

Abstract
Interferons inhibit viruses by inducing antiviral protein expression. One of the interferon-induced antiviral proteins, human Moloney leukemia virus 10 (MOV10), a superfamily 1 RNA helicase, has been shown to inhibit retroviruses and several RNA viruses. However, it remains undetermined whether MOV10 also inhibits DNA viruses, including hepatitis B virus (HBV). Here, we report that MOV10 dramatically reduces the levels of intracellular HBV DNA, resulting in significant inhibition of both the HBV experimental strain and the clinical isolates. Mechanistic experiments revealed that MOV10 interacts with HBV RNA and blocks the early step of viral reverse transcription, thereby impairing viral DNA synthesis, without affecting viral gene expression and pregenomic RNA encapsidation. Moreover, mutation of the helicase domain of MOV10 caused loss of binding to HBV RNA and of the anti-HBV activity. Together, our results indicate that MOV10 restricts HBV replication, insights that may open new avenues to the development of anti-HBV therapeutics.
Keywords: hepatitis B virus (HBV, Hep B); DNA viruses; innate immunity; RNA helicase; viral replication; liver disease; Moloney leukemia virus 10 (MOV10); RNA silencing; viral reverse transcription; virus-host interactions
Introduction
Hepatitis B virus (HBV),3 a small enveloped hepatotropic DNA virus, infects only humans and chimpanzees. It is estimated that ∼2 billion people have been exposed to HBV, with more than 10% becoming chronic carriers (1). Chronic HBV infection greatly increases the risk of developing progressive liver diseases (2). HBV-related liver diseases, including liver fibrosis, cirrhosis, and hepatocellular carcinoma, account for about 1 million deaths per year worldwide (3). Hence, HBV infection represents a major threat to public health.
As the best-studied member of Hepadnaviridae, HBV has also been classified as a pararetrovirus, because HBV DNA genome is generated by reverse transcription of the pregenomic RNA (pgRNA) (4). The synthesis of viral genomic DNA requires encapsidation of the 3.5-kb pgRNA into nascent nucleocapsid, which depends on viral polymerase (Pol) which selectively recognizes pgRNA via an RNA stem-loop structure called the 5′ϵ encapsidation signal. Following encapsidation, pgRNA is reverse transcribed by the co-packaged HBV Pol into a 3.2-kb partial dsDNA within viral nucleocapsid (5). In addition to viral pgRNA and proteins, several host factors have been reported to regulate HBV DNA replication. For instance, heat shock protein 90 is incorporated into nucleocapsid and promotes viral DNA synthesis (6). In contrast, human cytidine deaminase APOBEC3G functions as a host restriction factor by inhibiting HBV reverse transcription (7, 8). It remains to be determined whether there exist other host factors which also modulate HBV reverse transcription.
Human Moloney leukemia virus 10 (MOV10) protein is a 110-kDa RNA helicase. It contains seven conserved helicase motifs (I, Ia, II, III, IV, V, and VI) in its C terminus and is classified as a member of the helicase superfamily 1 based on the DEAG signature sequence in its motif II (9). MOV10 was initially identified as a putative GTP-binding protein in a transgenic mouse strain, which carried one copy of integrated proviral genome of Moloney leukemia virus located in the MOV-10 gene locus (10, 11). The cellular functions of MOV10 protein remained unknown until it was found to associate with argonaute proteins in the RNA-induced silencing complex, and thus is required for microRNA-guided mRNA cleavage (12). Besides the critical role in RNA silencing, several recent reports have uncovered additional functions of MOV10, including suppression of retrotransposition of endogenous retroelements and promoting the degradation of UPF1-regulated mRNA transcripts (13–15).
Increasing experimental evidence has shown that MOV10 plays an active role in viral replication. In the case of hepatitis D virus, MOV10 promotes viral replication through enhancing RNA-directed transcription (16). However, for the other viruses that have been tested, MOV10 acts as a host restriction factor. Ectopic expression of MOV10 represses replication of a number of retroviruses, although the antiviral activity of endogenous MOV10 remains inconclusive (17–19). The antiretroviral activity of MOV10 is well-established for HIV type 1 (HIV-1) by three independent studies, which demonstrate that MOV10 is efficiently incorporated into HIV-1 virions and interferes with viral reverse transcription in the target cells (9, 20, 21). MOV10 has been further reported to inhibit retrotransposition of intracisternal a particles, an endogenous retrovirus in the mammalian genome (22).
Although HBV is a DNA virus, replication of its DNA genome involves reverse transcription, which raises the possibility that MOV10 may also inhibit HBV infection. Indeed, in this study, we observed that MOV10 restricted HBV replication by reducing the level of intracellular viral DNA. Furthermore, our data showed that MOV10 targeted the early step of HBV reverse transcription.
Results
MOV10 inhibits HBV DNA replication
To investigate the effect of MOV10 on HBV replication, an overlength 1.1-mer HBV construct was co-transfected into HepG2 cells together with different amounts of MOV10-expressing plasmids, and with constant amount of GFP-expressing plasmids to normalize the transfection efficiency. Three days after transient transfection, the transfected cells were harvested and the expressions of MOV10 and GFP proteins were examined by immunoblotting with the respective antibodies (Fig. 1A). To determine HBV replication, we first measured the hepatitis B surface antigen (HBsAg) in the extracellular culture medium and found that MOV10 had no significant effect on the level of secreted HBsAg (Fig. 1B). Next, intracellular HBV DNA was extracted and subsequently analyzed by Southern blotting and quantitative real-time PCR (qPCR), respectively. Interestingly, we found that overexpression of MOV10 reduced HBV DNA levels in a dose-dependent manner (Fig. 1, C and D). Because the data of qPCR were consistent with that of Southern blotting (Fig. 1, C and D), we employed qPCR method to detect HBV DNA in following experiments. Furthermore, we analyzed HBV DNA levels and the infectivity of released viral particles in the extracellular culture medium. We observed similar degree of inhibition by MOV10 (Fig. 1, E–G), suggesting MOV10 inhibits intracellular viral DNA synthesis, not virus release.
