

Abstract
Photoreceptor phosphodiesterase 6 (PDE6) is the central effector of the visual excitation pathway in both rod and cone photoreceptors, and PDE6 mutations that alter PDE6 structure or regulation can result in several human retinal diseases. The rod PDE6 holoenzyme consists of two catalytic subunits (Pαβ) whose activity is suppressed in the dark by binding of two inhibitory γ-subunits (Pγ). Upon photoactivation of rhodopsin, the heterotrimeric G protein (transducin) is activated, resulting in binding of the activated transducin α-subunit (Gtα) to PDE6, displacement of Pγ from the PDE6 active site, and enzyme activation. Although the biochemistry of this pathway is understood, a lack of detailed structural information about the PDE6 activation mechanism hampers efforts to develop therapeutic interventions for managing PDE6-associated retinal diseases. To address this gap, here we used a cross-linking MS-based approach to create a model of the entire interaction surface of Pγ with the regulatory and catalytic domains of Pαβ in its nonactivated state. Following reconstitution of PDE6 and activated Gtα with liposomes and identification of cross-links between Gtα and PDE6 subunits, we determined that the PDE6–Gtα protein complex consists of two Gtα-binding sites per holoenzyme. Each Gtα interacts with the catalytic domains of both catalytic subunits and induces major changes in the interaction sites of the Pγ subunit with the catalytic subunits. These results provide the first structural model for the activated state of the transducin–PDE6 complex during visual excitation, enhancing our understanding of the molecular etiology of inherited retinal diseases.
Keywords: phosphodiesterases, G protein, phototransduction, allosteric regulation, mass spectrometry (MS), protein cross-linking, photoreceptor, integrative structural modeling, PDE6, transducin
Introduction
The photoreceptor cGMP phosphodiesterase (PDE6)4 is the central effector enzyme of the G protein–coupled visual transduction pathway in vertebrate rod and cone photoreceptors. PDE6 is exquisitely regulated by a cascade of reactions beginning with photoactivation of the visual pigment, opsin, and subsequent activation of the heterotrimeric G protein, transducin, in the signal-transducing outer segment of the photoreceptor cell. The activated transducin α-subunit (Gtα) then binds to the membrane-associated PDE6 and accelerates its hydrolytic activity to transiently lower cGMP levels in the photoreceptor outer segment. This results in the closure of cGMP-gated ion channels and hyperpolarization of the membrane, leading to synaptic transmission to other retinal neurons (1).
PDE6 belongs to the 11-member phosphodiesterase enzyme superfamily that shares a highly conserved catalytic domain responsible for the hydrolysis of the intracellular messengers cAMP and cGMP (2, 3). In addition to the C-terminal catalytic domain, the catalytic subunits of PDE6 consist of two N-terminal regulatory GAF (regulatory domain found in certain PDEs, bacterial adenylyl cyclases, and the bacterial transcription factor FhlaA) domains (GAFa and GAFb) that are also present in four other PDE families (4). However, PDE6 differs from the other 10 PDE families in several important respects: (a) unlike the other 10 homodimeric PDE families (as well as cone PDE6), rod PDE6 is composed of two different catalytic subunits, α and β, that form a heterodimer (Pαβ); (b) PDE6 catalysis is uniquely regulated by an intrinsically disordered, 9.7-kDa inhibitory γ-subunit (Pγ) that interacts with both the regulatory and catalytic domains of each catalytic subunit to form the nonactivated rod PDE6 holoenzyme (stoichiometry αβγγ); (c) rod and cone PDE6 are the only PDEs whose activation directly results from binding of a G protein, specifically the activated Gtα subunit; and (d) upon activation, PDE6 catalysis occurs at the diffusion-controlled limit, more than 2 orders of magnitude larger than the catalytic turnover rate of other PDE families (reviewed in Ref. 5).
Numerous biochemical approaches have been undertaken to understand the molecular mechanism by which Gtα binds to the nonactivated PDE6 holoenzyme and relieves the inhibitory constraint of Pγ on PDE6 catalysis. It has been conclusively demonstrated that in the nonactivated state of the PDE6 holoenzyme, the C-terminal portion of Pγ binds to the catalytic domain and blocks access of substrate to the enzyme active site (6–8). Catalytic activation of PDE6 is believed to result from interactions of the switch II–α3-helix region of Gtα with the C-terminal region of Pγ that displaces it from the catalytic pocket of PDE6 (9). This same region of Pγ also modulates the GTPase activity of Gtα (10) by potentiating the activity of RGS9 (regulator of G protein signaling 9) that binds to Gtα and Pγ in this inactivation complex (11). Additional sites of interaction between the Gtα and the N-terminal, polycationic, and glycine-rich regions of Pγ (reviewed in Refs. 12 and 13) have been implicated in regulating the efficacy with which Gtα is able to activate PDE6 (8, 13–15), as well as modulating the affinity of cGMP for noncatalytic binding sites in the GAFa domain of the PDE6 catalytic subunits (16–18).
Consistent with the demonstration of structural asymmetry in the binding interactions of the two Pγ subunits with the rod PDE6 Pαβ heterodimer (19, 20), complete activation of PDE6 by Gtα requires the binding of Gtα to two nonidentical binding sites on PDE6 (Ref. 21 and the references cited therein). Because of the limited information on structure–function relationships of PDE6 holoenzyme in its nonactivated and Gtα-activated states, the molecular sequence of events by which Gtα binds to PDE6 to relieve the inhibition of catalysis by Pγ at two different sites is not known.
Building on recent advances to determine the molecular architecture of the PDE6 holoenzyme at the atomic level using integrative structural modeling (20) and cryo-EM (22, 23), we present here a structural model for the nonactivated PDE6 holoenzyme that includes the complete interaction surface of its inhibitory Pγ subunits. We also provide a refined structural model for the membrane-associated structure of Gtα and its association with Pγ, as well as the complex of the activated G protein α-subunit (Gtα–GDP–ALF4−) with PDE6. In addition to elucidating the mechanistic basis of the first steps in visual signaling, this work provides insights into the molecular etiology of retinal diseases associated with mutations in transducin and PDE6.
Results
Solution structure of the PDE6 catalytic heterodimer
Upon comparing the 3.4 Å cryo-EM structure of the PDE6 holoenzyme (23) with our previous solution structure of PDE6 catalytic dimer determined by chemical cross-linking, identification of cross-linked peptides by mass spectrometric analysis, and integrative structural modeling (20), we observed that a number of distance restraints defined by our cross-linking results were inconsistent with the cryo-EM structure (e.g. cross-links in the β-subunit between residues 675 and 813 and between residues 675 and 815; Table 1). In addition, neither of the above-mentioned studies resolved the entire structure of the Pγ subunits that are tightly bound to the PDE6 catalytic dimer in its nonactivated state. We therefore performed integrative structural modeling of the bovine rod PDE6 holoenzyme using the cryo-EM structure of Gulati et al. (23) as a template (PDB code 6MZB), and the previously reported (20) and new cross-linking data for the PDE6 holoenzyme (Table 1) as inputs into the Integrated Modeling Platform (IMP) and Modeler (see “Experimental procedures”) to determine the complete structure for the tetrameric PDE6.
Table 1.
PDE6 holoenzyme intra- and intermolecular cross-linked peptides
Cross-linked peptides were identified following chemical cross-linking of 10–50 pmol of purified rod PDE6 holoenzyme as described under “Experimental procedures.” Except where indicated with superscripts, samples consisted of native PDE6 holoenzyme and were digested with trypsin prior to mass spectrometric analysis. Exp. m/z is the experimentally measured mass-to-charge ratio, z is the charge state of the peptide, and Δ is the accuracy measured in parts per million. The cross-linked peptides are defined as the protein subunit (pep1 and pep2) and amino acid residue number (aa1 and aa2) identified using the indicated cross-linker. In the aa1 column, the presence of a single-letter amino acid residue preceding the residue number indicates an amino acid substitution of the wild-type Pγ sequence at the site of cross-linking. In addition to the cross-links in this table, the PDE6 structural model included spatial constraints from cross-links reported previously for the PDE6 holoenzyme (20). BMH, 1,6-bismaleimidohexane; BMOE, bis-maleimidoethane; BS(PEG)9, PEGylated bis(sulfosuccinimidyl)suberate; Sulfo-MBS, m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester; Sulfo-SDA, sulfosuccinimidyl 4,4′-azipentanoate.
