 1) the point at which the change in membrane potential began to accelerate immediately before the AP and 2) the most depolarized potential reached by the largest subthreshold DSF recorded at RMP or during rheobase measurements (Odem et al., 2018).
2.7. Pharmacological agents
EPAC2 inhibitor ESI-05 was synthesized as described in Chen et al. (2013). EPAC activator 8-pCPT-2-O-Me-cAMP-AM (007-AM) was purchased from Tocris Bioscience (Bristol, England, UK) and EPAC 1 inhibitor CE3F4 from Cayman Chemicals (Ann Arbor, MI). Drugs were prepared in DMSO (Sigma-Aldrich, St Louis, MO) at a concentration of 20 mM for ESI-05 and CE3F4 and 10 mM for 007-AM. Reagents were then diluted in extracellular recording solution at a minimum dilution factor of 1/1000.
2.8. Behavioral testing
All tests were performed by the same blinded investigator. Mice were allowed to acclimate to the behavioral testing room for 30–60 min in their home cages with the investigator present before beginning testing.
2.8.1. Mechanical conflict (MC) test
The MC test (Harte et al., 2016, Odem et al., 2019) was used to assess voluntary avoidance of noxious probes as an operant measure of altered pain sensitivity. The commercially available MC test device (Mechanical Conflict-Avoidance System, Coy Lab Products, Grass Lake, MI, USA) consists of two rectangular chambers (each 16.5 cm wide by 21.5 cm deep by 15.25 cm high) connected by a narrow 30.4 cm long tunnel containing a dense array of sharp probes on the floor (Harte et al., 2016). We modified the tunnel floor for mice to a 30.4 cm by 3.8 cm array of 3 mm, more closely spaced, fine, blunted metal probes (tip diameter ~0.63 mm). In each test, a mouse was placed in the darkened first chamber with the gate closed to prevent access to the tunnel. After 30 s the chamber was illuminated by a bright light emitting diode with a mildly aversive mean illuminance of 442 fc at the compartment floor, intended to promote escape to the unlit chambers. After at least 15 s, when the mouse was facing the gate, the gate was lifted to allow free movement throughout the device for the remaining 3 min of the test. The test was video-recorded for subsequent analysis (Sony Handycam, HDR-XR260, recorded in HD). Videos were analyzed by a separate, blinded investigator for 1) latency to the first step onto the probe floor, 2) the total time on the probe floor, 3) the latency to the first crossing of the probes (all four paws in the dark chamber), 4) second crossing latency (front-paws re-entering the original chamber), and 5) total number of crossings. Latency measurements always began when the gate was fully raised. In our preliminary testing we observed that repeated trials significantly decreased the number of crossings and increased the latency to cross. Similar studies with rats suggested that a large component of the motivation to cross the aversive probes is the strong drive of rodents to explore an unfamiliar context (Odem et al., 2019). To reduce possible habituation to the MC device, we modified our procedure to allow the mice to experience the device only twice, with a period of 3–4 weeks separating each exposure. Unlike previously published studies using the MC test (Harte et al., 2016, Shepherd and Mohapatra, 2018), the mice had not experienced the probes before their first crossing. When crossing a second time within a given test trial (presumably motivated by innate exploratory drive), the mice had recently experienced the aversive probes and could exhibit voluntary avoidance behavior by delaying or avoiding a second crossing.
2.8.2. Von Frey mechanical sensitivity test
Von Frey filaments were used to determine the 50% paw withdrawal threshold using the up-down method (Chaplan et al., 1994). Animals were placed in red plastic testing chambers (8.9 cm wide by 8.9 cm deep by 15.2 cm high) on a wire grid bottom and allowed to acclimate for 30 min with the investigator present. Von Frey filaments were applied for ~1 s through the grid perpendicular to the plantar surface of the hindpaw with sufficient force to buckle against the paw, with the tester allowing 30 s between stimuli (starting with a bending force of 0.4 g; range: 0.02–4.0 g, North Coast Medical, Inc., San Jose, CA, USA). The subsequent filament bending force was increased following a negative response, or decreased following a positive response, with a total of 10 stimuli per hindpaw. A positive response was recorded if the paw was quickly withdrawn, which was often accompanied by paw grooming or shaking behavior.
2.8.3. Hargreaves heat sensitivity test
The latency to withdraw from a radiant heat stimulus was measured using the Plantar Analgesia Meter (IITC Life Science Inc., Woodland Hills, CA, USA) as described previously (Hargreaves et al., 1988). Mice were placed in red plastic testing chambers (8.9 cm wide by 8.9 cm deep by 15.2 cm high) on a 30 °C heated glass surface and allowed to acclimate for 30 min with the investigator present. A light beam, set to an intensity 30% of its maximum value (found to evoke a positive response in 10 s at baseline in naïve mice), was aimed at the hindpaw plantar surface until evoking a positive response, or for a maximum of 20 s to prevent tissue damage. The latency to evoke a positive response (brisk hindpaw withdrawal) was recorded and each hindpaw was tested 5 times, with a 5-minute interval between stimuli. The median response latency was recorded after excluding the shortest and longest latencies.
2.8.4. Elevated plus maze anxiety test
The 40 cm high elevated plus maze (EPM) was used to measure differences in anxiety. It consisted of four, 12 cm wide arms: two enclosed by 40 cm high walls and two open (modeled upon the Stoelting Co’s EPM model, Wood Dale, IL). Individual mice were placed in the EPM center and allowed to move freely for 5 min. The test was video recorded and later analyzed for time spent in the open versus closed arms, as previously described (Acharjee et al., 2013, Nyuyki et al., 2018, Pellow et al., 1985).
2.8.5. Rotarod motor coordination test
This test was used to assess coordinated motor function in mice, as described previously (Rozas et al., 1997). Mice were placed on the rotating rod (Model ENV-576 M, Med Associates, Georgia, VT) facing the direction opposite to the rotation. Mice underwent 3 preliminary training tests spaced 10 min apart. Mice must be able to stay on a rod rotating at 4 rpm for 60 s before moving past training. During the actual test, mice were placed on a rotating rod accelerating linearly with time from 4 to 40 rpm over the course of 5 min. The fall latency was recorded automatically by photobeam sensors, with the maximum time being 5 min. Mice underwent three trials spaced 15 min apart.
2.8.6. Open field activity measure
The open field apparatus (OFA) (ENV-515, Med Associates, Inc., St. Albans, VT) consists of an activity chamber (43.2 cm wide by 43.2 cm deep by 30.5 cm high) with 16 infrared transmitters and receivers evenly positioned around the chamber’s periphery. Mice were placed in the activity chamber and allowed to move freely for 25 min, and data were collected by the software provided with the OFA. The OFA software registers movements within the chamber by recording photobeam interruptions. We analyzed the first 5 min within the chamber for ambulatory distance traveled and average velocity.
2.9. Statistical analysis
Data are presented as mean ± SEM or incidence (% of sampled neurons). P < 0.05 is considered statistically significant. All data sets were tested for normality by the Shapiro-Wilk test. Normally distributed data were tested with the parametric t-test or 1-way ANOVA, followed by the Holm-Sidak method of pairwise comparison. Non-parametric tests included the Mann-Whitney U test or Kruskal-Wallis test, followed by Dunn’s test for each pair-wise comparison. Data reported as incidence were compared by Chi square or Fisher’s exact test when appropriate. Bonferroni corrections were made after multiple comparisons. Statistical analyses were conducted using SigmaPlot (Systat Software, Inc., San Jose, CA) and Prism v7.04 (GraphPad Software, Inc., La Jolla, CA, USA).
3. Results
3.1. Activity of both EPAC1 and EPAC2 is required for persistent hyperactivity of dissociated rat nociceptors after SCI
The major goal of our study was to determine the roles of EPAC isoforms in maintaining an SCI-induced hyperactive state in primary nociceptors. Presumptive nociceptors were selected on the basis of small soma diameter (≤30 µm) and nonaccommodating properties (firing multiple APs at random intervals during activation by 2-second depolarizing currents at twice the rheobase value) (Odem et al., 2018). Previous studies have shown that ~70% of the nonaccommodating (NA) type of neurons sampled under our conditions are nociceptors based on capsaicin sensitivity and/or binding of isolectin B4 (IB4) (Bavencoffe et al., 2016, Bedi et al., 2010, Odem et al., 2018). We did not test a separate electrophysiologically defined type of presumptive nociceptor, the rapidly accommodating (RA) type, which only discharge a single AP at the beginning of a 2-second test depolarization at twice rheobase and never display SA (Odem et al., 2018). Consistent with these previous studies, 1–8 months after SCI 67% of sampled neurons isolated from injured male rats exhibited SA, versus only 12% isolated from naïve animals (Fig. 1A). The high incidence of SA after SCI was associated with significant electrophysiological alterations promoting hyperactivity, including depolarization of the RMP (−50 mV in SCI versus −55 in naïve rats, Fig. 1B), decreased AP voltage threshold (−35 mV in SCI versus −32 in naïve, Fig. 1C), and lowered rheobase (45pA in SCI versus 83 pA in naïve rats, Fig. 1D).
Fig. 1.