Figure 1.

Overexpression of MOV10 inhibits HBV replication in HepG2 cells. A, different amounts of MOV10-expressing plasmids (0.25 μg, 0.5 μg, 1 μg) were co-transfected into HepG2 cells together with HBV construct pCMV-HBV (0.5 μg), and with constant amount of GFP-expressing plasmids (0.1 μg) to normalize transfection efficiency. The expression of MOV10 (top) and GFP (bottom) were detected by Western blotting. B, the culture medium of transfected cells was tested for HBsAg with ELISA. C, Southern blot analysis of extracted intracellular HBV DNA. The positions of RC, double-strand linear (DSL), and ssDNA intermediates are indicated. D and E, intracellular (D) and extracellular (E) HBV DNA were measured by qPCR. F, the supernatants of transfected cells were collected at 3 and 6 days post transfection, respectively. Equivalent amounts of 50-fold concentrated supernatants were used to infect HepG2-hNTCP cells. F and G, the culture medium collected at 6 days post infection was subsequently tested for hepatitis B e antigen (F) and HBV DNA levels (G). Results from three separate experiments are shown as mean values ± S.D. *, p < 0.05 (Student's t test).
Next, we investigated the impact of endogenous MOV10 knockdown on HBV replication. To this end, we established a stable HepG2 cell line that was transduced with MOV10 shRNA. The decrease of MOV10 in this cell line was confirmed by the results of Western blotting (Fig. 2A). We found that MOV10 depletion had no significant effect on secreted HBsAg level (Fig. 2B), whereas intracellular HBV DNA levels increased by about 2-fold (Fig. 2C), suggesting endogenous MOV10 suppresses HBV replication.
Figure 2.
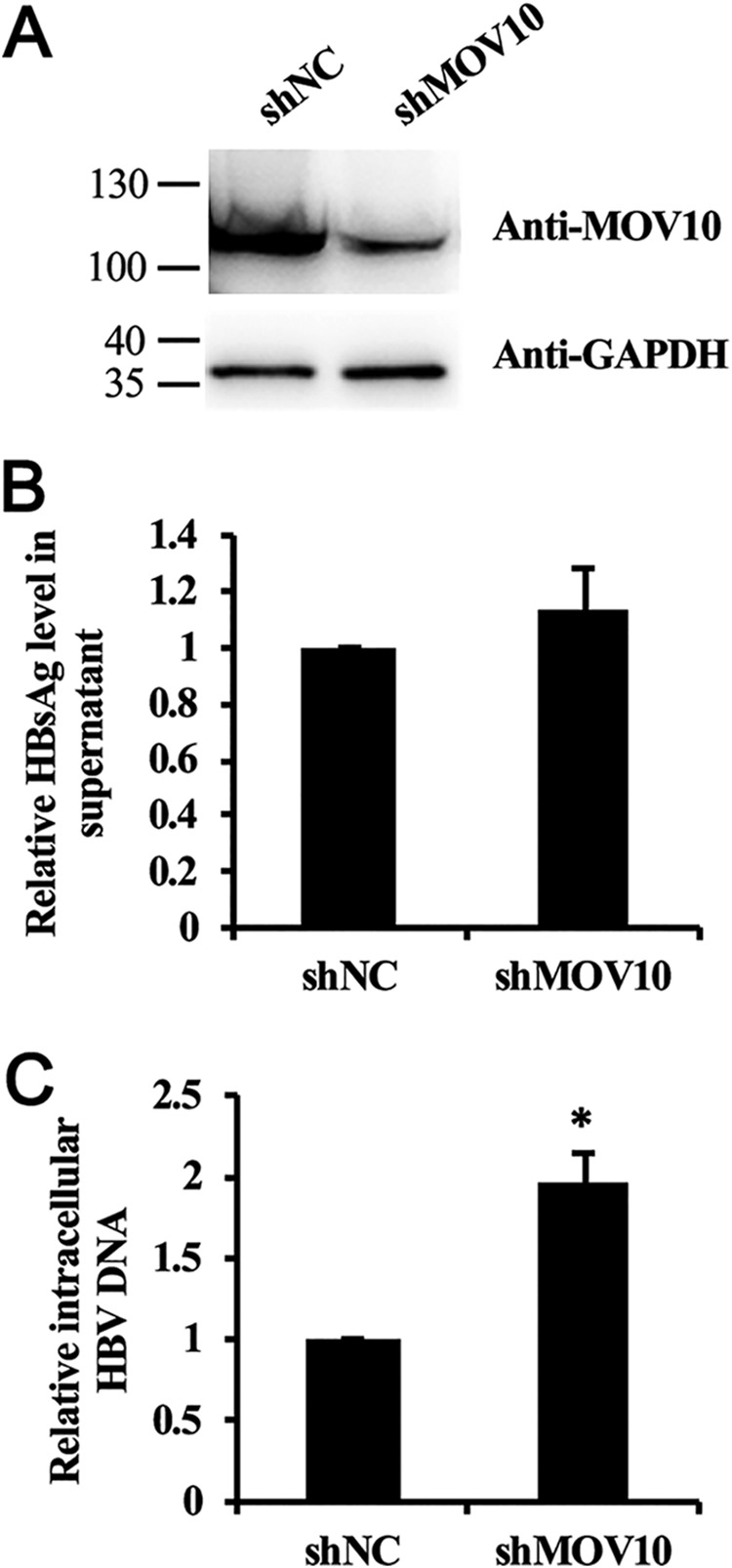
Knockdown of MOV10 increases HBV replication in HepG2 cells. HBV construct pCMV-HBV (0.5 μg) and GFP-expressing plasmids (0.1 μg) were transfected into control and stable MOV10-knockdown HepG2 cell lines, respectively. A, levels of endogenous MOV10 protein in control and stable MOV10-knockdown HepG2 cell lines were examined by Western blotting. B, the effect of MOV10 silencing on secreted HBsAg. C, the effect of MOV10 silencing on intracellular HBV DNA level. Intracellular HBV DNA was extracted and measured by qPCR. Results from three separate experiments are shown as mean values ± S.D. *, p < 0.05 (Student's t test).