| Exp. m/z | z | Δ | pep1 | aa1 | pep2 | aa2 | Cross-linker |
|---|---|---|---|---|---|---|---|
| ppm | |||||||
| 658.9788 | 3 | 7.4 | Pβ | 471 | Pβ | 475 | EDC |
| 431.8966 | 3 | −3.5 | Pβ | 675 | Pβ | 813 | EDC |
| 431.8969 | 3 | −2.8 | Pβ | 675 | Pβ | 815 | EDC |
| 405.4728 | 4 | 2.9 | Pβ | 823 | Pβ | 832 | EDC |
| 540.2943 | 3 | 2.3 | Pβ | 824 | Pβ | 832 | EDC |
| 606.6677 | 3 | 1.7 | Pβ | 825 | Pβ | 827 | Sulfo-SDA |
| 540.2935 | 3 | 0.81 | Pβ | 826 | Pβ | 828 | EDC |
| 530.9468 | 3 | 2.4 | Pβ | 826 | Pβ | 829 | Sulfo-SDA |
| 530.9463 | 3 | 1.4 | Pβ | 826 | Pβ | 831 | Sulfo-SDA |
| 567.6403 | 3 | −6.7 | Pβ | 826 | Pα/Pβ | 445/444 | Sulfo-SDA |
| 425.9827 | 4 | −5.2 | Pβ | 826 | Pα/Pβ | 442/441 | Sulfo-SDA |
| 606.6667 | 3 | 0.034 | Pβ | 826 | Pβ | 828 | Sulfo-SDA |
| 524.9417 | 3 | −7.8 | Pβ | 826 | Pα/Pβ | 444/443 | Sulfo-SDA |
| 573.6439 | 3 | 0.044 | Pβ | 826 | Pβ | 832 | Sulfo-SDA |
| 578.0868 | 4 | −6.9 | Pγ | 1a | Pβ | 78 | BS3 |
| 921.2301 | 4 | 6.6 | Pγ | C2b | Pβ | 84 | BMH |
| 678.3601 | 3 | −9.4 | Pγ | 4c | Pβ | 146 | EDC |
| 1034.3080 | 4 | 2.5 | Pγ | 7d | Pβ | 184 | BS(PEG)9 |
| 926.7150 | 4 | 0.73 | Pγ | C18e | Pα | 383 | Sulfo-MBS |
| 1113.0730 | 2 | −6.9 | Pγ | C18e | Pβ | 92 | BMH |
| 1027.5500 | 3 | 5.9 | Pγ | C18e | Pα | 233 | BMH |
| 653.0510 | 4 | −1.6 | Pγ | 31a | Pβ | 200 | Sulfo-MBS |
| 596.0509 | 4 | −15 | Pγ | 41c,f | Pα | 469 | Sulfo-SDA |
| 1063.2268 | 3 | −17 | Pγ | 44 | Pα/Pβ | 613/611 | BS3 |
| 800.6560 | 4 | −8.8 | Pγ | 44 | Pβ | 475 | BS3 |
| 498.4935 | 4 | −17 | Pγ | 52c,f | Pα/Pβ | 328/326 | EDC |
| 911.4194 | 3 | −15 | Pγ | K62c,f | Pβ | 450 | EDC |
| 911.4185 | 3 | −4.8 | Pγ | K62c,f | Pβ | 446 | EDC |
| 660.0972 | 4 | −1.3 | Pγ | K62c,d | Pα/Pβ | 394 | EDC |
| 879.7957 | 3 | 0.78 | Pγ | K62c,d | Pα/Pβ | 393 | EDC |
| 478.5120 | 4 | 0.34 | Pγ | K65c,g | Pα | 767 | EDC |
| 670.0686 | 4 | −13 | P | C68g | Pβ | 839 | BMOE |
a Sample consisted of Pαβ reconstituted with recombinant, wildtype rod Pγ.
b Pαβ reconstituted with Pγ2C/68S.
c Trypsin/Asp-N double digest.
d Pαβ reconstituted with Pγ58K/62K/65K/73K.
e Pαβ reconstituted with Pγ18C/68S.
f Pαβ reconstituted with Pγ62K/65K/73K/79K.
g Pαβ reconstituted with Pγ53K/62K/65K/73K.
As shown in Fig. 1A, the cross-link–refined solution structure of the PDE6 holoenzyme fits well within the cryo-EM envelope (23), with the spatial restraints imposed by the cross-linking results generating a more compact arrangement of structural elements, as well as providing predicted structures for missing elements in the cryo-EM structure (Fig. S2A). Comparisons at the level of individual domains of our cross-link–refined PDE6 solution structure with the cryo-EM structure (23) identified several significant differences in conformation (Fig. S2 (structures) and Fig. S3 (root-mean-square deviation plots)): (a) The N-terminal region preceding the GAFa domain in our structural model contains additional α-helical elements (Fig. S2B), consistent with the hypothesis (23) that this region may contribute to dimerization of the catalytic subunits. (b) Whereas the GAFa domains showed relatively small differences in secondary structure when compared with the cryo-EM structure (Fig. S2C), the GAFb domains of the PDE6 solution structure exhibited greater dissimilarity (Fig. S2D). Our cross-linking restraints identified conformational differences in several loop structures of GAFb, including the β1/β2 loop that contains a novel α2/α3 helix (Fig. S2D). This loop is in close proximity to the catalytic domain and as previously suggested may play a role in intersubunit allosteric communication (23, 24). (c) the catalytic domains of our structural model also exhibited significant differences compared with the cryo-EM structure, particularly in the flexible H-loop and M-loop regions near the enzyme active site and in the α16 helix (Fig. S2F). Cross-links in the C-terminal region (Table 1 and Ref. 20) imposed spatial restraints to the conformation of the α15 and α16 helices in our model that displaced these two helices toward the center of the catalytic domain (and are likely to contribute to the observed conformation of the H- and M-loops), as well as defining additional α-helical segments (Cα1 and Cα2) in the C-terminal region. The fact that the C termini of the PDE6 catalytic subunits are prenylated (25) and membrane-associated under our experimental conditions likely accounts for the structural differences we observe in the catalytic domain and C terminus. Together, these observations emphasize the importance of relying on this lower-resolution, chemical cross-linking/MS approach to define both flexible structural elements (e.g. loops) and protein conformations unique to the membrane-associated state that are often challenging to obtain from high-resolution, unbiased structural methods such as X-ray crystallography and cryo-EM.
Figure 1.

Integrative structural model of the PDE6 holoenzyme. The structural model of rod PDE6 holoenzyme (αβγγ) was determined by using the cryo-EM structure 6MZB (23) as a template and applying spatial restraints determined by chemical cross-linking of purified bovine rod PDE6 (Table 1 and Ref. 20). In the model, PDE6 subunits are colored as follows: α-subunit (Pα), cyan; β-subunit (Pβ), green; Pγ subunit primarily associated with α-subunit (Pγ(Pα)), red; and Pγ subunit primarily associated with β-subunit (Pγ(Pβ)), deep purple. A, superimposition of the template cryo-EM map (EMD-9297) with the cross-link–refined structural model of nonactivated PDE6 holoenzyme. B, asymmetric interactions of Pγ with the Pαβ catalytic dimer extending from the cGMP-binding GAFa domain to the GAFb domain and then crossing over to the catalytic domain to the site of inhibition of catalysis. Each Pγ subunit primarily interacts with one catalytic subunit. The two images are rotated 180°. C, interaction surface of the Pγ(Pα) subunit with the PDE6 catalytic dimer. Pγ(Pα) residues interacting with the catalytic dimer are shown as main-chain atom spheres: red, residues interacting with the α-subunit; pink, residues interacting with the β-subunit; and yellow, Pγ residues that interact with both catalytic subunits. Noninteracting Pγ(Pα) residues are shown as red loops and α-helix. The catalytic subunit interacting residues are shown as a surface representation (α-subunit, dark cyan; β-subunit, dark green). D, interaction surface of the Pγ(Pβ) subunit with Pαβ. The interaction surface of the Pγ(Pβ) subunit (180° rotation in C) is depicted in which the deep purple, light purple, and orange spheres represent interactions with the β-subunit, α-subunit, or both catalytic subunits, respectively.
Each intrinsically disordered Pγ subunit forms multiple interactions with both PDE6 catalytic subunits
To map the entire interaction surface of Pγ with the PDE6 catalytic dimer, we performed cross-linking experiments with a variety of chemical cross-linkers, as well as using several site-directed mutants of Pγ that were reconstituted with Pαβ. The 21 new intermolecular cross-links between Pγ and the α- or β-subunits (Table 1) along with previously reported cross-links (20) and the cryo-EM structure of two fragments of Pγ (23) permitted visualization for the first time of the molecular architecture of the entire PDE6 holoenzyme. Fig. 1B shows that the overall topology of the each Pγ subunit is similar, originating at the noncatalytic cGMP binding pocket in the GAFa domain of one catalytic subunit and terminating at the enzyme active site of the same catalytic subunit. Interestingly, the C-terminal region of Pγ—consisting of an α-helix and a C-terminal “cap”—is similar overall to the crystal structure of a Pγ fragment complexed with a PDE5/6 chimera (7). Although the N-and C-terminal regions of Pγ assume a predominantly linearly extended conformation, the midregion of Pγ exists in a random coil conformation.
Analysis of the interaction surface of Pγ with the catalytic subunits (Fig. 1, C and D) reveals marked differences in the number and types of interactions of each Pγ with the two catalytic subunits. One Pγ subunit (designated Pγ(Pα)) follows the trajectory of the α-subunit (Fig. 1C), with approximately one-half of its 87 residues forming an interaction surface in the GAFa, GAFb, and catalytic domains, ending at the active site of the α-subunit. Nine Pγ(Pα) residues interact with the β-subunit in its GAFa and GABb domains, with four of the nine being in close proximity to both catalytic subunits. The second Pγ subunit (designated Pγ(Pβ)) has an even greater interaction surface with the catalytic dimer (Fig. 1D), with 89% of its residues interacting with Pαβ. Pγ(Pβ) interactions with Pαβ include 62 residues of the β-subunit and 30 residues of the α-subunit, with 15 of these residues being in close proximity to both catalytic subunits. The large number of Pγ(Pβ) interactions with the α-subunit is most evident in the GAFb domain where the Pγ(Pβ) subunit comes into contact with the α-subunit GAFb domain (leftward projection in Fig. 1D), as well as multiple interactions of Pγ(Pβ) with the central α-helical “backbone” of both catalytic subunits. This complex network of interactions of both Pγ subunits with both catalytic subunits localized predominantly in the GAFb domains of the catalytic dimer may represent the structural basis for allosteric communication between the α- and β-subunits during transducin activation of PDE6 (see “Discussion”).