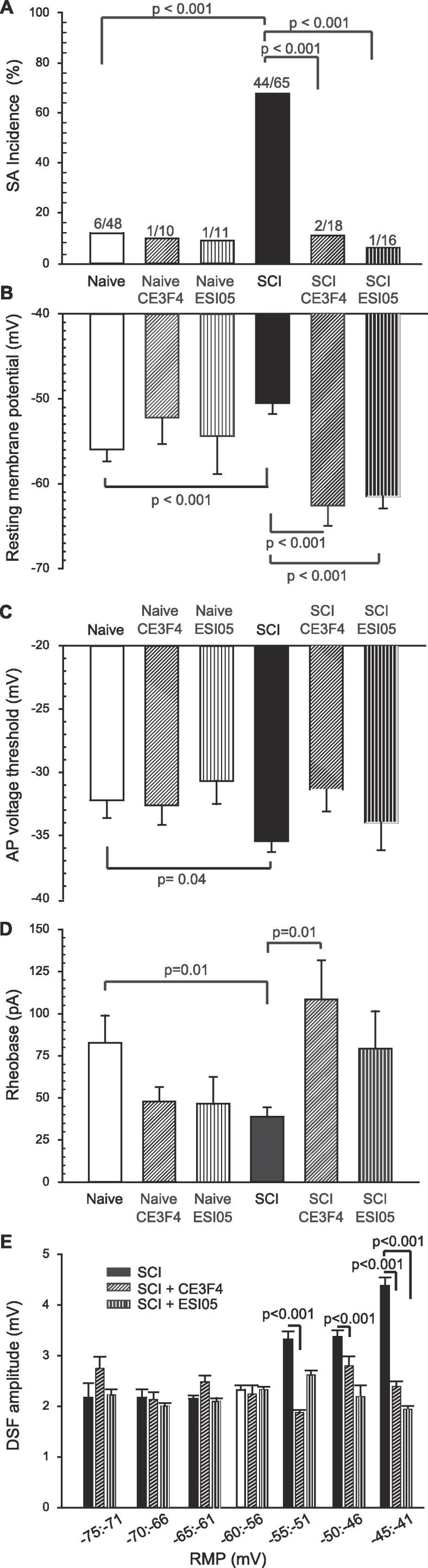
EPAC1 or EPAC2 activity maintains SCI-induced hyperexcitability in dissociated small diameter rat DRG neurons recorded by whole-cell patch clamp 18–30 h after dissociation. DRG neurons were pretreated with either 10 µM CE3F4 or 5 µM ESI-05 for 15–20 min before recording. (A) Inhibition of EPAC1 or 2 attenuated the incidence of SCI-induced SA. The ratio above each bar denotes the number of neurons with SA/the number of neurons sampled. Statistical comparisons of SA incidence were made with Bonferroni-corrected Fisher’s exact tests on the indicated pairs. (B) Inhibition of EPAC1 or 2 reversed SCI-induced depolarization of RMP. (C) Inhibition of EPAC1 or 2 did not reverse SCI-induced reduction of AP voltage threshold. (D) Inhibition of EPAC1 attenuated the SCI-induced decrease in rheobase. Data shown as mean ± SEM. Overall significance determined with one way ANOVA (or Kruskal-Wallis for non-parametric data), followed by multiple comparisons with Dunn’s method. Control Naïve vs SCI rats were compared by Mann-Whitney U test. (E) Inhibition of EPAC1 or EPAC2 decreased the amplitude of DSFs recorded at rest in DRG neurons from SCI rats, especially at more depolarized RMPs. DSFs were binned according to RMP. Data are represented as mean ± SEM. The indicated statistical comparisons were performed with Kruskal-Wallis test followed by multiple comparisons with Dunn’s method for each trio of data at each bin of RMP. ANOVA, analysis of variance; DRG, dorsal root ganglion; DSF, depolarizing spontaneous fluctuation; EPAC, exchange protein activated by cAMP; RMP, resting membrane potential; SA, spontaneous activity; SCI, spinal cord injury; SEM, standard error of the mean.
Previous studies have indicated that activity of either EPAC1 or EPAC2 can contribute to hyperexcitability in isolated sensory neurons (see Introduction). In nociceptors isolated from SCI rats, we found that pretreatment with either the EPAC1-selective inhibitor CE3F4 (10 µM) (Courilleau et al., 2012, Sonawane et al., 2017) or the EPAC2-specific inhibitor ESI-05 (5 µM) (Tsalkova et al., 2012) for 15 min before and during recording significantly decreased the incidence of SA (Fig. 1A), and hyperpolarized the RMP (Fig. 1B). Action potential voltage threshold in DRG neurons isolated from SCI rats was not significantly affected (Fig. 1C), while the rheobase increased after CE3F4 treatment (Fig. 1D). Neither drug had a significant effect on the incidence of SA, RMP, rheobase, or AP voltage threshold in isolated DRG neurons from naïve rats (Fig. 1). SCI-induced enhancement of depolarizing spontaneous fluctuations of membrane potential (DSFs) may bridge the gap between RMP and AP voltage threshold to trigger APs. Analysis of DSFs showed that the largest amplitudes occurred at more depolarized RMPs in nociceptors isolated from SCI rats, most notably at RMPs between −45 and −41 mV (Fig. 1E), as shown previously by Odem et. al. (Odem et al., 2018). CE3F4 treatment significantly decreased DSF amplitudes at RMPs between −55 and −41 mV while ESI-05 decreased DSFs between −45 and −41 mV (Fig. 1E). Thus, EPAC1 and EPAC2 are both necessary for SCI-induced SA promoted by 2 of the 3 general electrophysiological alterations that can drive SA (Odem et al., 2018): depolarization of resting membrane potential and an increase in the amplitudes of the DSFs. EPAC activity does not appear to be important for the third alteration that can promote SA - reduction in AP voltage threshold (Odem et al., 2018).
Finding major contributions of EPAC activity to depolarized RMP, enhanced DSFs, and SA after SCI raised the question of whether selective activation of EPAC in neurons isolated from naïve animals would be sufficient to induce a hyperactive state similar to the one induced by SCI. Pretreatment for 10–30 min with EPAC activator 007-AM (10 µM) failed to significantly increase the incidence of SA (Fig. 2A). Nevertheless, the EPAC activator significantly depolarized RMP (Fig. 2B), with an apparent trend for rheobase to decrease (Fig. 2C). Surprisingly, given that EPAC inhibitors fail to increase AP voltage threshold (Fig. 1C), 007-AM significantly reduced AP voltage threshold (Fig. 2D). In addition, 007-AM treatment appeared to increase DSF amplitudes in neurons with RMPs between −45 and −41 mV (Fig. 2E). Despite these hyperexcitable effects, EPAC stimulation not only failed to increase the incidence of neurons with SA at RMP (Fig. 2A) but also failed to increase the incidence of neurons exhibiting ongoing activity (OA) when artificially depolarized to −45 mV (Fig. 2F). These results indicate that both EPAC isoforms are necessary for maintenance of nociceptor hyperactivity after SCI, and they are consistent with evidence that activity of other cell signals (notably, PKA; Bavencoffe et al., 2016) is needed to enhance the incidence of SA and OA.
Fig. 2.

Activation of EPAC1 and 2 has a slight effect on DRG neurons isolated from naïve rats, depolarizing the RMP and hyperpolarizing AP voltage threshold. DRG neurons harvested from naïve rats were pretreated with 10 µM 8-pCPT-2-O-Me-cAMP-AM (007-AM) for 10–15 min before recording. (A) EPAC activation did not significantly affect SA incidence within neurons isolated from naïve rats. The ratio above each bar denotes the number of neurons with SA/the number of neurons sampled. Statistical comparison of SA was performed with Fisher’s exact test. (B) EPAC activation significantly depolarized RMP, with a trend toward a reduction in rheobase (C) and a significant hyperpolarization of AP voltage threshold. (D) Statistical comparisons of data (represented as mean ± SEM) were made by Mann Whitney U test. (E) EPAC activation significantly increased the DSF amplitudes at rest only for cells exhibiting RMP values between −45 and −41 mV. DSFs were binned according to initial voltage. Statistical comparison was performed by Kruskal-Wallis test followed by multiple comparisons with Dunn’s method for each duo of data at each bin of RMP. (F) EPAC activation did not significantly affect OA incidence. The ratio above each bar denotes the number of neurons with OA/the number of neurons sampled. Statistical comparison of OA was performed with Fisher’s exact test. DRG, dorsal root ganglion; DSF, depolarizing spontaneous fluctuation; EPAC, exchange protein activated by cAMP; n.s., non-significant; RMP, resting membrane potential; SA, spontaneous activity; SCI, spinal cord injury; SEM, standard error of the mean.
3.2. SCI induces behavioral hypersensitivity in wild-type, EPAC2−/−, and EPAC1−/− mice
Our finding in rats that SA was reduced by inhibition of either EPAC1 or EPAC2, combined with reports that EPAC2 protein levels increase in the DRG after peripheral inflammation (Gu et al., 2016, Matsuda et al., 2017, Vasko et al., 2014, Wang et al., 2007), and that PGE2-induced nociceptor sensitization involves EPAC2 (Vasko et al., 2014), led us to hypothesize that EPAC2 in primary nociceptors plays a significant role in maintaining chronic pain after SCI. To test this hypothesis, we conducted a comparative study using mouse models with genetic deletion of EPAC2 (EPAC2−/−) or EPAC1 (EPAC1−/−).
As previously shown for EPAC1−/− mice (Russart et al., 2018), we first confirmed that EPAC2−/− mice did not exhibit major differences from wild-type mice in general behavioral functions (Lee et al., 2015, Srivastava et al., 2012) but see also (Zhou et al., 2016): anxiety, as tested by time spent in the open arms of the elevated plus maze (EPM); motor coordination, as tested by latency to fall off the rotarod; and general activity level, as tested by average velocity within an activity box (Table 1). In addition, von Frey mechanical sensitivity tests and Hargreaves heat sensitivity tests were used to assess basal mechanical and thermal sensitivity, respectively. The mechanical withdrawal threshold and the thermal withdrawal latency did not differ significantly between genotypes, suggesting that EPAC2 deletion does not result in mechanical or heat hyposensitivity (Table 1).