To answer whether above findings were reproducible in other hepatoma cell lines, we performed the same experiments in Huh7 cells and observed similar inhibitory effect of MOV10 on HBV DNA replication (Fig. S1, A–G), which supports that MOV10-mediated inhibition of HBV is not cell type–specific.
Finally, we examined the anti-HBV activity of MOV10 with four more HBV-expressing DNA constructs which were cloned from the sera of chronic hepatitis B patients. These four HBV-expressing DNA constructs carry 1.3-mer the HBV genomes of two major different genotypes, B (HBV-B) and C (HBV-C), and the DNA construct pCMV-HBV we tested in the above experiments contains the HBV genome of genotype D. The results showed that MOV10 reduced the intracellular DNA levels of all four clinical HBV isolates (Fig. 3). Taken together, these data indicate that MOV10 restricts HBV replication by reducing the level of intracellular viral DNA.
Figure 3.
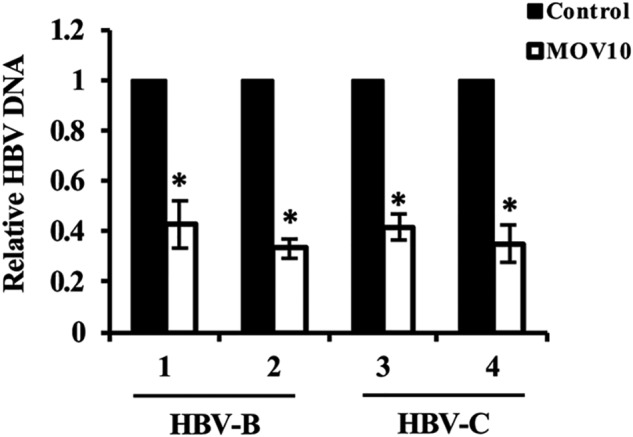
Activity of MOV10 on HBV isolates from chronic hepatitis B patients. The HBV-expressing constructs carrying 1.3-mer the HBV genomes (two genotype B and two genotype C) were cloned from the sera of chronic hepatitis B patients. These HBV constructs and GFP-expressing plasmids were co-transfected into HepG2 cells along with the MOV10-expressing plasmid, or control vector. Three days after transfection, intracellular HBV DNA was extracted and analyzed by qPCR. Results from three separate experiments are shown as mean values ± S.D. *, p < 0.05 (Student's t test).
Inhibition of HBV DNA replication by MOV10 helicase mutants
We next asked whether the RNA helicase activity of MOV10 is required for the inhibition of HBV replication. Among the seven conserved helicase motifs of MOV10, motif I and motif II are Walker A (GKT) and Walker B (DEAG) motifs, respectively. These two motifs are essential for the ATPase activity (23), thus mutating either of them inactivates the helicase activity of MOV10 (15). Accordingly, two MOV10 helicase mutants, MOV10 (KR) and MOV10 (EQ), were generated by changing the amino acids Lys-531 and Glu-647 to Arg and Gln, respectively (Fig. 4A). Expression of these two MOV10 mutants was confirmed by immunoblotting (Fig. 4B). Analysis of intracellular HBV DNA revealed that anti-HBV activities of both MOV10 mutants were attenuated compared with the WT MOV10, with the MOV10 (EQ) mutant exhibiting relatively stronger anti-HBV activity than the MOV10 (KR) mutant (Fig. 4C). This observation is in agreement with previous reports showing significantly reduced activities of these two mutants against HIV-1 and LINE-1 (14, 24).
Figure 4.
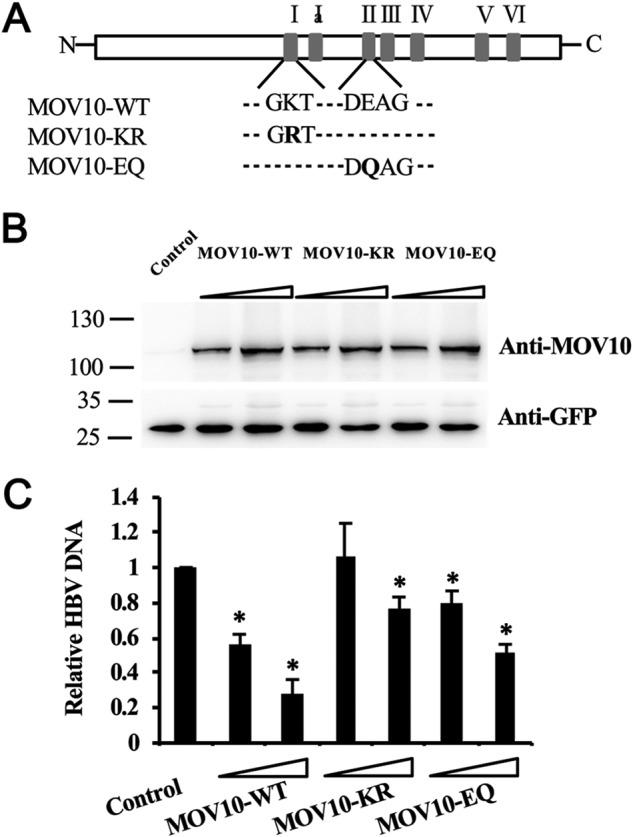
Inhibition of HBV replication by MOV10 helicase mutants. A, schematic illustration of WT MOV10 and two helicase mutants, MOV10-KR and MOV10-EQ. B, different amounts of MOV10-WT, MOV10-KR or MOV10-EQ vectors (0.5 μg, 1 μg) were co-transfected into HepG2 cells together with HBV construct pCMV-HBV (0.5 μg), and with constant amount of GFP-expression plasmids (0.1 μg) to normalize the transfection efficacy. The expression of MOV10 (top) and GFP (bottom) were detected by Western blotting. C, the effect of MOV10 and its two helicase mutants on HBV genome replication. Intracellular HBV DNA were extracted and measured by qPCR. Results from three separate experiments are shown as mean values ± S.D. *, p < 0.05 (Student's t test).