Structure of membrane associated Gtα and its interactions with soluble Pγ
We first carried out cross-linking experiments with activated Gtα attached to liposomes to determine the solution structure of membrane-associated Gtα. Experiments were carried out with Gtα–GDP–AlF4− for which a crystal structure is available (PDB code 1TAD). For the N-terminal α-helix (αN), which is missing from this crystal structure (and proposed to have conformational flexibility) (26), we used as a template the structure of the αN helix that was determined for the inactive transducin heterotrimer (PDB code 1GOT). With the αN helix and the Gtα–GDP–AlF4− structures as templates and the intramolecular Gtα cross-links that we identified (Table 2), a model of the membrane-associated, activated Gtα–GDP–AlF4− subunit of transducin was created (Fig. 2A). Intramolecular cross-links (Lys18 to Lys267 and Glu21 to Lys275; Table 2) between the αN helix and the Ras-like GTPase subdomain of Gtα imposed spatial constraints that are reflected in a major shift of the αN helix toward the αF/β2 loop region that is part of the interface between the GTPase subdomain and the helical insertion subdomain. This shift brings the αN helix in proximity with the nucleotide-binding site. We conclude that the structural model shown in Fig. 2A better represents the membrane-associated, solution structure of Gtα in that it takes into account the N-terminal acylation of Gtα responsible for its association with rod outer segment membranes in vivo.
Table 2.
Intra- and intermolecular cross-linked peptides of membrane-associated Gtα–GDP– AlF4− and Pγ
Cross-linked peptides were identified following chemical cross-linking of either lipobead-associated Gtα–GDP–AlF4− or Gtα–GDP–AlF4− incubated with a 2-fold stoichiometric excess of purified Pγ and analyzed as described under“Experimental procedures.” The abbreviations are defined in the legend to Table 1. All Gtα intramolecular cross-links were detected in both the absence and the presence of Pγ.
| Exp. m/z | z | Δ | pep1 | aa1 | pep2 | aa2 | Cross-linker |
|---|---|---|---|---|---|---|---|
| ppm | |||||||
| 451.2427 | 4 | 9.2 | Gtα | 16 | Gtα | 20 | EDC |
| 451.2424 | 4 | −9.8 | Gtα | 16 | Gtα | 25 | EDC |
| 539.2929 | 3 | −9.1 | Gtα | 17 | Gtα | 20 | BS3 |
| 451.2498 | 4 | 6.6 | Gtα | 17 | Gtα | 21 | EDC |
| 487.2670 | 3 | −9.4 | Gtα | 17 | Gtα | 22 | EDC |
| 723.7675 | 3 | 6.4 | Gtα | 17 | Gtα | 31 | BS3 |
| 434.9019 | 3 | −7.8 | Gtα | 18 | Gtα | 26 | EDC |
| 434.9022 | 3 | −7.1 | Gtα | 18 | Gtα | 26 | EDC |
| 637.7287 | 3 | −9 | Gtα | 18 | Gtα | 31 | BS3 |
| 565.9201 | 5 | −9.4 | Gtα | 18 | Gtα | 31 | BS3 |
| 345.2063 | 4 | 3.3 | Gtα | 18 | Gtα | 267 | BS3 |
| 899.8458 | 3 | 7.3 | Gtα | 20 | Gtα | 31 | BS3 |
| 415.5742 | 3 | −1.3 | Gtα | 20 | Gtα | 205 | BS3 |
| 468.2516 | 3 | −9.5 | Gtα | 21 | Gtα | 275 | Sulfo-SDA |
| 733.7522 | 3 | −8 | Gtα | 24 | Gtα | 31 | EDC |
| 899.8322 | 3 | −7.9 | Gtα | 25 | Gtα | 31 | BS3 |
| 455.7341 | 4 | −8.3 | Gtα | 25 | Gtα | 189 | EDC |
| 576.6672 | 3 | −8.3 | Gtα | 26 | Gtα | 31 | EDC |
| 330.5199 | 3 | −3.6 | Gtα | 26 | Gtα | 205 | EDC |
| 658.3810 | 3 | −9.1 | Gtα | 39 | Gtα | 47 | EDC |
| 817.4389 | 4 | −10 | Gtα | 169 | Gtα | 176 | EDC |
| 459.9397 | 3 | 4.2 | Gtα | 267 | Gtα | 275 | DSS |
| 446.2453 | 3 | 8 | Gtα | 267 | Gtα | 342 | EDC |
| 504.2656 | 2 | −3 | Gtα | 98 | Pγ | 39 | BS3 |
| 975.9864 | 2 | −10 | Gtα | 129 | Pγ | 25 | BS3 |
| 469.5794 | 3 | 7.2 | Gtα | 203 | Pγ | 39 | BS3 |
| 440.9130 | 3 | −10 | Gtα | 203 | Pγ | 45 | BS3 |
Figure 2.

Structural model of Gtα–GDP–AlF4− and its interaction with Pγ in solution. A, the structural model of Gtα was determined using the 1TAD crystal structure as the template (36) and refined with spatial restraints imposed from cross-linking results in the absence or presence of Pγ (Table 2). Structural elements that were unchanged in the cross-link–refined model are represented in green, with the conformational change of the αN helix shown in brown (for the crystal structure) and blue (for the cross-link modified solution structure). Also shown is the docked structure of Pγ (red) with Gtα–GDP–AlF4− based on the observed cross-linking results when Gtα associated with lipobeads was incubated with a 2-fold molar excess of Pγ. Note that no significant changes in Gtα conformation were observed upon Pγ binding. B, a comparison of the conformation of the central region of Pγ (residues 24–44, depicted as a gradient from blue to red spheres) when bound to Gtα or to the PDE6 catalytic subunits.
Previous biochemical studies have identified two major regions of Pγ that bind to activated Gtα, namely the polycationic central region of Pγ and the C-terminal half of Pγ (8, 11–13, 27–29). To determine the topological relationship of Gtα with Pγ, we incubated liposome-associated Gtα–GDP–AlF4− (see “Experimental procedures”) with purified Pγ and conducted cross-linking analyses of the protein band migrating at the apparent molecular mass expected for a 1:1 complex of Pγ and Gtα (∼50 kDa). We identified five intermolecular, cross-linked peptides spanning residues 25–45 of the central region of the Pγ molecule (Table 2) that interact with both the helical subdomain and the switch II region of the GTPase subdomain of Gtα (Fig. 2A). This 20-amino acid segment of Pγ interacts on the opposite face of the Gtα subunit from the interface of Gtα with the PDE6 catalytic domain (see below). As seen in Fig. 2B, Pγ assumes a highly extended linear structure when bound to Gtα compared with the conformation of the same region of Pγ bound to the PDE6 α- or β-subunits. (Although there is structural evidence that the C-terminal half of Pγ binds to the PDE6-facing side of Gtα (11, 30), our inability to observe cross-linked peptides between Gtα and this region of Pγ arises from the absence of amino acid residues in the C-terminal half of Pγ capable of generating cross-linked peptides for mass spectrometric detection (20).) No significant changes in the tertiary structure of Gtα were detected upon Pγ binding.
Molecular architecture of the G protein–effector activation complex
Full activation of PDE6 by Gtα is greatly enhanced when both proteins are associated with either rod outer segment membranes or are reconstituted with phospholipid bilayers (31). To determine the structure of the transducin–PDE6 complex in its membrane-associated state, we therefore preincubated purified proteins with cationic phospholipid vesicles that have been shown to enhance PDE6 activation by Gtα (32). To restrict our analysis to only membrane-associated Gtα and PDE6, we prepared liposome-coated silica beads (“lipobeads”; see “Experimental procedures” and Fig. S5) that allowed for sedimentation of membrane-associated proteins for further analysis. Using this method, ∼90% of the PDE6 holoenzyme was pulled down in the lipobeads pellet (Fig. S5C), and under these conditions we observed at least 80% of maximal activation of PDE6 catalysis by transducin (Fig. S5B).
We first assessed whether all of the cross-linked peptides we observed between Gtα and the PDE6 catalytic subunits could be accounted for by a single Gtα-binding site per Pαβ. Table 3 shows that three PDE6 α-subunit–specific and three β-subunit–specific cross-links with Gtα ruled out a single binding site per Pαβ, consistent with biochemical studies (21). When 10 cross-linked peptides between Gtα and the α- or β-subunit of PDE6 in Table 3 were used as distance restraints for input into the IMP workflow, we found no single structural model that was able to accommodate all of the cross-link restraints. Instead, we observed two major classes of structural models with different cross-links that violated the distance restraints. The predominant set of structural models was generated by omitting the two cross-links between Gtα and the GAFb domains (Gtα9-Pα442/Pβ440 and Gtα24-Pα330/Pβ328; Table 3); the remaining eight cross-links permitted docking of Gtα to two similar—but not identical—sites on the α- and β-subunit catalytic domains (Fig. 3A).
Table 3.