Table 1.
Behavioral measures from uninjured (naïve) wild-type and EPAC2−/− mice.
| Behavioral test | Wild-type | EPAC2−/− | P value | Test |
|---|---|---|---|---|
| Von Frey (g) | 2.5 ± 0.3 (18) | 3.1 ± 0.5 (11) | 0.281 | MW |
| Hargreaves (s) | 9.3 ± 0.5 (18) | 8.2 ± 0.4 (11) | 0.159 | t-test |
| Rotarod (s) | 171.5 ± 18.3 (16) | 138.3 ± 14.2 (7) | 0.274 | t-test |
| Activity box average velocity (cm/min) | 41.8 ± 3.4 (16) | 39.7 ± 2.1 (7) | 0.699 | t-test |
| Time on open arms of EPM (s) | 184.1 ± 10.6 (16) | 174.5 ± 25.5 (7) | 0.867 | MW |
| Mechanical conflict (MC) test | ||||
| Number of crossings | 8.7 ± 0.5 (18) | 9.4 ± 1.2 (11) | 0.585 | t-test |
| Time on probes (s) | 40.6 ± 4.3 (18) | 42.4 ± 5.4 (11) | 0.805 | t-test |
| Latency to 1st crossing (s) | 38.9 ± 5.6 (18) | 42.0 ± 8.6 (11) | 0.875 | MW |
| Latency to 2nd crossing (s) | 52.1 ± 6.1 (18) | 57.5 ± 9.5 (11) | 0.822 | MW |
Behavioral tests were run before surgery on naïve WT and EPAC2−/− female mice. Each value is the mean ± SEM (number of mice tested). Tests: paired t-test (t-test) and non-parametric Mann Whitney U test (MW). EPM, elevated plus maze.
Reliance on an enhancement of brisk withdrawal reflexes as a measure of increased pain sensitivity is problematic, especially after SCI, as sensitization of hindlimb reflexes can be part of spastic syndromes, and reflexive measures may not capture the motivational/affective dimensions of pain (Baastrup et al., 2010, Odem et al., 2019, Yezierski and Vierck, 2010). In contrast, an operant mechanical conflict (MC) test is a paradigm in which voluntary behavior reveals the aversiveness of a rough substrate that animals must cross to explore an unfamiliar setting and to escape from an aversive bright light (Harte et al., 2016, Odem et al., 2019, Pahng et al., 2017). When tested in the MC device, naïve wild type and EPAC2−/− mice crossed the aversive probes a similar number of times, and they spent about the same amount of time on the probes (Table 1). The first and second crossing latency was not significantly different between genotypes. The lack of a difference between the mouse genotypes in their reactions to the aversive probes is consistent with previous studies that found no clear differences between wild-type and EPAC2−/− mice in many behavioral properties (Lee et al., 2015, Srivastava et al., 2012), although explicit investigation of pain-like behavior in EPAC2−/− mice has not been reported.
Because the MC test depends upon proper plantar placement, weight support, and stepping, we required the injured mice to recover to a BMS score which reflected sufficient motor function to readily cross the probes, defined as ≥3 on the BMS (Basso et al., 2006). One day post-surgery, sham-operated mice exhibited complete locomotor function and received a score of 9, whereas SCI mice exhibited slight to no ankle movements and received a score of 0 or 1 (Fig. 3A). Unexpectedly, at 3 weeks post-SCI, only the female mice showed acceptable recovery of plantar placement and stepping behavior, as indicated by a BMS score of 3–4. The male SCI mice only showed active movements of the ankle joints and thus received BMS scores of 2, regardless of genotype (Fig. 3A). Because of this substantial gender difference in locomotor recovery, we only included female test subjects in the following behavioral experiments.
Fig. 3.

SCI induces a similar increase in pain-like behaviors of wild-type, EPAC1−/− and EPAC2−/− mice, as revealed by an operant mechanical conflict test. (A) Sex-specific differences in locomotor recovery post-SCI in mice. Gender significantly affected locomotor recovery post-SCI in mice. The BMS experiments were performed weekly after surgery, and significant differences between males and females were observed as early as 2 weeks post-injury. Mice were monitored during 4 min observation sessions, during which their movements were assessed according to the methodology and BMS scale originally published in (Basso et al., 2006). Statistical comparison was made using 2-way repeated measures ANOVA followed by Sidak tests. (B) Behavioral tests were performed on female mice 3–4 weeks after surgery. SCI significantly decreased the total number of crossings across aversive probes in the mechanical conflict device, and (C) increased the latency to the second complete crossing. Data are shown as mean ± SEM. 2-way ANOVA was used for comparisons of total number of crossings as data were parametric. For comparisons of latencies of second complete crossing, unpaired t-test was used to compare wild-type sham to wild-type SCI mice, as well as wild-type SCI mice to EPAC1−/− while Mann-Whitney U test was performed to compare wild-type SCI mice to EPAC1−/− SCI mice. ANOVA, analysis of variance; BMS, Basso Mouse Scale; EPAC, exchange protein activated by cAMP; KO, knock-out; n.s., non-significant; SCI, spinal cord injury; SEM, standard error of the mean.
The operant MC test, which reflects affective-motivational and cognitive-evaluative dimensions of pain (Harte et al., 2016, Odem et al., 2019, Pahng et al., 2017) revealed enhanced pain-avoidance behavior after SCI and no effects of deleting EPAC2 or EPAC1. SCI wild-type and EPAC2−/− or EPAC1−/− mice crossed the probes less often than sham-operated animals (Fig. 3B), and took significantly longer to complete the second crossing (Fig. 3C), indicating similarly increased pain-avoidance behavior in wild type, EPAC2−/− and EPAC1−/− SCI mice. Together, these data indicate that SCI increases sensitivity to noxious mechanical stimuli under our conditions, and that the enhanced pain-avoidance behavior is not prevented by deletion of either EPAC2 or EPAC1 alone.
3.3. Nociceptor hyperactivity in mice is blocked only by reducing the activity of both EPAC isoforms simultaneously
We had expected that EPAC2−/− mice would be protected against pain-like behaviors, as an EPAC2 inhibitor was able to mitigate SCI-induced SA in rats. Unexpectedly, EPAC2−/−, EPAC1−/− and wild-type mice demonstrated similar enhancement of pain-avoidance behavior after SCI. The lack of effect on pain-related behavior came as a surprise because previous work suggested that EPAC1 plays a major role in persistent inflammatory and neuropathic pain in rodent models (Eijkelkamp et al., 2013, Singhmar et al., 2016). To investigate implications of these apparently discrepant results, we used genetic and pharmacological approaches to determine EPAC1 and EPAC2 contributions to SCI-induced hyperactivity in mouse nociceptors.
Previously we found that presumptive nociceptors (as indicated by capsaicin sensitivity, IB4 binding, and soma diameter ≤30 μm) dissociated from rats comprised two distinct electrophysiological types, with ~70% being nonaccommodating (NA type) and ~30% rapidly accommodating (RA type) (Odem et al., 2018). In contrast, in mice we found that 94% of all DRG neurons with soma diameter ≤30 μm were the NA type and only 6% were the RA type. Here, we only report data from NA neurons. No significant electrophysiological differences were found between neurons isolated from wild-type naïve and sham-operated mice (Table 2), so both conditions were combined for neurons from wild-type mice in subsequent analyses. Data collected from male and female mice were also pooled because no significant differences between genders were found in wild-type naïve/sham and SCI conditions, with the exception of a modest difference in RMP (−54.5 ± 0.7 mV in males versus −58.2 ± 0.7 mV in females, Table 3).
Table 2.
Comparison of electrophysiological measures of excitability in neurons isolated from wild-type naïve and sham-operated mice.
| Measure | Naïve | Sham | P value | Test |
|---|---|---|---|---|
| SA incidence (%) | 5.8 (4/69) | 2.4 (1/41) | 0.649 | Fisher |
| OA incidence (%) | 24.1 (14/58) | 17.1 (7/41) | 0.461 | Fisher |
| RMP (mV) | −56.9 ± 0.7 (69) | −56.6 ± 0.8 (41) | 0.724 | MW |
| AP voltage threshold (mV) | −33.6 ± 0.8 (67) | −35.4 ± 1.0 (41) | 0.409 | MW |
| Rheobase (pA) | 39.4 ± 3.4 (69) | 30.6 ± 3.4 (41) | 0.163 | MW |
For incidence measures each % value is accompanied by a ratio of the number of neurons with SA or OA over the number of neurons sampled. Other values are the mean ± SEM (number of cells sampled). Tests: Fisher's exact test (Fisher), non-parametric Mann Whitney U test (MW). AP, action potential; OA, ongoing activity; RMP, resting membrane potential; SA, spontaneous activity.
Table 3.
Electrophysiological measures of excitability in male and female wild-type mice in naïve/sham and SCI conditions.
| Measure | Male | Female | P value | Test | |
|---|---|---|---|---|---|
| Naïve/Sham | SA incidence (%) | 4.8 (2/42) | 4.4 (3/68) | 1.000 | Fisher |
| OA Incidence (%) | 19.4 (3/31) | 22.1 (15/68) | 0.169 | Fisher | |
| RMP (mV) | −54.5 ± 0.7 (42) | −58.2 ± 0.7 (68) | <0.001 | MW | |
| AP Voltage Threshold (mV) | −34.2 ± 1.0 (40) | −34.4 ± 0.8 (68) | 0.438 | MW | |
| Rheobase (pA) | 33.0 ± 4.1 (42) | 38.0 ± 3.0 (68) | 0.152 | MW | |
| SCI | SA incidence (%) | 48.9 (22/45) | 37.5 (18/48) | 0.300 | Fisher |
| OA Incidence (%) | 77.8 (35/45) | 75.0 (36/48) | 0.810 | Fisher | |
| RMP (mV) | −52.4 ± 1.2 (45) | −53.5 ± 1.1 (48) | 0.465 | t-test | |
| AP Voltage Threshold (mV) | −37.8 ± 1.0 (45) | −37.1 ± 0.8 (47) | 0.509 | MW | |
| Rheobase (pA) | 24.9 ± 4.3 (45) | 21.0 ± 2.1 (48) | 0.738 | MW |
For incidence measures each % value is accompanied by a ratio of the number of neurons with SA or OA over the number of neurons sampled. Other values are the mean ± SEM (number of cells sampled). Tests: Fisher's exact test (Fisher), non-parametric Mann Whitney U test (MW), parametric unpaired t test (t-test). AP, action potential; OA, ongoing activity; RMP, resting membrane potential; SA, spontaneous activity.