MOV10 does not affect HBV gene expression, pgRNA encapsidation, and DNA stability
We next determined which step of HBV replication was inhibited by MOV10. MOV10 has been reported to regulate gene expression (12, 15). Therefore, it may exert its antiviral activity by directly reducing viral RNA levels. To test this possibility, we co-transfected HepG2 cells with the 1.1-mer HBV construct along with the MOV10-expressing plasmid, or control vector. Three days after transfection, the total RNA of transfected cells were extracted and the levels of viral RNAs were measured by qPCR. The data showed that MOV10 had no significant effect on the levels of the total and the 3.5 kb HBV RNA (Fig. 5A). We next analyzed viral protein expression by Western blotting and detected no significant difference in the hepatitis B core antigen (HBcAg) expression (Fig. 5B), which is consistent with the observation that MOV10 did not affect the levels of secreted HBsAg (Figs. 1B and 2B), suggesting that MOV10 does not affect the expression of viral proteins.
Figure 5.

MOV10 does not affect HBV gene expression, pgRNA encapsidation, and DNA stability. A, the effect of MOV10 on HBV RNA expression. MOV10-expressing plasmids (1 μg), or control vector were co-transfected into HepG2 cells together with HBV constructs pCMV-HBV (0.5 μg) and GFP-expressing plasmids (0.1 μg). HBV RNA were extracted and measured by qPCR. The co-transfected GFP gene was served as internal control. B, the effect of MOV10 on HBcAg production. The expression of HBcAg (top), MOV10 (middle), and GFP (bottom) were detected by Western blotting. C, the effect of MOV10 on pgRNA encapsidation. The cytoplasmic encapsidated pgRNA was extracted and determined by qPCR. D, the effect of MOV10 on intracellular HBV DNA stability. The experimental scheme is shown on the top. Two days after transient transfection, the cells were treated with 100 μm lamivudine. Levels of intracellular HBV DNA in the cells at different time points were measured by qPCR. Results from three separate experiments are shown as mean values ± S.D. *, p < 0.05 (Student's t test).
Next, we examined whether MOV10 affects HBV pgRNA encapsidation. To specifically detect the effect of MOV10 on pgRNA encapsidation, 1 day after transfection, we treated the cells with 100 μm lamivudine to block viral reverse transcription, the step following pgRNA encapsidation. Three days after transfection, we extracted and analyzed total viral RNAs and encapsidated pgRNAs, respectively. The qPCR results showed that the encapsidation efficiency was not changed by MOV10 overexpression (Fig. 5C), suggesting MOV10 inhibits HBV genome replication downstream of pgRNA encapsidation, either by blocking viral DNA synthesis, or promoting degradation of the synthesized HBV DNA.
To determine whether MOV10 accelerates the degradation of HBV DNA, we treated the transfected cells at day 3 after transfection with high dose of lamivudine (100 μm) to block further DNA synthesis. Then, we measured the intracellular HBV DNA levels at different time points as indicated (Fig. 5D). Our data showed that the decay kinetics of HBV DNA remained the same, regardless of MOV10 expression (Fig. 5D). In summary, these data support that MOV10 reduces intracellular HBV DNA levels through inhibiting viral DNA synthesis.
MOV10 impairs HBV reverse transcription
Next, we examined which step of viral DNA synthesis was affected by MOV10. The synthesis of HBV genomic DNA is initiated by reverse transcription of viral pgRNA into minus-strand DNA, followed by copying minus-strand DNA into the plus-strand DNA (25). To specifically measure the effect of MOV10 on viral reverse transcription, we employed an RH-defective polymerase HBV mutant, which is unable to degrade the pgRNA during reverse transcription, resulting in accumulation of nascent minus-strand DNA and the absence of plus-strand DNA (7). We co-transfected HepG2 cells with WT or RH-defective polymerase mutant together with control or MOV10-expressing plasmids. Three days after transfection, intracellular viral DNA was extracted and analyzed by Southern blotting and qPCR, respectively. Consistent with previous report (7), RH-defective polymerase mutant synthesized double-strand DNA-RNA hybrids, which migrate between ssDNA and relaxed circular (RC) DNA (Fig. 6A). MOV10 reduced levels of intracellular minus-strand DNA by about 3-fold, which is similar to its effect on the synthesis of WT HBV DNA (Fig. 6, A and B). Furthermore, to examine the effect of MOV10 on elongation of minus-strand DNA synthesis, we analyzed extracted intracellular minus-strand DNA by qPCR with different sets of primers (Fig. 6C). The data of qPCR analysis with different sets of primers showed similar reduction of HBV DNA (Fig. 6C), suggesting MOV10 blocks very early steps of viral reverse transcription, not elongation of minus-strand DNA.
Figure 6.

MOV10 impairs HBV reverse transcription. A, HepG2 cells were co-transfected with the wide type, or RH-defective HBV construct (0.5 μg) along with the MOV10-expressing plasmid (1 μg), or control vector. A and B, three days after transfection, extracted intracellular HBV DNA was analyzed by Southern blotting (A) and qPCR (B). The positions of RC, double-strand linear (DSL), and ssDNA intermediates are indicated. C, schematic presentation of HBV pgRNA and (−)DNA that was synthesized by RH-defective Pol. Positions of the sequences amplified by qPCR are indicated with numbered black lines. Intracellular (−) DNA was analyzed by qPCR with different sets of primers. Results from three separate experiments are shown as mean values ± S.D. *, p < 0.05 (Student's t test).
MOV10 interacts with HBV RNA
Because our data suggest that MOV10 inhibits HBV DNA synthesis at the reverse transcription step, we investigated whether MOV10 interacts with viral Pol or viral RNA. We first examined the interaction between MOV10 and viral Pol by performing immunoprecipitation. Human cytidine deaminase APOBEC3G, which has been reported to interact with HBV Pol (26), was used as a positive control. Our data did not show any detectable interaction between MOV10 and HBV Pol, whereas APOBEC3G co-precipitated with Pol, as expected (Fig. 7A).