Intermolecular cross-linked peptides of the activated complex of Gtα–GDP–AlF4− with PDE6 holoenzyme
Cross-linked peptides were identified following chemical cross-linking of a mixture of Gtα–GDP–AlF4− and PDE6 holoenzyme attached to lipobeads and analyzed as described under “Experimental procedures.” The abbreviations are defined in the legend to Table 1. All identified intra- and intermolecular cross-links involving PDE6 catalytic subunits were identical to those observed in the holoenzyme structure (Table 1 and Ref. 20) and omitted here.
| Exp. m/z | z | Δ | pep1 | aa1 | pep2 | aa2 | Cross-linker |
|---|---|---|---|---|---|---|---|
| ppm | |||||||
| 947.4468 | 2 | 3.7 | Gtα | 9 | Pα/Pβ | 442/440a | EDC |
| 673.5722 | 4 | −4.1 | Gtα | 10 | Pβ | 826 | BS(PEG)5 |
| 447.0946 | 8 | 7.8 | Gtα | 10 | Pα | 854b | BS3 |
| 684.3533 | 3 | −3.1 | Gtα | 17 | Pα | 551b | BS3 |
| 545.3019 | 2 | 2.4 | Gtα | 17 | Pα/Pβ | 808/806 | EDC |
| 674.0325 | 3 | −1 | Gtα | 17 | Pβ | 817 | Sulfo-SDA |
| 558.3024 | 4 | −8.9 | Gtα | 20 | Pα/Pβ | 807/805 | BS3 |
| 1360.241 | 2 | 4.3 | Gtα | 20 | Pα/Pβ | 620/618 | BS3 |
| 812.4367 | 3 | 5.7 | Gtα | 24 | Pα/Pβ | 330/328a | Sulfo-SDA |
| 752.024 | 3 | −7.6 | Gtα | 25 | Pα | 309c | BS3 |
| 775.0531 | 3 | −3.9 | Gtα | 128 | Pα/Pβ | 807/805b | BS3 |
| 569.994 | 3 | −7.8 | Gtα | 275 | Pβ | 307c | BS3 |
| 381.2 | 3 | −9.3 | Gtα | 98 | Pγ | 41 | EDC |
| 332.469 | 4 | 1.7 | Gtα | 275 | Pγ | 29 | BS3 |
| 337.8635 | 3 | −7.3 | Pα/Pβ | 328/326 | Pγ | 25 | EDC |
| 413.2165 | 5 | −14 | Pα | 551 | Pγ | 29 | BS3 |
a Cross-links that were omitted from specific structural models during docking of Gtα to catalytic domain.
b Cross-links that were omitted from specific structural models during docking Gtα to GAFb domain.
c Cross-links that were omitted from specific structural models from computational modeling due to loop flexibility.
Figure 3.

Model of Gtα–GDP–AlF4− docked to the Pαβ catalytic dimer. PDE6 holoenzyme and Gtα–GDP–AlF4− bound to lipobeads (see “Experimental procedures”) were exposed to chemical cross-linkers, and the identified cross-linked peptides between Gtα and PDE6 subunits (Table 3) were then used as spatial restraints for integrative structural modeling. Two predominant clusters of models of the Gtα–Pαβ complex were generated, one with Gtα docked to the two catalytic domains (with distance violations for Gtα24-Pα330/Pβ328, Gtα9-Pα442, and Gtα9-Pβ440) and the other with Gtα docked to the GAFb domains (with distance violations for Gtα10-Pα854, Gtα17-Pα551, and Gtα128-Pα807/Pβ817). Because of insufficient cross-links for Pγ in the activated complex, the inhibitory subunit is not shown. A, structural model of association of Gtα–GDP–AlF4− to the α-subunit (Gtα(Pα), orange) and to the β-subunit (Gtα(Pβ), blue) catalytic domains. B, detailed view of the Gtα GTPase subdomain interface with the α-subunit catalytic domain, with the interaction surface of Gtα colored red and the α- and β-subunit interacting residues colored magenta and brown, respectively. The black sphere indicates Gtα Gln200. C, alternate docking of Gtα to the GAFb domains of the Pαβ catalytic dimer (with the same orientation as in A).
Closer examination of the interface of Gtα with the PDE6 α-subunit catalytic domain (Fig. 3B) revealed that the GTPase subdomain of this Gtα molecule (including the switch II region and the αN helix) interacts with the α14 helix, the M-loop region (implicated in regulating Pγ occlusion of the active site) (7), and the α15 and α16 helices of the α-subunit catalytic domain, in excellent agreement with previous mutagenesis studies (33). Interestingly, the αB helix of this Gtα molecule interacts with the adjacent PDE6 β-subunit in the linker region between the GAFb and β-subunit catalytic domain (Fig. 3B). The interaction surface of the second Gtα with the β-subunit was generally similar to the α-subunit, but with a greater surface of interaction reflecting additional interactions with the region of the β-subunit linking the GAFb and catalytic domains. For both Gtα subunits, the N-terminal α-helix has significant surface interactions with the catalytic domains. With the cross-links in Table 3, we were unable to observe any significant changes in Gtα–GDP–AlF4− conformation upon interaction with Pαβ. The same was true for the PDE6 catalytic dimer where the overall root-mean-square deviations for each catalytic subunit was ∼1.0 A when comparing the nonactivated and transducin-activated conformations of each PDE catalytic subunit (Fig. S4). Interestingly, the interaction surface of Gtα with PDE6 catalytic subunits is similar to that observed for the complex of a membrane-bound adenylyl cyclase with the Gs α-subunit, particularly in the switch II and α3 helix regions of the GTPase domain (34).
A second cluster of structural models of Gtα-activated PDE6 docking with the GAFb domain (Fig. 3C) was identified when three different cross-links at the bottom of the Pαβ catalytic domains (Gtα10-Pα854, Gtα17-Pα551, and Gtα128-Pα807/Pβ805) were omitted during the structural modeling (Table 3). Although insufficient cross-linking data for the Pγ subunit precluded structural modeling of Pγ in the transducin-activated PDE6 complex, the same central region of Pγ that binds to purified Gtα in an extended conformation (Fig. 2A) is associated with the GAFb domain of nonactivated PDE6 (Fig. 1, C and D) and likely promotes Gtα binding to the GAFb domain shown in Fig. 3C. This second binding site for Gtα is supported by biochemical studies indicating a role for the central region of Pγ in facilitating Gtα activation of PDE6 catalysis (8), as well as enhancing the dissociation of cGMP from GAFa noncatalytic binding sites (13).
Discussion
This paper reports the first complete structural models for the PDE6 holoenzyme (Fig. 1), the activated α-subunit of transducin in a complex with the inhibitory γ-subunit of PDE6 (Fig. 2), and the fully activated state of PDE6 in a complex with two transducin α-subunits (Fig. 3)—all in their membrane-associated state that mimics the localization of the transducin–PDE6 protein complex on photoreceptor outer segment disk membranes. Together, these structural models advance our understanding of the mechanism of visual excitation in rod photoreceptors by revealing the asymmetric surface of interaction between each Pγ subunit and the Pαβ catalytic dimer, as well as the different sites of interaction of Gtα with PDE6 and the major conformational changes that the Pγ subunits must undergo upon transducin activation of PDE6 in the phototransduction pathway.
Chemical cross-linking combined with mass spectrometric analysis (35) has enabled us to refine the secondary, tertiary, and quaternary structure of PDE6 in its nonactivated and transducin-activated states. It is important to emphasize that our ability to carry out integrative structural modeling of PDE6 in its nonactivated and activated states was enabled by having the atomic-level crystal structure for Gtα–GDP–AlF4− (36) and a high-resolution cryo-EM structure for PDE6 holoenzyme (23). The distance restraints imposed by cross-linked residues within and between proteins comprising the nonactivated and activated states of PDE6 permitted us to dock Gtα subunits to each catalytic subunit of PDE6, thereby providing a structural basis for the allosteric mechanism for G protein–coupled activation of PDE6 during visual excitation—including the functional asymmetry of the PDE6 holoenzyme that underlies the requirement for successive binding of two Gtα molecules for full enzyme activation (Refs. 21 and 37 and references cited therein). This cross-linking/mass spectrometric approach also permitted visualization of flexible regions of the PDE6 catalytic and inhibitory subunits that were poorly resolved by cryo-EM (23), as well as structural elements not available in the existing crystal structures for Gtα.
Our integrative structural modeling of PDE6 reveals the multiple intersubunit interactions that underlie the multifaceted allosteric regulation of this G protein-activated enzyme: (a) each Pγ subunit interacts with both PDE6 catalytic subunits, with lateral, cross-subunit communication likely transmitted through the GAFb domains where a number of Pγ residues are in close proximity to both catalytic subunits (Fig. 1, C and D); (b) the β-subunit exhibits greater interactions with Pγ than the α-subunit, consistent with two classes of binding sites for Pγ with Pαβ (18); and (c) in addition to the extensive Pαβ dimerization surface, direct allosteric communication may occur between the β1/β2 loop in the GAFb domain of one catalytic subunit and the catalytic domain of the other subunit (23), as well as between the catalytic domains and C-terminal regions of the two subunits (Fig. 1B).
Defining the molecular architecture of the transducin–PDE6–activated complex permitted structural verification of the stoichiometry of two Gtα subunits bound to the PDE6 catalytic subunits in its fully activated state, as well as unexpectedly revealing two distinct sites of interaction of Gtα with the GAFb (Fig. 3C) and catalytic domains (Fig. 3, A and B) of the PDE6 catalytic subunits. The observation that each Gtα subunit has sites of interaction with both the α- and β-subunits of PDE6 is consistent with a cooperative activation mechanism in which the binding of the first Gtα induces conformational changes in Pαβ that alter the ability of the second Gtα subunit to bind to and trigger full enzyme activation (21, 37).
For the model for G protein activation of the central effector enzyme of the visual signaling pathway, Fig. 4 presents a model consistent with our experimental results for the light-induced activation of PDE6 holoenzyme by transducin that involves the sequential binding of two Gtα subunits that results in both Gtα subunits releasing the inhibitory constraint of Pγ from its interactions with each PDE6 catalytic domain to cause full activation of PDE6 (37).
Figure 4.