Neurons isolated from wild-type mice 8–10 weeks after SCI showed a significant increase in the incidence of SA at RMP (Fig. 4A, B). In addition, each of the three intrinsic electrophysiological alterations that in principle can promote SA were found after SCI: depolarization of RMP (Fig. 4C), hyperpolarization of AP voltage threshold (Fig. 4D), and increased DSF amplitude (see below). SCI-induced hyperexcitability also manifested as a decrease in rheobase (Fig. 4E).
Fig. 4.

Inhibition of both EPAC1 and 2 isoforms is necessary to mitigate SCI-induced hyperexcitability in dissociated mouse DRG neurons. Small to medium-diameter DRG neurons (≤30 µm) harvested from lumbar levels were recorded by whole-cell patch clamp 18–30 h after dissociation. Neurons were pretreated with either vehicle, 10–20 µM CE3F4 or 5 µM ESI-05 for 15–20 min before recording. (A) Representative 10-second recordings obtained from neurons at RMP. Arrows indicate two of the larger DSFs. (B) Nociceptors isolated from EPAC1−/− or EPAC2−/− mice were not protected against increased incidence of SCI-induced SA; additional pharmacological inhibition of the complementary isoform was required to bring SA incidence towards a level comparable to neurons isolated from naïve/sham mice. The ratio above each bar denotes the number of neurons with SA/the number of neurons sampled. Statistical comparisons of SA incidence were made with Bonferroni-corrected Fisher’s exact tests on the indicated pairs. (C) SCI-induced depolarization of the RMP in wild-type and EPAC2−/− mice; additional inhibition of the EPAC1 isoform was required to mitigate SCI-induced depolarization within neurons isolated from EPAC2−/− mice. (D) SCI induced significant hyperpolarization of AP voltage threshold in wild-type and EPAC2−/− mice (trending within EPAC1−/− SCI mice); additional inhibition of the EPAC1 isoform did not cause a further significant change in the SCI-induced AP voltage threshold hyperpolarization within nociceptors from EPAC2−/− mice. (E) SCI decreased rheobase in wild-type and EPAC2−/− mice (trending within EPAC1−/− SCI mice); additional inhibition of the EPAC1 isoform did not cause a further significant change in the SCI-induced decrease in rheobase within nociceptors from EPAC2−/− mice. Comparisons of data (mean ± SEM) were made by t-test (for wild-type data), or Kruskal-Wallis followed by Dunn’s method for pairwise comparisons AP, action potential; DRG, dorsal root ganglion; EPAC, exchange protein activated by cAMP; KO, knock-out; MP, membrane potential; NA, nonaccommodating; n.s., non-significant; SA, spontaneous activity; SCI, spinal cord injury; SEM, standard error of the mean; WT, wild-type.
To examine the contribution of each EPAC isoform to SCI-induced hyperexcitability, we performed pharmacological inhibition of each isoform separately and then with combined inhibitors on nociceptors from WT mice. Neither pretreatment with CE3F4 (10 µM) or ESI-05 (5 µM) reversed the SCI-induced increase in SA incidence, depolarization of RMP, reduction in AP voltage threshold, or reduction in rheobase (Fig. 4B-E). On the other hand, combined treatment with the two EPAC inhibitors significantly reduced the SA incidence and depolarized the AP voltage threshold to levels comparable to those in neurons from uninjured mice (Fig. 4B-E).
These results suggest that SCI-induced SA in mice requires EPAC activity, but EPAC1 and EPAC2 activity are redundant so that activity of either isoform can fulfill this requirement in the absence of the other. To further test this hypothesis, we performed patch-clamp recordings on nociceptors isolated from EPAC2−/− and EPAC1−/− mice. In both genetic models, we observed a significant increase in SA incidence after SCI compared to naïve animals (Fig. 4B). Deletion of EPAC2 did not significantly alter the effects of SCI on electrical properties as compared to WT. Interestingly, even though neurons from EPAC1−/− SCI mice exhibited SA, they failed to show significant depolarization of RMP, hyperpolarization of AP voltage threshold, or reduction in rheobase (Fig. 4C-E), although in each case a trend for a change in the predicted direction could be seen. As a final test of the hypothesis that either EPAC1 or EPAC2 can fulfill the requirement for EPAC activity to maintain SCI-induced SA, we combined pharmacological inhibition of one isoform with genetic deletion of the other isoform. Treatment of nociceptors isolated from EPAC2−/− SCI mice with the EPAC1 inhibitor, CE3F4, modestly decreased the incidence of SA, eliminating statistical significance when compared to the naive EPAC2−/− mice (Fig. 4B). The converse experiment, using nociceptors from EPAC1−/− SCI mice with the EPAC2 inhibitor ESI-05, eliminated the difference in SA compared to the naive EPAC1−/− mice and caused a significant decrease in SA compared to the SCI EPAC1−/− mice (Fig. 4B). The differences between naive and SCI groups in AP voltage threshold (Fig. 4D) and rheobase (Fig. 4E) also were eliminated by combining pharmacological inhibition of one EPAC isoform with genetic deletion of the other isoform (with the exception of rheobase in EPAC2−/− mice). One unpredicted result was that CE3F4 treatment of SCI nociceptors from EPAC2−/− mice further depolarized rather than hyperpolarized the RMP (Fig. 4C). Nonetheless, the SA findings and most of the other findings in this set of experiments support the hypothesis that EPAC1 and EPAC2 activity have redundant functions in mice related to the maintenance of SCI-induced SA and associated hyperexcitability.
Irregular DSFs were recently recognized as modifiable electrophysiological phenomena contributing significantly to nociceptor SA and OA (and related pain) (Odem et al., 2018), and they have not been described previously in mouse nociceptors. We found that DSFs in mouse NA neurons (Fig. 4A) appear indistinguishable from those in rat NA neurons. Neurons from wild-type mice showed no significant effect of sham surgery on DSF amplitudes measured at RMP or when the neuron was artificially depolarized to −45 mV, whereas SCI increased DSF amplitudes when neurons were tested at either of these membrane potentials (Fig. 5A). Importantly, SCI-induced increases in DSF amplitude were not prevented by genetic deletion of EPAC2 or EPAC1 (Fig. 5A).
Fig. 5.

SCI increases the amplitude of DSFs. (A) SCI surgery, but not sham, induced an increase in mean DSF amplitudes recorded at RMP (left) and at −45 mV (right) in presumptive nociceptors isolated from wild-type, EPAC1−/− and EPAC2−/− mice. (B) EPAC1 or EPAC2 deletion had little to no effect on the voltage-dependence of the DSF. (C) Incidence of large subthreshold DSFs (≥8 mV) found in SCI WT mice was not significantly changed in EPAC1−/− or EPAC2−/− SCI mice at RMP and at −45 mV. Suprathreshold DSFs were excluded from analysis. Data are represented as mean ± SEM. Statistical comparisons were performed with Kruskal-Wallis followed by Dunn’s method for pairwise comparisons and Bonferroni-corrected Fisher’s exact test for comparisons of incidences. DSF, depolarizing spontaneous fluctuation; EPAC, exchange protein activated by cAMP; KO, knock-out; SCI, spinal cord injury; SEM, standard error of the mean.
One feature of rat DSFs that contributes to their effectiveness in driving SA is the increase in their amplitude at depolarized RMPs under hyperexcitable conditions (Odem et al., 2018). We found that this voltage dependence of DSFs also occurs after SCI in wild-type mice and in both the EPAC2 and the EPAC1 knockouts, with little difference between genotypes observed, especially in the most depolarized range of subthreshold RMPs (-50 to −41 mV) (Fig. 5B). Large DSFs generated at RMPs within this voltage range would be expected often to exceed AP threshold (which was reduced after SCI, see Fig. 4D). To test whether genetic deletion of EPAC2 or EPAC1 significantly reduces the incidence of large DSFs that are likely to reach AP threshold we selected 8 mV as a minimum amplitude for large DSFs (which would match the difference between the midpoint of −45 mV for the depolarized subthreshold range and the AP threshold of −37 mV after SCI). All the APs generated during SA at RMP or during OA at the −45 mV holding potential appeared to be triggered by DSFs, but to ensure that DSF amplitudes were measured precisely, only large subthreshold DSFs were included in this analysis. No significant differences were found between the incidence of large DSFs after SCI in neurons from wild-type mice and from EPAC2−/− or EPAC1−/− mice at RMP or when depolarized to −45 mV, although there was a possible trend for reduced incidence of large DSFs at RMP in the EPAC1−/− mice (Fig. 5C). The lack of significant differences in large DSFs between wild-type and either EPAC knockout condition reinforces the evidence supporting redundancy between EPAC1 and EPAC2 activity in promoting SA.