Figure 7.

MOV10 interacts with HBV RNA. A, MOV10 does not interact with HBV Pol. 293T cells were co-transfected with the MOV10- or APOBEC3G-expressing plasmids along with the FLAG-Pol-expressing plasmids, or empty vector. Immunoprecipitation was performed with anti-FLAG antibodies. B, endogenous MOV10 interacts with HBV RNA. HepG2 cells were transfected with HBV construct pCMV-HBV. RIP assay were performed with IgG, or anti-MOV10 antibodies. The immunoprecipitated HBV 3.5kb RNA was measured by qPCR. The housekeeping gene GAPDH was used as internal control. C, HepG2 cells were co-transfected with HBV construct pCMV-HBV and MOV10-WT, MOV10-KR, or MOV10-EQ vectors. RIP assay were performed with anti-FLAG antibodies. The immunoprecipitated HBV 3.5kb RNA was measured by qPCR. The housekeeping gene GAPDH was used as internal control. Results from three separate experiments are shown as mean values ± S.D. *, p < 0.05 (Student's t test).
We next investigated whether MOV10 binds to HBV RNA by conducting RNA immunoprecipitation (RIP) analysis. We used anti-MOV10 antibody and anti-Flag antibody to precipitate endogenous and FLAG-tagged MOV10, respectively. Co-immunoprecipitated RNA was extracted and analyzed by qPCR. These results showed that both endogenous and exogenous MOV10 associated with HBV RNA (Fig. 7, B and C). Furthermore, we also performed RIP assay to assess the interaction of MOV10 mutants, MOV10 (KR), and MOV10 (EQ), with HBV RNA. Interestingly, these two mutants, with attenuated anti-HBV activities, also have decreased HBV RNA–binding capacity compared with WT MOV10. Consistent with their relative anti-HBV activities, the MOV10 (EQ) mutant had stronger interaction with viral RNA compared with the MOV10 (KR) mutant (Fig. 7C).
MOV10 restricts HBV infection in HBV-susceptible HepG2-NTCP cells
Finally, we tested anti-HBV activity of MOV10 in HepG2-hNTCP cells. We transduced HepG2-hNTCP cells with lentivirus expressing MOV10 or MOV10 shRNA to establish the MOV10-overexpressing or knockdown cell lines. The expression of MOV10 in these cell lines was examined by immunoblotting (Fig. 8A). Then, the equivalent amounts of HBV virions were used to infect these cell lines. One week after infection, intracellular HBV DNA in infected cells was extracted and analyzed by qPCR. The results showed that HBV DNA was reduced by about 70% in the MOV10-overexpressing cell line (Fig. 8B). In contrast, HBV replication was increased by about 3-fold upon MOV10 knockdown (Fig. 8B). Interestingly, we also observed a moderate effect of MOV10 on the levels of HBsAg and viral RNA (Fig. 8, C and D). Then, we measured HBV covalently closed circular DNA (cccDNA) and found that the effect of MOV10 on HBsAg and viral RNA was mainly attributed to its effect on cccDNA formation (Fig. 8E), which indicates MOV10 also plays a role at early stage of the HBV life cycle.
Figure 8.
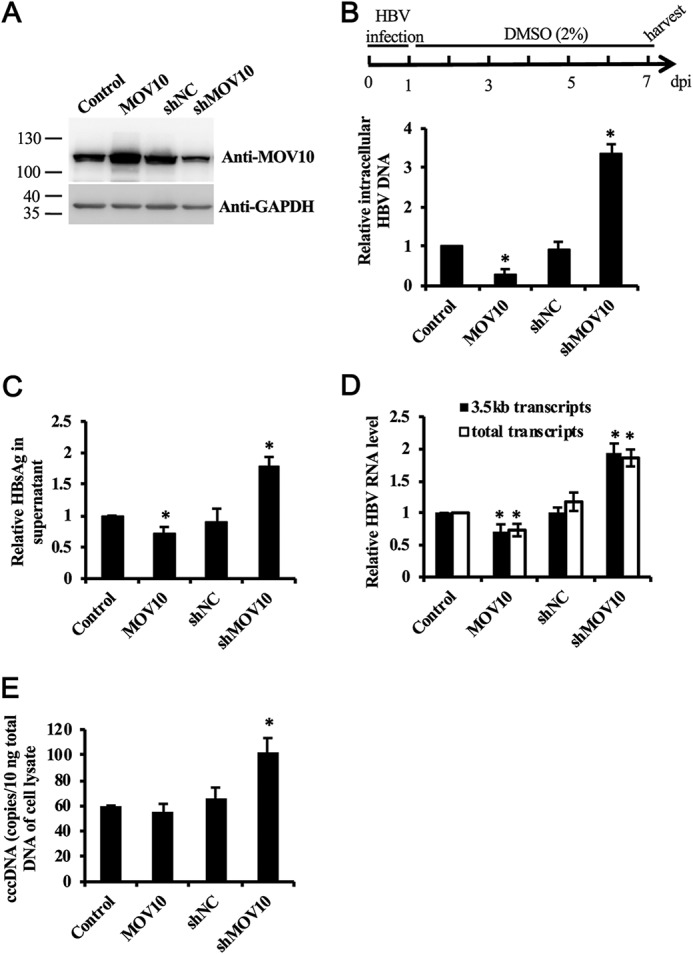
MOV10 restricts HBV infection in HBV-susceptible HepG2-NTCP cells. A, levels of MOV10 in the control, MOV10-overexpressing, and MOV10-knockdown HepG2-hNTCP cell lines were examined by Western blotting. B–D, 1 week after infection, intracellular HBV DNA (B), RNA (D), and cccDNA (E) in the infected cells were extracted and analyzed by qPCR. C, the level of HBsAg in supernatant was measured by ELISA. Results from three separate experiments are shown as mean values ± S.D. *, p < 0.05 (Student's t test).