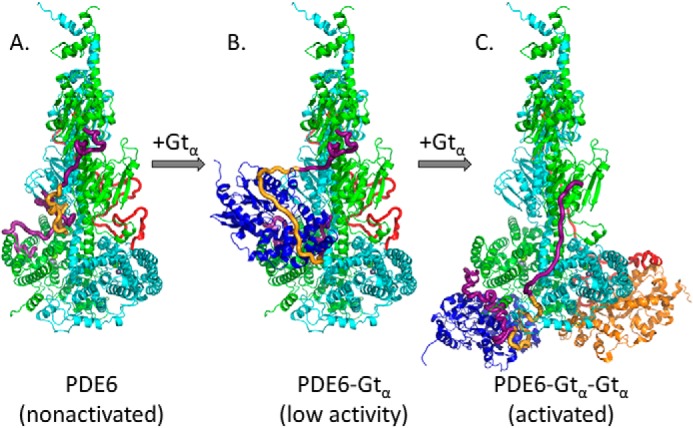
Proposed model for the activation of PDE6 by transducin during visual excitation. A, in the dark-adapted condition, the PDE6 holoenzyme is inhibited by its Pγ subunits occluding the enzyme active site (Fig. 1B, rotated 90°). B, the first light-activated Gtα subunit is proposed to initially bind to the GAFb docking site (see Fig. 3C) without causing significant catalytic activation of PDE6 (21). The Pγ subunit was docked to this complex using the following information: (a) the central region of Pγ (gold) was docked using the cross-links obtained for the Gtα–Pγ complex (Table 2) in conjunction with the cross-links used to dock Gtα to the GAFb domain (Table 3); (b) lacking cross-linking data for the N-terminal region of Pγ in the activated complex, this region of Pγ (purple) relied on PDE6 holoenzyme cross-links, and thus its topology only differs from Fig. 1 to the extent needed to accommodate cross-link spatial restraints imposed by the Pγ central region; and (c) in the absence of Pγ cross-links for its C-terminal region in the activated complex, we modeled this region of Pγ (purple) interacting with Gtα using the crystal structure of Pγ (residues 50–87) bound to a chimeric G protein (PDB code 1FQJ) (11). C, upon binding of a second Gtα, PDE6 becomes fully activated as both Gtα subunits dock to the catalytic domains and displace the C-terminal region of Pγ from the enzyme active sites. To accommodate the binding of the central region of Pγ to the helical face (Table 2) and the C-terminal region of Pγ to the GTPase face of Gtα (Table 3 and Ref. 11), a major displacement of the N-terminal Pγ residues from the GAFa domains must occur.
Upon light activation of the phototransduction cascade, nonactivated PDE6 holoenzyme (Fig. 4A) is proposed to form initial interactions between the central region of Pγ (associated with the GAFb domains) and an activated Gtα subunit (Fig. 3C), resulting in the central region of Pγ becoming significantly more extended (Fig. 4B). In this model, the binding of Gtα to this central region of Pγ does not require major displacement of either the N- or C-terminal regions of Pγ from its holoenzyme conformation. Upon binding of a second Gtα, Fig. 4C depicts a relocation of the first Gtα from the GAFb to the catalytic domain, along with binding of the second Gtα to the catalytic domain of the other catalytic subunit (Fig. 4C)—resulting in full enzyme activation. As a consequence of Gtα binding to the central and C-terminal regions of Pγ when docked to the catalytic domains, our model requires that the N-terminal region of Pγ dissociates from its interactions with the GAFa domain (Fig. 4C). This structural model for sequential activation of PDE6 is supported by prior biochemical and structural studies of Gtα interactions with PDE6 subunits in the activated complex (11, 33, 38–40). The required displacement of Pγ from the GAFa domains is also consistent with a lowered affinity of cGMP to its GAFa-binding sites upon transducin activation of rod PDE6 (8, 13, 17), as well as offering insights into differences in how rod and cone PDE6 may be activated by transducin (15). Experimental support for the model in Fig. 4 is currently under investigation, including validating the GAFb domain as an initial docking site for one or both Gtα subunits, identifying whether the α- or β-subunit preferentially binds the first Gtα, the allosteric communication pathway leading to binding of the second Gtα subunit, and the significance of cGMP occupancy of the GAFa-binding sites for the activation, recovery, and light adaptation stages of visual transduction.
In addition to advancing a structural basis for understanding the initial events in the visual signaling pathway, structural elucidation of PDE6 in its nonactivated and transducin-activated states offers insights into the molecular etiology of pathogenic mutations in these proteins and possible therapeutic interventions. For example, having characterized the interaction surface of Gtα-PDE6, it is now evident that a missense mutation impairing GTPase activity (Q200E; black sphere in Fig. 3B) that is responsible for autosomal dominant congenital stationary night blindness (41) is located at the interface between the Gtα switch II region and the PDE6 catalytic domain where Pγ regulation of catalytic activation occurs. Based on our structural model of Gtα interactions with the central region of Pγ (Fig. 2), we hypothesize that the disease-causing D129G mutation in Gtα (42) eliminates an ionic interaction with Pγ Lys26 (<4 Å apart) that participates in the binding of activated transducin to PDE6 holoenyzme. Because somatic mutations in PDE6 catalytic subunit genes have been implicated in various cancers (Ref. 43) and references cited therein), knowledge of the atomic-level structure of PDE6 may become relevant should a causative link be established between PDE6 mutations and tumorigenesis.
Given that abnormal accumulation of cGMP is believed to be the causative factor in many retinal degenerative diseases (44), understanding the structural organization of PDE6 and the protein–protein interactions that regulate its activity may provide insights into development of allosteric regulators of PDE6 analogous to those being developed for other members of the PDE family of enzymes (45, 46).
Experimental procedures
Materials
Bovine retinas were purchased from W. L. Lawson, Inc. The Mono Q, HiTrap SP Sepharose FF, HiTrap Blue HP, and Superdex 200 columns were from GE Healthcare. The C18 reverse-phase column (Proto 300, 4.6 × 250 mm) was from Thermo Fisher Scientific. The primers for Pγ mutagenesis and plasmid purification kits were from Invitrogen and Qiagen, respectively. The QuikChange II site-directed mutagenesis kit was from Agilent Technologies. Phospholipids and the Mini-Extruder were from Avanti Polar Lipids. Trypsin and Asp-N were purchased from Promega. Silica particles (70-nm diameter, plain) were obtained from Advance Scientific. Chemical cross-linkers were from Thermo Fisher, and all other reagents were from Millipore-Sigma, Thermo-Fisher, or VWR.
Preparation of purified PDE6
Rod PDE6 holoenzyme (subunit composition, αβγγ) was isolated from bovine rod outer segments and purified by anion-exchange and gel-filtration chromatography as described previously (47). The Pαβ catalytic dimer was prepared from purified PDE6 holoenzyme by limited trypsin proteolysis to selectively degrade the Pγ subunits; the time course of proteolytic activation of PDE6 catalysis was empirically determined to ensure that >90% of the Pγ subunit was degraded without altering the apparent molecular mass of the catalytic subunits on SDS-PAGE. Pαβ was then repurified by Mono Q chromatography (47). Purified PDE6 preparations were stored in 20 mm HEPES, pH 7.5, 100 mm NaCl, 2 mm MgCl2 (HNM buffer) plus 50% glycerol at −20 °C until use. Just prior to the experiment, the protein was buffer-exchanged and adjusted to the indicated concentration for the cross-linking reaction.
PDE6 catalysis of cGMP hydrolysis was quantified using a coupled-enzyme assay with colorimetric detection of Pi (48). The PDE6 concentration was estimated based on the rate of cGMP hydrolysis of trypsin-activated PDE6 and knowledge of the kcat of the enzyme (5600 mol cGMP hydrolyzed per mol Pαβ per second) (49).
Preparation of persistently activated transducin α-subunit
Gtα was selectively extracted from PDE6-depleted rod outer segment membranes by adding either 50 μm GTPγS or 100 μm GTP to the ROS membranes and recovering the solubilized Gtα following centrifugation of the membranes. Gtα was subsequently purified by affinity chromatography on a HiTrap Blue HP column (50), followed by Superdex 200 gel-filtration chromatography to remove residual PDE6. The concentration of Gtα was determined by a colorimetric protein assay (51) using bovine γ-globulin as a standard. Purified Gtα was stored at −20 °C in 50% glycerol supplemented with 50 μm of GTPγS or GDP until use. Prior to a cross-linking experiment, the Gtα-GTPγS or Gtα-GDP was buffer-exchanged into the appropriate cross-linking buffer. In the case of GDP-bound Gtα, the Gtα was incubated with 30 μm AlCl3 and 10 mm NaF for 15 min on ice to form the activated Gtα–GDP–AlF4− complex (52).
Expression and purification of Pγ mutants
Pγ site-directed mutants were created with the codon-optimized WT bovine rod Pγ sequence as the template and the QuikChange II site-directed mutagenesis kit to introduce amino acid substitutions. The pET11a plasmids with the sequence-verified Pγ mutant sequences were transformed into Escherichia coli BL21(DE3) cells and grown at 37 °C in 2-YT medium to an A600 of ∼0.6. Then 0.5 mm isopropyl β-d-1-thiogalactopyranoside was added, and the cells were incubated at 30 °C for 6 h. The recombinant Pγ protein was purified from the cell extract using a HiTrap SP column followed by C18 reverse-phase HPLC (53). The apparent molecular mass and purity (>95%) of the recombinant Pγ protein was verified by SDS-PAGE. Pγ cysteine mutants were prepared as described previously (20). All Pγ mutants were observed to inhibit Pαβ catalysis over the same concentration range as WT Pγ.
Preparation of liposomes and lipobeads to study interactions of transducin with PDE6
Large unilamellar vesicles and sucrose-loaded vesicles (consisting of an 80:20 molar ratio of 1,2-dioleoyl-sn-glycero-3-phosphocholine and 1,2-dioleoyl-3-trimethylammonium-propane) were initially utilized to improve the efficiency of transducin activation of PDE6 (32), closely following established procedures (54). To further improve the ability to quantitatively sediment PDE6 and Gtα attached to the liposomes (and to eliminate soluble proteins), we adapted an existing method to prepare silica bead–supported liposomes (i.e. lipobeads) (55) for membrane association of PDE6 and Gtα. The ability to pulldown PDE6 (∼90% PDE bound) and the enhancement of PDE6 activation by Gtα (up to 95% of maximum activation) were equivalent for all of the above liposome preparations.