Interestingly, although significant SA was found in neurons after SCI in wild-type, EPAC2−/− and EPAC1−/− mice, the overall incidence of SA in mice after SCI was lower than in rats; mice of all genotypes had an SA incidence of 40%, while rats in this study had an SA incidence of 67% (Fisher’s exact test p < 0.0001). SA in vitro probably indicates only part of the pain-related functions of ongoing discharge in nociceptors because it is likely that ongoing discharge (OA) is also promoted in vivo by exposure to humoral signals that depolarize RMP at the cellular sites of AP generation (Walters, 2019). This possibility has received support by an in vitro model, where extrinsic inputs support OA in neurons from naive rats when treated with a low concentration of serotonin combined with artificial depolarization of RMP to −45 mV (Odem et al., 2018). We asked whether, in isolated mouse neurons, prior SCI combined with depolarization of RMP to −45 mV would reveal SCI-dependent OA, and whether this OA would be reduced by genetically deleting EPAC, or by combined genetic and pharmacological interventions that concurrently reduce the activity of both isoforms. SCI dramatically increased the incidence of OA at −45 mV in neurons isolated from wild-type, EPAC1−/−, and EPAC2−/− mice (Fig. 6A, B). Furthermore, neither EPAC1 nor EPAC2 knockout mice were protected against SCI-induced increases in OA. Similar to our observations on SA incidence in Fig. 4, treatment with an EPAC inhibitor of neurons from mice in which the opposite isoform was genetically deleted led to a large, significant decrease in the incidence of OA (Fig. 6B). Together with the results summarized in Fig. 4, these findings provide strong evidence that EPAC1 and EPAC2 in mouse nociceptors either have redundant functions or can be induced to compensate for the deleted isoform.
Fig. 6.

Inhibition of both EPAC1 and 2 is necessary to mitigate SCI-induced OA in presumptive mouse nociceptors. To measure extrinsically driven OA, small DRG neurons were artificially depolarized to −45 mV by current injection for 30–60 s. DRG neurons isolated from EPAC1−/− or EPAC2−/− mice were not protected against SCI-induced OA; additional inhibition of the complementary isoform was required to reverse the effect of the injury. (A) Representative 10-second recordings obtained from neurons artificially held at −45 mV. APs are clipped at 0 mV to allow sufficient magnification for clear display of DSFs. (B) Incidence of OA measured at a holding potential of −45 mV. The ratio above each bar denotes the number of neurons with OA/the number of neurons sampled. Statistical comparisons of OA incidence were made with Bonferroni-corrected Fisher’s exact tests on the indicated pairs. DRG, dorsal root ganglion; EPAC, exchange protein activated by cAMP; KO, knock-out; OA, ongoing activity; MP, membrane potential; SCI, spinal cord injury; WT, wild-type.
3.4. EPAC1 expression levels are unchanged in rat and EPAC2−/− mice post SCI
Previous reports have described conflicting effects of various injuries on EPAC expression levels in rat DRGs, with CFA-induced inflammation reported to increase only Epac2 mRNA and protein levels in DRG neurons (Vasko et al., 2014) or both EPAC1 and 2 protein levels in the DRG (Gu et al., 2016), and a skin/muscle incision and retraction injury model reported to only increase EPAC1 protein in the DRG (Cao et al., 2016). Our electrophysiological data from rat neurons suggest that both EPAC1 and EPAC2 expression might be affected by SCI. However, we found no significant differences in EPAC1 or EPAC2 protein levels in DRGs from naïve, sham-operated, and SCI rats (Fig. 7A, B). Previous work in a mouse model of spinal nerve transection (Eijkelkamp et al., 2013) found increased Epac1 mRNA expression in DRGs. This suggested that SCI might increase EPAC1 protein expression in DRGs from wild-type mice. Again, however, we did not observe significant increases in either EPAC1 or EPAC2 protein levels after SCI in wild-type or EPAC2−/− mice (Fig. 7C, D). Nor did we observe an increase in EPAC1 protein levels in DRGs from naïve EPAC2−/− mice compared to wild-type mice (1.0 +/- 0.4; standard deviation; n = 3). No change of EPAC2 protein levels have been reported by other groups in EPAC1−/− mice in lungs (Oldenburger et al., 2014), hippocampus (Yan et al., 2013, Zhou et al., 2016) and pancreas (Song et al., 2013).
Fig. 7.

EPAC1 and 2 expression levels are unchanged after SCI in DRGs isolated from mice and rats. (A) EPAC1 and 2 expression levels were not significantly different in naïve versus SCI rats. (B) Bar graph represents band density for EPAC1 or EPAC2 levels normalized to total actin, with levels in the first lane (Naïve) set to 1. Naïve n = 4; Sham n = 5; SCI n = 3. Comparisons of data (mean ± SEM) were made by One-way ANOVA or Kruskal-Wallis followed by Dunn’s method for pairwise comparisons. (C) EPAC1 and 2 expression levels were not significantly different in wild-type sham versus SCI mice or EPAC2−/− naïve versus SCI mice. (D) Bar graph represents band density for EPAC1 or EPAC2 levels normalized to total protein as measured by stain-free imaging, with levels in the first lane of each blot set to 1. Wild-type Sham n = 4; wild-type SCI n = 5: EPAC2−/− naïve n = 7; EPAC2−/− SCI n = 6 Comparisons of data (mean ± SEM) were made by t-test. ANOVA, analysis of variance; DRG, dorsal root ganglion; EPAC, exchange protein activated by cAMP; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; n.s., non-significant; SCI, spinal cord injury; SEM, standard error of the mean; WT, wild-type.
These protein expression data suggest that functional redundancy rather than knockout- or injury-induced increases in expression of EPAC isoforms explains the lack of effects of EPAC1 or EPAC2 knockout on pain-avoidance behavior after SCI and the failure of interventions that target either isoform individually (in contrast to dual targeting) to attenuate SCI-induced hyperactivity in nociceptors.
4. Discussion
Here we demonstrate the importance of both EPAC isoforms (EPAC1 and EPAC2) in maintaining hyperactivity in probable nociceptors after SCI in two different rodent species. In rats, inhibition of either EPAC isoform was sufficient to mitigate SCI-induced hyperactivity, indicating that activity in both is necessary for the hyperactivity. In mice, knockout of neither EPAC1 nor EPAC2 was sufficient to ameliorate SCI-induced hyperactivity, which our evidence suggests is because of redundant signaling by the two EPAC isoforms (Fig. 8).
Fig. 8.

Comparison of the contributions of cAMP effectors in rat and mouse nociceptors to the maintenance of SCI-induced hyperactivity (SA and OA) and links to ongoing pain. In rat nociceptors, pharmacological inhibition (blue x's) of either EPAC isoform is sufficient to block the hyperactivity. In mouse nociceptors, block of both EPAC isoforms is necessary to block the hyperactivity, either by combining two EPAC inhibitors or an inhibitor for one isoform with genetic deletion of the second isoform (red x). The necessity of PKA in rat nociceptors for hyperactivity after SCI was reported previously (Bavencoffe et al., 2016), while the necessity of HCN channels for similar hyperexcitability has been reported in other injury models (Bernal and Roza, 2018, Djouhri et al., 2015, Djouhri et al., 2018, Emery et al., 2011, Young et al., 2014) and observed after SCI (A.G. Bavencoffe, C.W. Dessauer and E.T. Walters, unpublished observations). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4.1. Roles of EPAC1 and EPAC2 in maintaining nociceptor hyperactivity in rats
Recent work has yielded conflicting views of the roles of EPAC1 versus EPAC2 in pain signaling in rats (Cao et al., 2016, Gu et al., 2016, Vasko et al., 2014). Gu et al. (2016) observed increased expression of both isoforms in the DRG during CFA-induced inflammation, while others have demonstrated an increase in only EPAC2 expression using the same rat inflammatory model (Vasko et al., 2014). In contrast, EPAC1 expression increases in DRGs in a postsurgical pain model, and inhibition of EPAC1 yields a robust reduction of pain-like behaviors (Cao et al., 2016), which has not been reported for EPAC2 inhibition. Here we show that while neither EPAC1 nor EPAC2 protein levels increased after SCI, both EPAC isoforms play important roles in SCI-induced hyperactivity in small dissociated DRG neurons that are primarily nociceptors. Selective pharmacological inhibition of either EPAC isoform completely reversed SCI-induced SA in rat neurons. In addition, each inhibitor significantly reversed the SCI-induced depolarization of RMP and the increase in DSF amplitude measured at relatively depolarized RMPs. Much weaker, although functionally parallel, effects were indicated for the EPAC inhibitors on rheobase and AP threshold. Thus, in rat nociceptors, inhibition of either EPAC isoform can affect multiple aspects of nociceptor excitability to reduce SA, indicating an essential role for both EPAC1 and EPAC2 in maintaining persistent hyperactivity after SCI. However, we also found that pharmacological activation of both EPACs is not sufficient by itself to induce SA or OA, despite depolarizing the RMP, reducing the AP threshold, and possibly increasing DSF amplitudes. This result is consistent with an important role for other cell signals, notably PKA (Bavencoffe et al., 2016), working with EPACs to maintain nociceptor hyperactivity.
4.2. Redundant nociceptor functions for EPAC1 and EPAC2 in mice
Previous studies have reported that EPAC1−/− mice are protected against inflammatory (Eijkelkamp et al., 2013, Singhmar et al., 2016, Wang et al., 2013) and neuropathic pain (Eijkelkamp et al., 2013, Singhmar et al., 2018), but possible roles for EPAC2 are unknown. We show that EPAC2−/− mice do not differ from their wild-type counterparts for general behavioral functions as found previously for EPAC1−/− mice (Russart et al., 2018). While our results are in accord with previous publications (Lee et al., 2015, Srivastava et al., 2012), another study reported that EPAC2−/− mice exhibit signs of anxiety (Zhou et al., 2016). While this result differs from our observations, the anxiety tests were not the same; we used the elevated plus maze, while Zhou et al. opted for the open field test.