Discussion
RNA helicases play an important role in the host innate response against viral infection (19). Several RNA helicases have been reported to restrict HBV replication either by functioning as innate sensors to trigger the host antiviral response, or as direct antiviral effectors targeting distinct stages of HBV replication (27–30). In the present study, we have identified another RNA helicase MOV10, which restricts HBV replication. Our data show that MOV10 represses HBV replication by dramatically reducing the intracellular viral DNA levels. In addition to the genotype D strain, which is commonly used in HBV research, four HBV strains (two genotype B and two genotype C), which were cloned from the sera of chronic hepatitis B patients, were used to test the anti-HBV activity of MOV10. Our results demonstrate that MOV10 possesses broad activity against different HBV genotypes.
In this study, we also tried to identify the stages of the HBV replicative cycle that are affected by MOV10. We first examined the effect of MOV10 on viral gene expression. Our data show that MOV10 has no significant effect on the expressions of HBV RNAs and viral proteins, which is similar to the findings with intracisternal a particles and HIV-1 (9, 22). However, it is worthy to note that we used co-transfected GFP gene instead of endogenous GAPDH as the internal control. In some of our experiments, we observed slight decrease of viral and GFP expression in MOV10 overexpressing cells (data not shown). This minor effect could simply result from different transfection efficiency. However, we cannot rule out the possibility that MOV10 nonspecifically affects both exogenous HBV and GFP levels. Similarly, Burdick et al. (21) reported that overexpression of MOV10 could reduce the steady-state levels of both HIV-1 protein and GFP, but not endogenous α-tubulin. In any case, the primary inhibitory effect of MOV10 on HBV DNA production does not result from affecting viral gene expression.
Next, we examined the effect of MOV10 on encapsidation of viral pgRNA as well as stability of synthesized HBV DNA. Our data show that both processes are not affected by MOV10. Moreover, by utilizing an RH-defective HBV mutant, we show that MOV10 reduces levels of minus-strand viral DNA, suggesting that MOV10 reduces the intracellular viral DNA levels most likely by interfering with viral reverse transcription. The exact mechanism of MOV10-mediated inhibition of reverse transcription remains unclear. It is possible that MOV10 exerts its effect by steric hindrance or structure changes in viral RNA as proposed by Lu et al. (22). However, even though we did not observe interactions between HBV Pol and MOV10, we cannot rule out a possible interference of MOV10 with Pol function, e.g. affecting alignment of Pol prior to reverse transcription.
In addition to retroviruses, MOV10 has been reported to modulate replication of several RNA viruses, including hepatitis C virus, hepatitis D virus, and influenza virus (16–18), all of which replicate their genomes by RNA-dependent RNA polymerases. It is thus possible that MOV10 modulates viral replication by affecting the RNA-directed transcription or reverse transcription; both processes utilize RNA as the template. To our knowledge, there is no report showing that MOV10 interacts with viral polymerase. Hence, MOV10, as a RNA-binding protein, probably interferes with the RNA-directed transcription and reverse transcription by binding to the viral RNA. This hypothesis is supported by our data showing that MOV10 interacts with HBV RNA, but not HBV polymerase. Furthermore, we showed that the anti-HBV activity of MOV10 and its helicase mutants, MOV10 (KR) and MOV10 (EQ), are correlated with their ability to bind to viral RNA. MOV10 (EQ), despite its defect in the helicase activity, still exhibited considerable HBV RNA–binding capacity and restriction activity against HBV, suggesting RNA-binding capacity of MOV10, but not its helicase activity, is correlated with the inhibition of HBV replication. Consistent with our findings, Izumi et al. (24) reported the R730A/N731A MOV10 mutant, which is defective in interaction with HIV-1 RNA, did not inhibit HIV-1 infectivity. Ma et al. (31) reported that MOV10 has no effect on HBV DNA levels. This discrepancy with our finding could result from the different experimental protocols and systems used in these two studies. The detailed causes await further investigation.
In conclusion, we report that MOV10 inhibits HBV replication. In particular, our data suggest that MOV10 blocks HBV DNA synthesis by targeting the early step of reverse transcription. Given that HBV reverse transcription takes place inside the nascent viral core particles, further studies are warranted to examine the incorporation of MOV10 into HBV nucleocapsid. Moreover, the precise mechanism of MOV10-mediated inhibition of reverse transcription remains to be elucidated.
Experimental procedures
Cell culture
The human hepatoblastoma cell line HepG2 was cultured in Eagle's minimal essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS) and 2 mm l-glutamine. The human hepatocellular carcinoma cell line Huh7 and human embryonic kidney cell line 293T were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS and 2 mm l-glutamine. Stable cell line HepG2-hNTCP, HepG2-hNTCP-Control, and HepG2-hNTCP-MOV10 were generated as described previously (32). To engineer cell lines HepG2-shNC, HepG2-shMOV10, Huh7-shNC, Huh7-shMOV10, HepG2-hNTCP-shNC, and HepG2-hNTCP-shMOV10, 293T cells were co-transfected with ViraPower Lentiviral Packaging Mix (Thermo Fisher Scientific) and pLKD-shNC, or pLKD-shMOV10 to produce the lentiviral pseudoparticles, which were subsequently used to infect HepG2, Huh7, or HepG2-hNTCP cells. Stably transduced cell lines were selected in complete EMEM or DMEM medium containing 2.5 μg/ml puromycin.
Plasmids
pCMV-HBV contains the WT HBV 1.1-mer overlength genomic sequence (GenBank: MF967563.1) driven by the cytomegalovirus (CMV) immediate-early promoter (33). The pFLAG/HA-MOV10 expresses an N-terminal FLAG/HA-tagged human MOV10 helicase (14). The plasmids expressing the mutants MOV10 (KR) and MOV10 (EQ) were created as described previously (14). HBV Pol expression plasmid p3×FLAG-Pol was kindly provided by Professor Jieliang Chen (34). The plasmid pCMV-HBV-RH− carrying RNase H (RH)-defective polymerase mutant was constructed as reported previously (7). To generate the pCDH-MOV10 DNA construct, the MOV10 cDNA was amplified and inserted into pCDH-CMV-EF1-GFP vector (System Biosciences). shRNA-expressing plasmids pLKD-shNC and pLKD-shMOV10 were purchased from Obio Technology. The target sequence of pLKD-shMOV10 is 5′-GAAACCCTGTGGTG ACCAA-3′.