Lipobeads were prepared by first washing 5 mg of 70-nm silica beads several times with HNM buffer followed by centrifugation for 3 min at 15,000 × g. The bead pellet was then resuspended in HNM buffer. 1,2-Dioleoyl-sn-glycero-3-phosphocholine and 1,2-dioleoyl-3-trimethylammonium-propane were mixed at a molar ratio of 80:20 in chloroform, evaporated, and resuspended in HNM buffer containing the lipobeads to a final phospholipid concentration of 500 μm. Unilamellar vesicles coating the silica particles were formed by extruding the mixture 15 times through a 0.1-μm polycarbonate membrane using a Mini-Extruder (Fig. S5A).
Chemical cross-linking, in-gel digestion, and MS analysis
Chemical cross-linking reactions were carried out following the manufacturer's protocols for each cross-linker. For cross-linking reactions with BS3, DSS, sulfosuccinimidyl 4,4′-azipentanoate, or m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester, proteins were cross-linked in HNM buffer; for EDC cross-linking reactions, 100 mm MES buffer, pH 6.5, was used. After the cross-linking reaction was quenched, proteins were precipitated with TCA, separated by SDS-PAGE, and visualized with Coomassie Brilliant Blue G-250. For the case of the nonactivated PDE6 holoenzyme, a 50-fold molar excess of the cross-linker was used, closely following the protocol of our previous study (20).
To carry out cross-linking reactions with the complex of activated Gtα and PDE6 holoenzyme, PDE6 holoenzyme (10–50 pmol) was mixed with a 500-fold molar excess of Gtα–GDP–AlF4− or Gtα-GTPγS along with 0.6 mg of lipobeads. The mixture was incubated at room temperature for 1 h and then spun at 10,000 × g for 1.5 min (Fig. S5). Unbound proteins in the supernatant fraction (∼10% of the total PDE6 and ∼50% of the Gtα) were discarded, and the lipobead-associated proteins were resuspended and cross-linked for 1 h with the following molar excess of cross-linker relative to PDE6: BS3 or DSS (500-fold), m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester (100-fold), sulfosuccinimidyl 4,4′-azipentanoate (100-fold), or EDC (1000-fold). Following quenching of the cross-linking reaction with 20 mm Tris, pH 7.5, the samples were spun at 5,000 × g for 1.5 min, resuspended in SDS-PAGE sample buffer, and loaded onto NuPAGE 4–12% Bis-Tris gels. Protein bands on the gel were visualized with Coomassie Brilliant Blue G-250.
Cross-linked products were in-gel digested and analyzed by LC-MS and LC-MS/MS as described previously (20), except that we also used Asp-N to generate peptide fragments. For Asp-N digestions, 3 ng of Asp-N were added to the gel pieces and incubated for 18 h at 37 °C. For proteolytic digestions with both enzymes, 300 ng of trypsin was added to the gel pieces for 4 h, then 3 ng of Asp-N was added, and samples were incubated for an additional 18 h. The tryptic peptides were extracted as described (20), and Asp-N or double-digested peptide samples were extracted using 50% acetonitrile and 7% formic acid.
One-microliter aliquots of the concentrated peptides were injected into the Dionex Ultimate 3000 RSLC nano UHPLC system (Dionex Corporation, Sunnyvale, CA) and separated by a PepMap RSLC column (75 μm × 25 cm, 100 Å, 2 μm) at a flow rate of 450 nl/min (mobile phase A: 0.1% formic acid in H2O, mobile phase B: 0.1% formic acid in 80% acetonitrile). The eluant was directed into the nano-electrospray ionization source of an LTQ Orbitrap XL mass spectrometer (Thermo Scientific). LC-MS data were acquired in an information-dependent acquisition mode. Full MS spectra were acquired in the Orbitrap (m/z 315–2000). The five most intense ions were selected for collision-induced dissociation in the linear ion trap for MS/MS data acquisition (24).
Identification of cross-linked peptides
Peak lists were created using RawConverter version 1.1.0.19 for input into Protein Prospector (version 5.14.0 or newer). The data were initially searched against the full Swiss-Prot database (version 2013.6.27 or newer) to verify the absence of contaminating proteins, and then the search was restricted to bovine PDE6 subunits (P11541, P23439, and P04972) and to bovine Gtα (P04695). In experiments in which mutant proteins were used, all protein sequences were input as user-defined proteins. Trypsin or a trypsin/Asp-N double digestion was selected with three to five maximum missed cleavages with precursor and fragment mass tolerances of 15 ppm and 0.7 Da, respectively. A maximum of two variable modifications were allowed, including: acetyl (N terminus); acetyl + oxidation (N-terminal methionine); glutamine to pyro-glutamine; methionine-loss (N-terminal methionine); methionine loss + acetyl (N-terminal methionine); oxidation (methionine); and specific cross-linker modifications (when appropriate). This search process also allowed for a single mass modification based on the cross-linker used: for cross-linkers selectable in Protein Prospector (bis-maleimidoethane, DSS/BS3, and EDC), the preset mass modifications were used; for user-defined cross-links, the default values were used.
Cross-linked peptides were identified using an integrated module in Protein Prospector, using a previously described strategy (20, 56) in which a given score is credited for each fragment matched with the score weighting dependent on the type of ion type matched These scores were then converted to expectation values by determining the distribution of scores for random answers and calculating a probability and then an expectation value of a given score being in this distribution. Samples scoring above 10 for a trypsin digest or 5 for a double digestion were validated based on manual inspection of the spectra. Only results where the score difference was greater than zero confirmed that the cross-linked peptide match was better than a single peptide match alone and were therefore considered. Expectation values were calculated based on matches to single peptides and thus were treated as another score rather than as a statistical measure of reliability. False discovery rates were assessed by comparing the cross-linking data to available structures (57) (PDB codes 6MZB for PDE6 and 1TAD for Gtα).
Integrative structural modeling of PDE6, Gtα, and the Gtα-PDE6–activated complex
Integrative structural modeling was performed using the open-source Integrated Modeling Platform (58) and Modeler (59) in an iterative manner. To perform rigid body docking of protein subunits, IMP was carried out in 2 × 104 Metropolis Monte-Carlo sampling steps with a high temperature of 2.0, a low temperature of 0.5, and with a new system configuration following each step. The top 100 scoring models were generated and saved, and IMP was then used to perform clustering on the top 100 models to aid in model selection. The best fitting model was run in Modeler using the same cross-linking restraints to further refine the model, evaluate stereochemical quality, and fill in the missing atoms. Secondary structure identification was initially determined by PyMOL version 2.3 (Schrodinger) and further refined and validated with Coot (60).
The Pαβ catalytic dimer refinement was performed using the PDE6 cryo-EM structure (23) as the template (PDB code 6MZB). Structural model refinements used the spatial restraints imposed by cross-linked peptides we identified in samples of native and reconstituted PDE6 catalytic subunits, as described previously (20). The domain boundaries and secondary structure assignments for the PDE6 catalytic subunits are given in the supporting information (Fig. S1). Analysis of the root-mean-square deviations of our structural model with other available structures was carried out using Visual Molecular Dynamics software version 1.9.3 (61).
To model the PDE6 holoenzyme (Fig. 1), the refined Pαβ model was used as a single, unchanging rigid body, and each Pγ subunit was treated as eight separate rigid bodies consisting of residues 2–30, 31, 38–41, 44–45, 52–53, 58–62, 68, and 70–87. This approach circumvented the lack of uniform cross-linking data for the entire Pγ subunit. Two of the rigid bodies (residues 2–30 and 70–87) were based on the Pγ structure and topology obtained from the PDE6 cryo-EM structure (23). The remaining Pγ peptide fragments were generated in silico (http://www.arguslab.com/arguslab.com/ArgusLab.html),5 assuming a linearly extended conformation. IMP was then used to dock the Pγ fragments. Subsequently, Modeler was used with the same cross-linking constraints to fill in the missing portions of Pγ, as well as to add the missing atoms to each subunit.
The structure of membrane-associated Gtα–GDP–AlF4− was obtained using the X-ray crystal structure of Gtα–GDP–AlF4− (PDB code 1TAD) (36) as the primary template and imposing distance restraints from cross-linked peptides we identified, as described above. Because the cross-linking data of purified Gtα included cross-links from the N-terminal α-helix of Gtα that is not included in the 1TAD crystal structure, the Gtα structure was refined by including the N-terminal helix (amino acids 1–27) obtained from the transducin heterotrimer structure (PDB code 1GOT) with the 1TAD structure as two rigid bodies for conducting integrative modeling.
The structure of Pγ docked to Gtα–GDP–AlF4− (Fig. 2) was performed by treating Gtα–GDP–AlF4− as a rigid body and dividing the central region of Pγ into three rigid bodies consisting of residues 25, 39–41, and 45 in IMP. Modeler was used to refine the structure and add missing atoms to the model.
The structure of the activated complex of Gtα and the Pαβ catalytic dimer (Fig. 3) was docked using the previously described structures as templates. Pαβ was treated as a single rigid body, and two Gtα–GDP–AlF4− structures were included in the modeling in IMP, followed by refinement with Modeler.
Data availability
The input data files, modeling scripts, and output models can be accessed at https://github.com/rcotelab/Irwin-et-al-2019.5 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (62) with the data set identifier PXD015989.