Here, we show that knockout of either EPAC1 or EPAC2 alone fails to mitigate SCI-induced pain-like behavior in mice. While this result might be explained by minimal contributions of EPAC2 to SCI-induced pain, it also could be explained either by compensatory enhancement of expression of the other EPAC isoform or by redundancy of the signaling functions of the two EPACs for maintaining nociceptor hyperactivity. EPAC proteins show functional redundancy in the brain related to long-term potentiation, spatial learning, and social interactions (Yang et al., 2012), while we know of no reports of significant compensatory changes in EPAC protein expression reported after knockout of one isoform (Srivastava et al., 2012). Our finding of no significant changes in EPAC1 protein after EPAC2 knockout provides evidence against the possibility of compensatory increases in expression of the other EPAC isoform after genetic deletion of one isoform. In addition, we found no significant alterations in the protein expression of either EPAC isoform in wild-type mice produced by SCI.
Our electrophysiological findings support redundant (although not identical) functions of EPAC1 and EPAC2 in maintaining hyperactivity in mouse nociceptors. Redundancy is strongly indicated by several complementary findings. First, treatment with selective inhibitors of EPAC1 activity or EPAC2 activity that we found to be effective in reversing SA in rat nociceptors failed to reverse SCI-induced SA and OA in mouse nociceptors. Second, knockout of neither EPAC1 nor EPAC2 significantly attenuated SCI-induced SA or OA. Third, individually applied EPAC inhibitors as well as the individual EPAC knockouts had relatively little effect on other hyperexcitable properties that promote SA and OA after SCI, including depolarized RMP, reduced AP voltage threshold, and (for the knockouts) increased DSF amplitude and increased frequency of large DSFs. Fourth, combined application of the EPAC1 and EPAC2 inhibitors strongly attenuated SA and OA as well as most of the other hyperexcitable properties. Fifth, combining pharmacological inhibition of one EPAC isoform with genetic deletion of the other isoform nearly eliminated the SCI-induced SA and OA as well as most of the other alterations induced by SCI. Thus, we conclude that, while EPAC signaling plays very similar roles in maintaining SCI-induced nociceptor hyperactivity in mice and rats, the two isoforms have redundant roles in mouse nociceptors whereas they have individually essential roles in rat nociceptors.
The functions of EPAC1 and EPAC2 in maintaining nociceptor hyperactivity in mice are similar but not identical. We found significant differences after SCI in rheobase and AP voltage threshold in neurons isolated from EPAC2−/− mice but not from EPAC1−/− mice. This suggests that EPAC1 may contribute more than EPAC2 to SCI-induced effects on nociceptors, which may help to explain previously reported differences in the contributions of EPAC1 and EPAC2 to inflammatory and neuropathic pain models in rodents. An interesting question is whether differences in the functions of EPAC1 and EPAC2 might contribute to the differences we found between our selected rodent species in the electrophysiological effects of SCI. Notably, compared to rats, wild-type mice exhibited a substantially lower incidence of SA in neurons and less depolarization of RMP in neurons after SCI (compared to naïve, 10% depolarization in SCI rats versus 6% in SCI mice, p < 0.032, Mann Whitney).
4.3. SCI enhances pain-avoidance behavior in mice despite EPAC1 or EPAC2 deletion
An important question is how EPAC-dependent regulation of nociceptor excitability affects pain-related behavior. Reliance on tests measuring brisk withdrawal reflexes to assess pain in animals has received increasing scrutiny because of the tenuous relationship between reflex responses and the affective component of pain. Reflexive measures for SCI pain are especially problematic because hyperreflexia occurs in spastic syndromes secondary to SCI, independent of increases in pain (Baastrup et al., 2010, Yezierski and Vierck, 2010). As an alternative to determine the effects of EPAC1 or EPAC2 genetic deletion on pain sensitivity, we opted for an operant mechanical conflict (MC) test that assesses voluntary behavior revealing the affective-motivational and cognitive-evaluative dimensions of pain (Harte et al., 2016, Pahng et al., 2017). Our results with the MC test revealed significant effects of SCI on mouse pain-avoidance behavior. As described in the methods, we modified the conventional MC test (Harte et al., 2016) for use with mice (Shepherd and Mohapatra, 2018, Zhou and Carlton, 2012) with a novel design and test procedure that took advantage of the innate exploratory drive of the mouse to reveal enhanced avoidance of aversive probes long after SCI. These results indicate that SCI produced persistent mechanical hyperalgesia. Consistent with our electrophysiological evidence for redundancy or SCI-induced compensation by the other isoform of EPAC after SCI, deletion of neither EPAC1 nor EPAC2 in mice had any apparent effect on enhanced pain avoidance induced by SCI.
4.4. EPAC and PKA signaling in nociceptors during chronic pain
Signaling by cAMP-PKA has been well established as a driver of sensory neuron sensitization (Bavencoffe et al., 2016, Efendiev et al., 2013, Li et al., 2019, Song et al., 2006, Villarreal et al., 2009). Much less is known about the roles of another downstream effector, EPAC (Hucho and Levine, 2007). In nociceptors isolated from SCI rats, we found a decrease in hyperactivity after EPAC inhibition paralleling that seen in our previous study of PKA contributions (Bavencoffe et al., 2016). Notably different, however, was the profound hyperpolarization of RMP after EPAC inhibition. Since EPAC activation by 007-AM did not induce a hyperexcitable state comparable to the one observed after SCI, it is possible that both cAMP-dependent signaling pathways, PKA and EPAC, work in tandem to promote different components of nociceptor hyperexcitability, with EPAC potentially playing a larger role in regulating resting membrane potential (Fig. 8). Other studies have suggested that cAMP signaling biased towards either PKA or EPAC-PKCε promotes different phases of pain (Eijkelkamp et al., 2010, Huang et al., 2015, Wang et al., 2007). Several examples of synergy between PKA and EPAC have been reported previously (Hewer et al., 2011, Yu et al., 2017), including one proposing cooperative roles in sensory neuron sprouting and neurite extension in the spinal cord after SCI (Wei et al., 2016).
The compensatory and/or redundant relationships between EPAC1 and 2 suggested by our results (Fig. 8) have implications when considering cooperative roles of EPAC and PKA, especially in the context of targeted pain therapies. While knockout of EPAC1 has been shown to protect against persistent neuropathic and inflammatory allodynia (Eijkelkamp et al., 2013, Singhmar et al., 2016), EPAC2 interacts with multiple signaling complexes coordinating neurotransmission and exocytosis (Shibasaki et al., 2004, Shibasaki et al., 2007, Ster et al., 2007), which probably play a role in nociceptive transmission and excitability in pain pathways. Both isoforms could also potentially influence any regeneration that occurs after nerve injury, as EPAC1 contributes to primary axon development, while mature neurons control dendrite stability and outgrowth through elevated EPAC2 levels (Murray and Shewan, 2008). In addition, despite employing different means of translocation, EPAC1 and 2 can both translocate to the plasma membrane to activate Rap and signal to downstream components involved in pain signaling, such as PKCε (Li et al., 2006). Isoform-specific mechanisms could potentially be hijacked to compensate for the absence of either EPAC isoform and might affect synergistic signaling between EPAC and PKA.
In conclusion, both EPAC1 and EPAC2 contribute to nociceptor hyperactivity that promotes chronic pain after SCI in rats and mice. This finding adds to accumulating evidence that EPAC signaling may be a plausible therapeutic target for chronic pain. However, the functional redundancy of these EPAC isoforms must be considered when developing treatments to target EPAC effectively in nociceptors.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors thank Dr. Fang C. Mei for her expert technical assistance and for her management of the EPAC1 and 2 mouse colonies; she supplied all of the mice used in this project. We also thank Dr. Susan M. Carlton for the use of her custom MC test tunnel adaptor and Dr. Steven Katzen for his assistance with rat surgeries. This work was supported by National Institute of Neurological Diseases and Stroke Grant NS091759 to C.W. Dessauer and E.T. Walters, National Institute of Health Grant R35GM122536 to Xiaodong Cheng, and a grant from Mission Connect, a program of TIRR Foundation to C.W. Dessauer and J. Herrera.