Isolation and analyses of HBV DNA
For analysis of intracellular HBV DNA replicative intermediates, transfected cells were lysed with lysis buffer (10 mm Tris, pH 7.4, 50 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40) on ice for 40 min. The nuclei were removed from the cells lysates by centrifugation at 12,000 × g for 10 min. Contaminated plasmids in the lysates were digested by DNase I (70 units) treatment at 37 °C for 6 h. Then, the treated cell lysate was added with equal volume 2× reaction buffer (200 mm NaCl, 20 mm Tris, pH 8.0, 50 mm EDTA, pH 8.0, 1% SDS) with proteinase K (final concentration of 0.5 mg/ml) and incubated at 37 °C with shaking overnight. The mixture was extracted with phenol-chloroform twice, and precipitated by sodium acetated ethanol containing 1 μl glycogen (20 mg/ml), and finally dissolved in 50 μl Tris-EDTA buffer.
To measure extracellular HBV DNA, the supernatant was collected 3 days after transfection, then mixed with same volume 20% PEG 8000, and incubated at 4 °C overnight, followed by centrifugation at 4 °C, 12,000 × g for 15 min. The pellet was suspended in 1× DNase I buffer (40 mm Tris, pH 7.5, 8 mm MgCl2), then treated with DNase I (70 units) at 37 °C for 6 h. HBV DNA was extracted as described above.
Extracted HBV DNA was analyzed by either qPCR or Southern blotting. Southern blotting was conducted as described previously (35), with the following modification. HBV DNA was detected by hybridization with a digoxigenin-labeled (Roche) riboprobe specific for minus-strand HBV DNA. qPCR analysis was performed by using SYBR Premix Ex Taq kit (Taraka) with primers listed in Table S1. All extracted DNA was also tested for DNA vector contamination by qPCR with primers as reported previously (36).
To quantify HBV cccDNA, total cellular DNA of HBV-infected cells was extracted as described previously (37). For selective cccDNA PCR, isolated DNA was treated with 10 units plasmid-safe DNase (Epicenter) in a 50 μl volume for 8 h at 37 °C, followed by DNase inactivation at 70 °C for 30 min. qPCR analysis was performed by using SYBR Premix Ex Taq kit (Taraka) with primers listed in Table S1. qPCR cycling program includes 95 °C for 30 s, followed by 40 cycles at 95 °C for 30 s, 65 °C for 25 s, and 72 °C for 45 s. HBV cccDNA copy numbers were calculated as described previously (37).
Isolation and analyses of HBV RNA
Total RNA was extracted from transfected cells with TRIzol Reagent (Invitrogen) following the manufacturer's instructions. Extraction of encapsidated pregenomic HBV RNA was performed according to the previous report (38, 39). Briefly, lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40) was added into the transfected cells and kept on ice for 30 min. The supernatant of the cell lysates was collected after centrifugation at 4 °C, 12,000 × g for 10 min, and then treated with 40 units of micrococcal nuclease (Transgen) at 37 °C for 15 min in the presence of 5 mm CaCl2 to remove free RNA. The protected encapsidated pgRNA was extracted with the TRIzol LS Reagent (Invitrogen). The extracted RNA was converted into cDNA using the PrimeScript RT reagent kit with guide DNA Eraser (Takara). HBV 3.5 kb transcripts and total HBV-specific transcripts were quantified by qPCR with primers as reported previously (37).
HBV preparation and infection
HepG2 cells were transfected with pCMV-HBV vector using Lipofectamine 3000 (Invitrogen) following the manufacturer's instructions. The supernatants of transfected cells were collected at 3 and 6 days post transfection, respectively. Viruses were concentrated by ∼50-fold using Amicon Ultra-15 (100 kDa) (Millipore) and quantified by qPCR. The concentrated virions were used to infect HepG2-hNTCP cells at 1000 genome equivalent/cell in the presence of 4% PEG 8000 for 24 h as described previously (40).
Western blotting
The cell lysates were resolved by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore). Then the membrane was probed with the primary antibodies, followed by incubation with horseradish peroxidase–conjugated secondary antibodies. The following antibodies were used: rabbit anti-MOV10 (catalogue number ab80613; Abcam), rabbit IgG (catalogue number ab172730; Abcam), mouse anti-FLAG (catalogue number F1804; Sigma), mouse anti-GFP (catalogue number TA-06; ZSGB-BIO), mouse anti-HA (catalogue number TA-04; ZSGB-BIO), and rabbit anti-HBcAg (catalogue number GB058629; Gene Tech).
RNA immunoprecipitation
Transfected cells were harvested and lysed (50 mm Tris, pH 7.4, 150 mm NaCl, 1% NonidetP-40, protease inhibitor mixture, 100 units/ml RNase inhibitor) at 3 days after transfection. The nuclei were removed by centrifugation at 12,000 × g for 10 min. The cell lysates were incubated with 1 μg of anti-FLAG antibody for 2 h at 4 °C, followed by the addition of 20 μl Protein A/G PLUS-Agarose (Santa Cruz) for overnight. The immunoprecipitated materials were washed four times with the lysis buffer and one time with PBS. The precipitated RNA was extracted using TRIzol (Invitrogen) and detected by qPCR with the same primers used for detecting HBV 3.5 kb transcripts. The housekeeping gene GAPDH was used as internal control.
Construction of replication-competent HBV vectors from the patients' sera
Serum samples were collected from four chronic hepatitis B patients, who were positive for hepatitis B e antigen and were not taking antiviral treatment. The total DNA from the patient's sera was isolated after treatment with proteinase K followed by phenol-chloroform extraction and ethanol precipitation as described above. The construction procedure of replication-competent HBV plasmids containing 1.3-fold HBV genomes from the sera was described previously (32).