Author contributions
M. J. I. and R. H. C. conceptualization; M. J. I., R. G., X.-Z. G., F. C., and R. H. C. data curation; M. J. I., R. G., X.-Z. G., K. B. C., F. C., and R. H. C. formal analysis; M. J. I., F. C., and R. H. C. supervision; M. J. I. and R. H. C. funding acquisition; M. J. I. and R. H. C. validation; M. J. I., R. G., X.-Z. G., K. B. C., F. C., and R. H. C. investigation; M. J. I., R. G., and X.-Z. G. visualization; M. J. I., R. G., X.-Z. G., K. B. C., F. C., and R. H. C. methodology; M. J. I. and R. G. writing-original draft; M. J. I. and R. H. C. project administration; M. J. I., R. G., X.-Z. G., F. C., and R. H. C. writing-review and editing.
Supplementary Material
Acknowledgment
We thank Sue Matte for assistance with the preparation of proteins used in this study.
This work was supported by NEI, National Institutes of Health Grant R01 EY05798 (to R. H. C.), NIGMS, National Institutes of Health Grant P20 GM113131 (to R. H. C.), National Science Foundation Grant CLF 1307367 (to F. C.), NICHD, National Institutes of Health Grant R01 HD093783 (to F. C.), and University of New Hampshire Collaborative Research Excellence grant (to R. H. C. and F. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S3 and Figs. S1–S5.
The mass spectrometric raw data and spectral libraries associated with this manuscript are available from ProteomeXchange with the accession number PXD015989.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PDE6
- photoreceptor PDE
- PDE
- phosphodiesterase
- Gtα
- transducin α-subunit (gene name GNAT1)
- Pαβ
- rod PDE6 catalytic heterodimer consisting of the α-subunit (Pα; gene name PDE6A) and the β-subunit (Pβ; PDE6B)
- Pγ
- PDE6 inhibitory subunit (gene name PDE6G)
- EDC
- 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- BS3
- bis(sulfosuccinimidyl)suberate
- DSS
- disuccinimidyl suberate
- PDB
- Protein Data Bank
- IMP
- Integrated Modeling Platform
- αN
- N-terminal α-helix.
References
- 1. Arshavsky V. Y., and Burns M. E. (2012) Photoreceptor signaling: supporting vision across a wide range of light intensities. J. Biol. Chem. 287, 1620–1626 10.1074/jbc.R111.305243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bender A. T., and Beavo J. A. (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 58, 488–520 10.1124/pr.58.3.5 [DOI] [PubMed] [Google Scholar]
- 3. Francis S. H., Blount M. A., and Corbin J. D. (2011) Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol. Rev. 91, 651–690 10.1152/physrev.00030.2010 [DOI] [PubMed] [Google Scholar]
- 4. Zoraghi R., Corbin J. D., and Francis S. H. (2004) Properties and functions of GAF domains in cyclic nucleotide phosphodiesterases and other proteins. Mol. Pharmacol. 65, 267–278 10.1124/mol.65.2.267 [DOI] [PubMed] [Google Scholar]
- 5. Cote R. H. (2006) Photoreceptor phosphodiesterase (PDE6): a G-protein-activated PDE regulating visual excitation in rod and cone photoreceptor cells. In Cyclic Nucleotide Phosphodiesterases in Health and Disease (Beavo J. A., Francis S. H., and Houslay M. D., eds) pp. 165–193, CRC Press, Boca Raton, FL [Google Scholar]
- 6. Granovsky A. E., Natochin M., and Artemyev N. O. (1997) The γ subunit of rod cGMP-phosphodiesterase blocks the enzyme catalytic site. J. Biol. Chem. 272, 11686–11689 10.1074/jbc.272.18.11686 [DOI] [PubMed] [Google Scholar]
- 7. Barren B., Gakhar L., Muradov H., Boyd K. K., Ramaswamy S., and Artemyev N. O. (2009) Structural basis of phosphodiesterase 6 inhibition by the C-terminal region of the γ-subunit. EMBO J. 28, 3613–3622 10.1038/emboj.2009.284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang X. J., Skiba N. P., and Cote R. H. (2010) Structural requirements of the photoreceptor phosphodiesterase γ-subunit for inhibition of rod PDE6 holoenzyme and for its activation by transducin. J. Biol. Chem. 285, 4455–4463 10.1074/jbc.M109.057406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Granovsky A. E., and Artemyev N. O. (2001) A conformational switch in the inhibitory γ-subunit of PDE6 upon enzyme activation by transducin. Biochemistry 40, 13209–13215 10.1021/bi011127j [DOI] [PubMed] [Google Scholar]
- 10. Slepak V. Z., Artemyev N. O., Zhu Y., Dumke C. L., Sabacan L., Sondek J., Hamm H. E., Bownds M. D., and Arshavsky V. Y. (1995) An effector site that stimulates G-protein GTPase in photoreceptors. J. Biol. Chem. 270, 14319–14324 10.1074/jbc.270.24.14319 [DOI] [PubMed] [Google Scholar]
- 11. Slep K. C., Kercher M. A., He W., Cowan C. W., Wensel T. G., and Sigler P. B. (2001) Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature 409, 1071–1077 10.1038/35059138 [DOI] [PubMed] [Google Scholar]
- 12. Guo L. W., and Ruoho A. E. (2008) The retinal cGMP phosphodiesterase γ-subunit: A chameleon. Curr. Protein Pept. Sci. 9, 611–625 10.2174/138920308786733930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X. J., Gao X. Z., Yao W., and Cote R. H. (2012) Functional mapping of interacting regions of the photoreceptor phosphodiesterase (PDE6) γ-subunit with PDE6 catalytic dimer, transducin, and regulator of G-protein signaling 9-1 (RGS9-1). J. Biol. Chem. 287, 26312–26320 10.1074/jbc.M112.377333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muradov H., Boyd K. K., and Artemyev N. O. (2010) Rod phosphodiesterase-6 PDE6A and PDE6B subunits are enzymatically equivalent. J. Biol. Chem. 285, 39828–39834 10.1074/jbc.M110.170068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X., Plachetzki D. C., and Cote R. H. (2019) The N termini of the inhibitory γ-subunits of phosphodiesterase-6 (PDE6) from rod and cone photoreceptors differentially regulate transducin-mediated PDE6 activation. J. Biol. Chem. 294, 8351–8360 10.1074/jbc.RA119.007520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cote R. H., Bownds M. D., and Arshavsky V. Y. (1994) cGMP binding sites on photoreceptor phosphodiesterase: role in feedback regulation of visual transduction. Proc. Natl. Acad. Sci. U.S.A. 91, 4845–4849 10.1073/pnas.91.11.4845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Norton A. W., D'Amours M. R., Grazio H. J., Hebert T. L., and Cote R. H. (2000) Mechanism of transducin activation of frog rod photoreceptor phosphodiesterase: allosteric interactions between the inhibitory γ subunit and the noncatalytic cGMP binding sites. J. Biol. Chem. 275, 38611–38619 10.1074/jbc.M004606200 [DOI] [PubMed] [Google Scholar]
- 18. Mou H., and Cote R. H. (2001) The catalytic and GAF domains of the rod cGMP phosphodiesterase (PDE6) heterodimer are regulated by distinct regions of its inhibitory γ subunit. J. Biol. Chem. 276, 27527–27534 10.1074/jbc.M103316200 [DOI] [PubMed] [Google Scholar]
- 19. Guo L. W., Grant J. E., Hajipour A. R., Muradov H., Arbabian M., Artemyev N. O., and Ruoho A. E. (2005) Asymmetric interaction between rod cyclic GMP phosphodiesterase γ subunits and αβ subunits. J. Biol. Chem. 280, 12585–12592 10.1074/jbc.M410380200 [DOI] [PubMed] [Google Scholar]
- 20. Zeng-Elmore X., Gao X. Z., Pellarin R., Schneidman-Duhovny D., Zhang X. J., Kozacka K. A., Tang Y., Sali A., Chalkley R. J., Cote R. H., and Chu F. (2014) Molecular architecture of photoreceptor phosphodiesterase elucidated by chemical cross-linking and integrative modeling. J. Mol. Biol. 426, 3713–3728 10.1016/j.jmb.2014.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qureshi B. M., Behrmann E., Schöneberg J., Loerke J., Bürger J., Mielke T., Giesebrecht J., Noé F., Lamb T. D., Hofmann K. P., Spahn C. M. T., and Heck M. (2018) It takes two transducins to activate the cGMP-phosphodiesterase 6 in retinal rods. Open. Biol. 8, 180075 10.1098/rsob.180075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z., He F., Constantine R., Baker M. L., Baehr W., Schmid M. F., Wensel T. G., and Agosto M. A. (2015) Domain organization and conformational plasticity of the G protein effector, PDE6. J. Biol. Chem. 290, 17131–17132 10.1074/jbc.A115.647636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gulati S., Palczewski K., Engel A., Stahlberg H., and Kovacik L. (2019) Cryo-EM structure of phosphodiesterase 6 reveals insights into the allosteric regulation of type I phosphodiesterases. Sci. Adv. 5, eaav4322. 10.1126/sciadv.aav4322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chu F., Hogan D., Gupta R., Gao X. Z., Nguyen H. T., and Cote R. H. (2019) Allosteric regulation of rod photoreceptor phosphodiesterase 6 (PDE6) elucidated by chemical cross-linking and quantitative mass spectrometry. J. Mol. Biol. 431, 3677–3689 10.1016/j.jmb.2019.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anant J. S., Ong O. C., Xie H. Y., Clarke S., O'Brien P. J., and Fung B. K. (1992) In vivo differential prenylation of retinal cyclic GMP phosphodiesterase catalytic subunits. J. Biol. Chem. 267, 687–690 [PubMed] [Google Scholar]
- 26. Zhang Z., Melia T. J., He F., Yuan C., McGough A., Schmid M. F., and Wensel T. G. (2004) How a G protein binds a membrane. J. Biol. Chem. 279, 33937–33945 10.1074/jbc.M403404200 [DOI] [PubMed] [Google Scholar]