References
- Acharjee S., Nayani N., Tsutsui M., Hill M.N., Ousman S.S., Pittman Q.J. Altered cognitive-emotional behavior in early experimental autoimmune encephalitis – Cytokine and hormonal correlates. Brain Behav. Immun. 2013;33:164–172. doi: 10.1016/j.bbi.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Baastrup C., Maersk-Moller C.C., Nyengaard J.R., Jensen T.S., Finnerup N.B. Spinal-, brainstem- and cerebrally mediated responses at- and below-level of a spinal cord contusion in rats: evaluation of pain-like behavior. Pain. 2010;151:670–679. doi: 10.1016/j.pain.2010.08.024. [DOI] [PubMed] [Google Scholar]
- Basso D.M., Beattie M.S., Bresnahan J.C. A sensitive and reliable locomotor rating scale for open field testing in rats. J. Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Basso D.M., Fisher L.C., Anderson A.J., Jakeman L.B., McTigue D.M., Popovich P.G. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J. Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bavencoffe A., Li Y., Wu Z., Yang Q., Herrera J., Kennedy E.J., Walters E.T., Dessauer C.W. Persistent Electrical Activity in Primary Nociceptors after Spinal Cord Injury Is Maintained by Scaffolded Adenylyl Cyclase and Protein Kinase A and Is Associated with Altered Adenylyl Cyclase Regulation. J. Neurosci. 2016;36:1660–1668. doi: 10.1523/JNEUROSCI.0895-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi S., Yang Q., Crook R., Du J., Wu Z., Fishman H., Grill R., Carlton S., Walters E. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J. Neurosci. 2010;30:14870–14882. doi: 10.1523/JNEUROSCI.2428-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal L., Roza C. Hyperpolarization-activated channels shape temporal patterns of ectopic spontaneous discharge in C-nociceptors after peripheral nerve injury. Eur. J. Pain. 2018 doi: 10.1002/ejp.1226. [DOI] [PubMed] [Google Scholar]
- Burke D., Fullen B.M., Stokes D., Lennon O. Neuropathic pain prevalence following spinal cord injury: A systematic review and meta-analysis. Eur. J. Pain. 2017;21:29–44. doi: 10.1002/ejp.905. [DOI] [PubMed] [Google Scholar]
- Cao S., Bian Z., Zhu X., Shen S.R. Effect of Epac1 on pERK and VEGF Activation in Postoperative Persistent Pain in Rats. J. Mol. Neurosci.: MN. 2016;59:554–564. doi: 10.1007/s12031-016-0776-x. [DOI] [PubMed] [Google Scholar]
- Chaplan S.R., Bach F.W., Pogrel J.W., Chung J.M., Yaksh T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen H., Tsalkova T., Chepurny O.G., Mei F.C., Holz G.G., Cheng X., Zhou J. Identification and characterization of small molecules as potent and specific EPAC2 antagonists. J. Med. Chem. 2013;56:952–962. doi: 10.1021/jm3014162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courilleau D., Bisserier M., Jullian J.C., Lucas A., Bouyssou P., Fischmeister R., Blondeau J.P., Lezoualc'h F. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J. Biol. Chem. 2012;287:44192–44202. doi: 10.1074/jbc.M112.422956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhamer J., Lucas J., Zelaya C., Nahin R., Mackey S., DeBar L., Kerns R., Von Korff M., Porter L., Helmick C. Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults – United States, 2016. MMWR Morb. Mortal. Wkly Rep. 2018;67:1001–1006. doi: 10.15585/mmwr.mm6736a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouhri L., Al Otaibi M., Kahlat K., Smith T., Sathish J., Weng X. Persistent hindlimb inflammation induces changes in activation properties of hyperpolarization-activated current (Ih) in rat C-fiber nociceptors in vivo. Neuroscience. 2015;301:121–133. doi: 10.1016/j.neuroscience.2015.05.074. [DOI] [PubMed] [Google Scholar]
- Djouhri L., Smith T., Ahmeda A., Alotaibi M., Weng X. Hyperpolarization-activated cyclic nucleotide-gated channels contribute to spontaneous activity in L4 C-fiber nociceptors, but not Abeta-non-nociceptors, after axotomy of L5-spinal nerve in the rat in vivo. Pain. 2018;159:1392–1402. doi: 10.1097/j.pain.0000000000001224. [DOI] [PubMed] [Google Scholar]
- Efendiev R., Bavencoffe A., Hu H., Zhu M.X., Dessauer C.W. Scaffolding by A-kinase anchoring protein enhances functional coupling between adenylyl cyclase and TRPV1 channel. J. Biol. Chem. 2013;288:3929–3937. doi: 10.1074/jbc.M112.428144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N., Linley J.E., Torres J.M., Bee L., Dickenson A.H., Gringhuis M., Minett M.S., Hong G.S., Lee E., Oh U. A role for Piezo2 in EPAC1-dependent mechanical allodynia. Nat. Commun. 2013;4:1682. doi: 10.1038/ncomms2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N., Wang H., Garza-Carbajal A., Willemen H., Zwartkruis F., Wood J., Dantzer R., Kelley K., Heijnen C., Kavelaars A. Low nociceptor GRK2 prolongs prostaglandin E2 hyperalgesia via biased cAMP signaling to Epac/Rap1, protein kinase Cepsilon, and MEK/ERK. J. Neurosci. 2010;30:12806–12815. doi: 10.1523/JNEUROSCI.3142-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery E.C., Young G.T., Berrocoso E.M., Chen L., McNaughton P.A. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. 2011;333:1462–1466. doi: 10.1126/science.1206243. [DOI] [PubMed] [Google Scholar]
- Finnerup N., Baastrup C. Spinal Cord Injury Pain: Mechanisms and Management. Curr. Pain Headache Rep. 2012;16:207–216. doi: 10.1007/s11916-012-0259-x. [DOI] [PubMed] [Google Scholar]
- Finnerup N.B. Pain in patients with spinal cord injury. Pain. 2013;154(Suppl 1):S71–S76. doi: 10.1016/j.pain.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Gu Y., Li G., Chen Y., Huang L.Y. Epac-protein kinase C alpha signaling in purinergic P2X3R-mediated hyperalgesia after inflammation. Pain. 2016;157:1541–1550. doi: 10.1097/j.pain.0000000000000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K., Dubner R., Brown F., Flores C., Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Harte S.E., Meyers J.B., Donahue R.R., Taylor B.K., Morrow T.J. Mechanical Conflict System: A Novel Operant Method for the Assessment of Nociceptive Behavior. PLoS ONE. 2016;11 doi: 10.1371/journal.pone.0150164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch M.N., Cushing T.R., Carlson G.D., Chang E.Y. Neuropathic pain and SCI: Identification and treatment strategies in the 21st century. J. Neurol. Sci. 2018;384:75–83. doi: 10.1016/j.jns.2017.11.018. [DOI] [PubMed] [Google Scholar]
- Herrera J.J., Chacko T., Narayana P.A. Histological correlation of diffusion tensor imaging metrics in experimental spinal cord injury. J. Neurosci. Res. 2008;86:443–447. doi: 10.1002/jnr.21481. [DOI] [PubMed] [Google Scholar]
- Herrera J.J., Sundberg L.M., Zentilin L., Giacca M., Narayana P.A. Sustained expression of vascular endothelial growth factor and angiopoietin-1 improves blood-spinal cord barrier integrity and functional recovery after spinal cord injury. J. Neurotrauma. 2010;27:2067–2076. doi: 10.1089/neu.2010.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewer R.C., Sala-Newby G.B., Wu Y.-J., Newby A.C., Bond M. PKA and Epac synergistically inhibit smooth muscle cell proliferation. J. Mol. Cell. Cardiol. 2011;50:87–98. doi: 10.1016/j.yjmcc.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.Y., Dai S.P., Chang Y.C., Sun W.H. Acidosis Mediates the Switching of Gs-PKA and Gi-PKCepsilon Dependence in Prolonged Hyperalgesia Induced by Inflammation. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0125022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T., Dina O., Levine J. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J. Neurosci. 2005;25:6119–6126. doi: 10.1523/JNEUROSCI.0285-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T., Levine J.D. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Lee K., Kobayashi Y., Seo H., Kwak J.H., Masuda A., Lim C.S., Lee H.R., Kang S.J., Park P., Sim S.E. Involvement of cAMP-guanine nucleotide exchange factor II in hippocampal long-term depression and behavioral flexibility. Mol. Brain. 2015;8:38. doi: 10.1186/s13041-015-0130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Asuri S., Rebhun J.F., Castro A.F., Paranavitana N.C., Quilliam L.A. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J. Biol. Chem. 2006;281:2506–2514. doi: 10.1074/jbc.M508165200. [DOI] [PubMed] [Google Scholar]
- Li Z.H., Cui D., Qiu C.J., Song X.J. Cyclic nucleotide signaling in sensory neuron hyperexcitability and chronic pain after nerve injury. Neurobiol. Pain. 2019;6 doi: 10.1016/j.ynpai.2019.100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M., Oh-Hashi K., Yokota I., Sawa T., Amaya F. Acquired Exchange Protein Directly Activated by Cyclic Adenosine Monophosphate Activity Induced by p38 Mitogen-activated Protein Kinase in Primary Afferent Neurons Contributes to Sustaining Postincisional Nociception. Anesthesiology. 2017;126:150–162. doi: 10.1097/ALN.0000000000001401. [DOI] [PubMed] [Google Scholar]
- Murray A., Shewan D. Epac mediates cyclic AMP-dependent axon growth, guidance and regeneration. Mol. Cell. Neurosci. 2008;38:578–588. doi: 10.1016/j.mcn.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Nyuyki K.D., Cluny N.L., Swain M.G., Sharkey K.A., Pittman Q.J. Altered Brain Excitability and Increased Anxiety in Mice With Experimental Colitis: Consideration of Hyperalgesia and Sex Differences. Front. Behav. Neurosci. 2018;12:58. doi: 10.3389/fnbeh.2018.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odem M.A., Bavencoffe A.G., Cassidy R.M., Lopez E.R., Tian J., Dessauer C.W., Walters E.T. Isolated nociceptors reveal multiple specializations for generating irregular ongoing activity associated with ongoing pain. Pain. 2018;159:2347–2362. doi: 10.1097/j.pain.0000000000001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odem M.A., Lacagnina M.J., Katzen S.L., Li J., Spence E.A., Grace P.M., Walters E.T. Sham surgeries for central and peripheral neural injuries persistently enhance pain-avoidance behavior as revealed by an operant conflict test. Pain. 2019 doi: 10.1097/j.pain.0000000000001642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburger A., Timens W., Bos S., Smit M., Smrcka A.V., Laurent A.C., Cao J., Hylkema M., Meurs H., Maarsingh H. Epac1 and Epac2 are differentially involved in inflammatory and remodeling processes induced by cigarette smoke. FASEB J. 2014;28:4617–4628. doi: 10.1096/fj.13-248930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahng A.R., Paulsen R.I., McGinn M.A., Edwards K.N., Edwards S. Neurobiological Correlates of Pain Avoidance-Like Behavior in Morphine-Dependent and Non-Dependent Rats. Neuroscience. 2017;366:1–14. doi: 10.1016/j.neuroscience.2017.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellow S., Chopin P., File S.E., Briley M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J. Neurosci. Methods. 1985;14:149–167. doi: 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- Pereira L., Cheng H., Lao D.H., Na L., van Oort R.J., Brown J.H., Wehrens X.H.T., Chen J., Bers D.M. Epac2 Mediates Cardiac β1-Adrenergic Dependent SR Ca(2+) Leak and Arrhythmia. Circulation. 2013;127:913–922. doi: 10.1161/CIRCULATIONAHA.12.148619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas G., Guerra M.J., Labandeira-Garcia J.L. An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res. Protocols. 1997;2:75–84. doi: 10.1016/s1385-299x(97)00034-2. [DOI] [PubMed] [Google Scholar]
- Russart K.L.G., Huk D., Nelson R.J., Kirschner L.S. Elevated aggressive behavior in male mice with thyroid-specific Prkar1a and global Epac1 gene deletion. Horm. Behav. 2018;98:121–129. doi: 10.1016/j.yhbeh.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shariati B., Thompson E.L., Nicol G.D., Vasko M.R. Epac activation sensitizes rat sensory neurons through activation of Ras. Mol. Cell. Neurosci. 2016;70:54–67. doi: 10.1016/j.mcn.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd A.J., Mohapatra D.P. Pharmacological validation of voluntary gait and mechanical sensitivity assays associated with inflammatory and neuropathic pain in mice. Neuropharmacology. 2018;130 doi: 10.1016/j.neuropharm.2017.11.036. 18–29.https: //doi.org/10.1016/j.neuropharm.2017.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki T., Sunaga Y., Fujimoto K., Kashima Y., Seino S. Interaction of ATP sensor, cAMP sensor, Ca2+ sensor, and voltage-dependent Ca2+ channel in insulin granule exocytosis. J. Biol. Chem. 2004;279:7956–7961. doi: 10.1074/jbc.M309068200. [DOI] [PubMed] [Google Scholar]
- Shibasaki T., Takahashi H., Miki T., Sunaga Y., Matsumura K., Yamanaka M., Zhang C., Tamamoto A., Satoh T., Miyazaki J. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddall P.J., McClelland J.M., Rutkowski S.B., Cousins M.J. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003:249–257. doi: 10.1016/S0304-3959(02)00452-9. [DOI] [PubMed] [Google Scholar]
- Singhmar P., Huo X., Eijkelkamp N., Berciano S.R., Baameur F., Mei F.C., Zhu Y., Cheng X., Hawke D., Mayor F. Critical role for Epac1 in inflammatory pain controlled by GRK2-mediated phosphorylation of Epac1. Proc. Natl. Acad. Sci. 2016;113:3036–3041. doi: 10.1073/pnas.1516036113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhmar P., Huo X., Li Y., Dougherty P.M., Mei F., Cheng X., Heijnen C.J., Kavelaars A. Orally active Epac inhibitor reverses mechanical allodynia and loss of intraepidermal nerve fibers in a mouse model of chemotherapy-induced peripheral neuropathy. Pain. 2018 doi: 10.1097/j.pain.0000000000001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonawane Y.A., Zhu Y., Garrison J.C., Ezell E.L., Zahid M., Cheng X., Natarajan A. Structure-Activity Relationship Studies with Tetrahydroquinoline Analogs as EPAC Inhibitors. ACS Med. Chem. Lett. 2017;8:1183–1187. doi: 10.1021/acsmedchemlett.7b00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.J., Mondal P., Li Y., Lee S.E., Hussain M.A. Pancreatic beta-cell response to increased metabolic demand and to pharmacologic secretagogues requires EPAC2A. Diabetes. 2013;62:2796–2807. doi: 10.2337/db12-1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X.J., Wang Z.B., Gan Q., Walters E.T. cAMP and cGMP contribute to sensory neuron hyperexcitability and hyperalgesia in rats with dorsal root ganglia compression. J. Neurophysiol. 2006;95:479–492. doi: 10.1152/jn.00503.2005. [DOI] [PubMed] [Google Scholar]
- Srivastava D.P., Jones K.A., Woolfrey K.M., Burgdorf J., Russell T.A., Kalmbach A., Lee H., Yang C., Bradberry M.M., Wokosin D. Social, communication, and cortical structural impairments in Epac2-deficient mice. J. Neurosci. 2012;32:11864–11878. doi: 10.1523/JNEUROSCI.1349-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ster J., De Bock F., Guerineau N.C., Janossy A., Barrere-Lemaire S., Bos J.L., Bockaert J., Fagni L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2519–2524. doi: 10.1073/pnas.0611031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T., Mei F.C., Li S., Chepurny O.G., Leech C.A., Liu T., Holz G.G., Woods V.L., Cheng X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc. Natl. Acad. Sci. 2012;109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko M.R., Habashy Malty R., Guo C., Duarte D.B., Zhang Y., Nicol G.D. Nerve growth factor mediates a switch in intracellular signaling for PGE2-induced sensitization of sensory neurons from protein kinase A to Epac. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0104529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal C.F., Sachs D., Funez M.I., Parada C.A., de Queiroz Cunha F., Ferreira S.H. The peripheral pro-nociceptive state induced by repetitive inflammatory stimuli involves continuous activation of protein kinase A and protein kinase C epsilon and its Na(V)1.8 sodium channel functional regulation in the primary sensory neuron. Biochem. Pharmacol. 2009;77:867–877. doi: 10.1016/j.bcp.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Walters E.T. Nociceptors as chronic drivers of pain and hyperreflexia after spinal cord injury: an adaptive-maladaptive hyperfunctional state hypothesis. Front. Physiol. 2012;3:309. doi: 10.3389/fphys.2012.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters E.T. Adaptive mechanisms driving maladaptive pain: how chronic ongoing activity in primary nociceptors can enhance evolutionary fitness after severe injury. Philos. Trans. R. Soc. B: Biol. Sci. 2019;374:20190277. doi: 10.1098/rstb.2019.0277. https://royalsocietypublishing.org/doi/abs/10.1098/rstb.2019.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Gu Y., Li G., Huang L. A critical role of the cAMP sensor Epac in switching protein kinase signalling in prostaglandin E2-induced potentiation of P2X3 receptor currents in inflamed rats. J. Physiol. 2007;584:191–203. doi: 10.1113/jphysiol.2007.135616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Heijnen C.J., van Velthoven C.T.J., Willemen H.L.D.M., Ishikawa Y., Zhang X., Sood A.K., Vroon A., Eijkelkamp N., Kavelaars A. Balancing GRK2 and EPAC1 levels prevents and relieves chronic pain. J. Clin. Investig. 2013;123:5023–5034. doi: 10.1172/JCI66241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D., Hurd C., Galleguillos D., Singh J., Fenrich K.K., Webber C.A., Sipione S., Fouad K. Inhibiting cortical protein kinase A in spinal cord injured rats enhances efficacy of rehabilitative training. Exp. Neurol. 2016;283:365–374. doi: 10.1016/j.expneurol.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Wu Z., Yang Q., Crook R.J., O'Neil R.G., Walters E.T. TRPV1 Channels Make Major Contributions to Behavioral Hypersensitivity and Spontaneous Activity in Nociceptors After Spinal Cord Injury. Pain. 2013;154:2130–2141. doi: 10.1016/j.pain.2013.06.040. [DOI] [PubMed] [Google Scholar]
- Yan J., Mei F.C., Cheng H., Lao D.H., Hu Y., Wei J., Patrikeev I., Hao D., Stutz S.J., Dineley K.T. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Mol. Cell. Biol. 2013;33:918–926. doi: 10.1128/MCB.01227-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Wu Z., Hadden J.K., Odem M.A., Zuo Y., Crook R.J., Frost J.A., Walters E.T. Persistent pain after spinal cord injury is maintained by primary afferent activity. J. Neurosci. 2014;34:10765–10769. doi: 10.1523/JNEUROSCI.5316-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Shu X., Liu D., Shang Y., Wu Y., Pei L., Xu X., Tian Q., Zhang J., Qian K. EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron. 2012;73:774–788. doi: 10.1016/j.neuron.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yezierski R.P., Vierck C.J. Reflex and pain behaviors are not equivalent: lessons from spinal cord injury. Pain. 2010;151:569–570. doi: 10.1016/j.pain.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Young G.T., Emery E.C., Mooney E.R., Tsantoulas C., McNaughton P.A. Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels. Pain. 2014;155:1708–1719. doi: 10.1016/j.pain.2014.05.021. [DOI] [PubMed] [Google Scholar]
- Yu X., Zhang Q., Zhao Y., Schwarz B.J., Stallone J.N., Heaps C.L., Han G. Activation of G protein-coupled estrogen receptor 1 induces coronary artery relaxation via Epac/Rap1-mediated inhibition of RhoA/Rho kinase pathway in parallel with PKA. PLoS ONE. 2017;12 doi: 10.1371/journal.pone.0173085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Ma S.L., Yeung P.K., Wong Y.H., Tsim K.W., So K.F., Lam L.C., Chung S.K. Anxiety and depression with neurogenesis defects in exchange protein directly activated by cAMP 2-deficient mice are ameliorated by a selective serotonin reuptake inhibitor. Prozac. Transl Psychiatry. 2016;6 doi: 10.1038/tp.2016.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Carlton S. A novel operant method to test acute mechanical hypersensitivity in mice using a modification of the Coy Mechanical Conflict-Avoidance System. Soc. Neurosci. Abstr. 2012 80.03. [Google Scholar]