- 27. Artemyev N. O., Rarick H. M., Mills J. S., Skiba N. P., and Hamm H. E. (1992) Sites of interaction between rod G-protein α-subunit and cGMP-phosphodiesterase γ-subunit: implications for phosphodiesterase activation mechanism. J. Biol. Chem. 267, 25067–25072 [PubMed] [Google Scholar]
- 28. Artemyev N. O., Mills J. S., Thornburg K. R., Knapp D. R., Schey K. L., and Hamm H. E. (1993) A site on transducin α-subunit of interaction with the polycationic region of cGMP phosphodiesterase inhibitory subunit. J. Biol. Chem. 268, 23611–23615 [PubMed] [Google Scholar]
- 29. Cunnick J., Twamley C., Udovichenko I., Gonzalez K., and Takemoto D. J. (1994) Identification of a binding site on retinal transducin a for the phosphodiesterase inhibitory γ subunit. Biochem. J. 297, 87–91 10.1042/bj2970087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grant J. E., Guo L. W., Vestling M. M., Martemyanov K. A., Arshavsky V. Y., and Ruoho A. E. (2006) The N terminus of GTPγS-activated transducin α-subunit interacts with the C terminus of the cGMP phosphodiesterase γ-subunit. J. Biol. Chem. 281, 6194–6202 10.1074/jbc.M509511200 [DOI] [PubMed] [Google Scholar]
- 31. Malinski J. A., and Wensel T. G. (1992) Membrane stimulation of cGMP phosphodiesterase activation by transducin: Comparison of phospholipid bilayers to rod outer segment membranes. Biochemistry 31, 9502–9512 10.1021/bi00154a024 [DOI] [PubMed] [Google Scholar]
- 32. Melia T. J., Malinski J. A., He F., and Wensel T. G. (2000) Enhancement of phototransduction protein interactions by lipid surfaces. J. Biol. Chem. 275, 3535–3542 10.1074/jbc.275.5.3535 [DOI] [PubMed] [Google Scholar]
- 33. Natochin M., Granovsky A. E., and Artemyev N. O. (1998) Identification of effector residues on photoreceptor G protein, transducin. J. Biol. Chem. 273, 21808–21815 10.1074/jbc.273.34.21808 [DOI] [PubMed] [Google Scholar]
- 34. Qi C., Sorrentino S., Medalia O., and Korkhov V. M. (2019) The structure of a membrane adenylyl cyclase bound to an activated stimulatory G protein. Science 364, 389–394 10.1126/science.aav0778 [DOI] [PubMed] [Google Scholar]
- 35. Chu F., Thornton D. T., and Nguyen H. T. (2018) Chemical cross-linking in the structural analysis of protein assemblies. Methods 144, 53–63 10.1016/j.ymeth.2018.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sondek J., Lambright D. G., Noel J. P., Hamm H. E., and Sigler P. B. (1994) GTPase mechanism of G-proteins from the 1.7-Å crystal structure of transducin α–GDP–AlF4−. Nature 372, 276–279 10.1038/372276a0 [DOI] [PubMed] [Google Scholar]
- 37. Lamb T. D., Heck M., and Kraft T. W. (2018) Implications of dimeric activation of PDE6 for rod phototransduction. Open Biol. 8, 180076 10.1098/rsob.180076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Skiba N. P., Bae H., and Hamm H. E. (1996) Mapping of effector binding sites of transducin α-subunit using Gat/Ga i1 chimeras. J. Biol. Chem. 271, 413–424 10.1074/jbc.271.1.413 [DOI] [PubMed] [Google Scholar]
- 39. Liu Y., Arshavsky V. Y., and Ruoho A. E. (1996) Interaction sites of the COOH-terminal region of the γ subunit of cGMP phosphodiesterase with the GTP-bound a subunit of transducin. J. Biol. Chem. 271, 26900–26907 10.1074/jbc.271.43.26900 [DOI] [PubMed] [Google Scholar]
- 40. Milano S. K., Wang C., Erickson J. W., Cerione R. A., and Ramachandran S. (2018) Gain-of-function screen of a-transducin identifies an essential phenylalanine residue necessary for full effector activation. J. Biol. Chem. 293, 17941–17952 10.1074/jbc.RA118.003746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szabo V., Kreienkamp H. J., Rosenberg T., and Gal A. (2007) p.Gln200Glu, a putative constitutively active mutant of rod a-transducin (GNAT1) in autosomal dominant congenital stationary night blindness. Hum. Mutat. 28, 741–742 10.1002/humu.9499 [DOI] [PubMed] [Google Scholar]
- 42. Naeem M. A., Chavali V. R., Ali S., Iqbal M., Riazuddin S., Khan S. N., Husnain T., Sieving P. A., Ayyagari R., Riazuddin S., Hejtmancik J. F., and Riazuddin S. A. (2012) GNAT1 associated with autosomal recessive congenital stationary night blindness. Invest. Ophthalmol. Vis. Sci. 53, 1353–1361 10.1167/iovs.11-8026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maryam A., Vedithi S. C., Khalid R. R., Alsulami A. F., Torres P. H. M., Siddiqi A. R., and Blundell T. L. (2019) The molecular organization of human cGMP specific phosphodiesterase 6 (PDE6): structural implications of somatic mutations in cancer and retinitis pigmentosa. Comput. Struct. Biotechnol. J. 17, 378–389 10.1016/j.csbj.2019.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Power M., Das S., Schutze K., Marigo V., Ekstrom P., and Paquet-Durand F. (2019) Cellular mechanisms of hereditary photoreceptor degeneration: focus on cGMP. Prog. Retin. Eye Res., in press 10.1016/j.preteyeres.2019.07.005 [DOI] [PubMed] [Google Scholar]
- 45. Omar F., Findlay J. E., Carfray G., Allcock R. W., Jiang Z., Moore C., Muir A. L., Lannoy M., Fertig B. A., Mai D., Day J. P., Bolger G., Baillie G. S., Schwiebert E., Klussmann E., et al. (2019) Small-molecule allosteric activators of PDE4 long form cyclic AMP phosphodiesterases. Proc. Natl. Acad. Sci. U.S.A. 116, 13320–13329 10.1073/pnas.1822113116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baillie G. S., Tejeda G. S., and Kelly M. P. (2019) Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat. Rev. Drug. Discov. 18, 770–796 10.1038/s41573-019-0033-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pentia D. C., Hosier S., Collupy R. A., Valeriani B. A., and Cote R. H. (2005) Purification of PDE6 isozymes from mammalian retina. Methods Mol. Biol 307, 125–140 [DOI] [PubMed] [Google Scholar]
- 48. Cote R. H. (2000) Kinetics and regulation of cGMP binding to noncatalytic binding sites on photoreceptor phosphodiesterase. Methods Enzymol. 315, 646–672 10.1016/S0076-6879(00)15873-2 [DOI] [PubMed] [Google Scholar]
- 49. Mou H., Grazio H. J. 3rd, Cook T. A., Beavo J. A., and Cote R. H. (1999) cGMP binding to noncatalytic sites on mammalian rod photoreceptor phosphodiesterase is regulated by binding of its g and d subunits. J. Biol. Chem. 274, 18813–18820 10.1074/jbc.274.26.18813 [DOI] [PubMed] [Google Scholar]
- 50. Ting T. D., Goldin S. B., and Ho Y.-K. (1993) Purification and characterization of bovine transducin and its subunits. In Photoreceptor Cells (Hargrave P. A., ed.) pp. 180–195, Academic Press, New York [Google Scholar]
- 51. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., and Klenk D. C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 10.1016/0003-2697(85)90442-7 [DOI] [PubMed] [Google Scholar]
- 52. Bigay J., Deterre P., Pfister C., and Chabre M. (1987) Fluoride complexes of aluminium or beryllium act on G-proteins as reversibly bound analogues of the γ phosphate of GTP. EMBO J. 6, 2907–2913 10.1002/j.1460-2075.1987.tb02594.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Artemyev N. O., Arshavsky V. Y., and Cote R. H. (1998) Photoreceptor phosphodiesterase: interaction of inhibitory γ subunit and cyclic GMP with specific binding sites on catalytic subunits. Methods 14, 93–104 10.1006/meth.1997.0568 [DOI] [PubMed] [Google Scholar]
- 54. Wensel T. G., He F., and Malinski J. A. (2005) Purification, reconstitution on lipid vesicles, and assays of PDE6 and its activator G protein, transducin. Methods Mol. Biol. 307, 289–313 [DOI] [PubMed] [Google Scholar]
- 55. Alkhammash H. I., Li N., Berthier R., and de Planque M. R. (2015) Native silica nanoparticles are powerful membrane disruptors. Phys. Chem. Chem. Phys. 17, 15547–15560 10.1039/C4CP05882H [DOI] [PubMed] [Google Scholar]
- 56. Chu F., Baker P. R., Burlingame A. L., and Chalkley R. J. (2010) Finding chimeras: a bioinformatics strategy for identification of cross-linked peptides. Mol. Cell. Proteomics 9, 25–31 10.1074/mcp.M800555-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Trnka M. J., Baker P. R., Robinson P. J., Burlingame A. L., and Chalkley R. J. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 13, 420–434 10.1074/mcp.M113.034009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Webb B., Lasker K., Velázquez-Muriel J., Schneidman-Duhovny D., Pellarin R., Bonomi M., Greenberg C., Raveh B., Tjioe E., Russel D., and Sali A. (2014) Modeling of proteins and their assemblies with the Integrative Modeling Platform. Methods Mol. Biol. 1091, 277–295 10.1007/978-1-62703-691-7_20 [DOI] [PubMed] [Google Scholar]
- 59. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 60. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 62. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The input data files, modeling scripts, and output models can be accessed at https://github.com/rcotelab/Irwin-et-al-2019.5 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (62) with the data set identifier PXD015989.