This study was performed according to the Declaration of Helsinki and approved by the institutional review board of Nanjing Drum Tower Hospital. Written informed consent was obtained from the patients.
Author contributions
T. L. and Q. S. methodology; T. L. writing-original draft; Q. S. and Y. L. resources; S. C. conceptualization; S. C. and Q. Z. writing-review and editing; Q. Z. funding acquisition; Q. Z. project administration.
Supplementary Material
This work was supported by National Natural Science Foundation of China Grant 81602400. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1 and Table S1.
- HBV
- hepatitis B virus
- cccDNA
- covalently closed circular DNA
- CMV
- cytomegalovirus
- pgRNA
- pregenomic RNA
- Pol
- polymerase
- qPCR
- quantitative PCR
- RC
- relaxed circular
- RH
- RNase H
- RIP
- RNA immunoprecipitation
- ssDNA
- single-stranded DNA.
References
- 1. Schweitzer A., Horn J., Mikolajczyk R. T., Krause G., and Ott J. J. (2015) Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 386, 1546–1555 10.1016/S0140-6736(15)61412-X [DOI] [PubMed] [Google Scholar]
- 2. Arzumanyan A., Reis H. M., and Feitelson M. A. (2013) Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer. 13, 123–135 10.1038/nrc3449 [DOI] [PubMed] [Google Scholar]
- 3. Lavanchy D. (2004) Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 11, 97–107 10.1046/j.1365-2893.2003.00487.x [DOI] [PubMed] [Google Scholar]
- 4. Nassal M. (2008) Hepatitis B viruses: Reverse transcription a different way. Virus. Res. 134, 235–249 10.1016/j.virusres.2007.12.024 [DOI] [PubMed] [Google Scholar]
- 5. Hirsch R. C., Lavine J. E., Chang L. J., Varmus H. E., and Ganem D. (1990) Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature 344, 552–555 10.1038/344552a0 [DOI] [PubMed] [Google Scholar]
- 6. Hu J., Toft D. O., and Seeger C. (1997) Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. The EMBO J. 16, 59–68 10.1093/emboj/16.1.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nguyen D. H., Gummuluru S., and Hu J. (2007) Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 81, 4465–4472 10.1128/JVI.02510-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonvin M., Achermann F., Greeve I., Stroka D., Keogh A., Inderbitzin D., Candinas D., Sommer P., Wain-Hobson S., Vartanian J. P., and Greeve J. (2006) Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology 43, 1364–1374 10.1002/hep.21187 [DOI] [PubMed] [Google Scholar]
- 9. Wang X., Han Y., Dang Y., Fu W., Zhou T., Ptak R. G., and Zheng Y. H. (2010) Moloney leukemia virus 10 (MOV10) protein inhibits retrovirus replication. J. Biol. Chem. 285, 14346–14355 10.1074/jbc.M110.109314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mooslehner K., Karls U., and Harbers K. (1990) Retroviral integration sites in transgenic Mov mice frequently map in the vicinity of transcribed DNA regions. J. Virol. 64, 3056–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mooslehner K., Müller U., Karls U., Hamann L., and Harbers K. (1991) Structure and expression of a gene encoding a putative GTP-binding protein identified by provirus integration in a transgenic mouse strain. Mol. Cell Biol. 11, 886–893 10.1128/MCB.11.2.886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meister G., Landthaler M., Peters L., Chen P. Y., Urlaub H., Lührmann R., and Tuschl T. (2005) Identification of novel argonaute-associated proteins. Curr. Biol. 15, 2149–2155 10.1016/j.cub.2005.10.048 [DOI] [PubMed] [Google Scholar]
- 13. Goodier J. L., Cheung L. E., and Kazazian H. H. Jr. (2012) MOV10 RNA helicase is a potent inhibitor of retrotransposition in cells. PLoS. Genet. 8, e1002941 10.1371/journal.pgen.1002941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li X., Zhang J., Jia R., Cheng V., Xu X., Qiao W., Guo F., Liang C., and Cen S. (2013) The MOV10 helicase inhibits LINE-1 mobility. J. Biol. Chem. 288, 21148–21160 10.1074/jbc.M113.465856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gregersen L. H., Schueler M., Munschauer M., Mastrobuoni G., Chen W., Kempa S., Dieterich C., and Landthaler M. (2014) MOV10 is a 5′ to 3′ RNA helicase contributing to UPF1 mRNA target degradation by translocation along 3′ UTRs. Mol. Cell 54, 573–585 10.1016/j.molcel.2014.03.017 [DOI] [PubMed] [Google Scholar]
- 16. Haussecker D., Cao D., Huang Y., Parameswaran P., Fire A. Z., and Kay M. A. (2008) Capped small RNAs and MOV10 in human hepatitis delta virus replication. Nat. Struct. Mol. Biol. 15, 714–721 10.1038/nsmb.1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schoggins J. W., Wilson S. J., Panis M., Murphy M. Y., Jones C. T., Bieniasz P., and Rice C. M. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 10.1038/nature09907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang J., Huang F., Tan L., Bai C., Chen B., Liu J., Liang J., Liu C., Zhang S., Lu G., Chen Y., and Zhang H. (2016) Host protein Moloney leukemia virus 10 (MOV10) acts as a restriction factor of influenza A virus by inhibiting the nuclear import of the viral nucleoprotein. J. Virol. 90, 3966–3980 10.1128/JVI.03137-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ranji A., and Boris-Lawrie K. (2010) RNA helicases: Emerging roles in viral replication and the host innate response. RNA Biol. 7, 775–787 10.4161/rna.7.6.14249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Furtak V., Mulky A., Rawlings S. A., Kozhaya L., Lee K., Kewalramani V. N., and Unutmaz D. (2010) Perturbation of the P-body component Mov10 inhibits HIV-1 infectivity. PLoS ONE 5, e9081 10.1371/journal.pone.0009081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burdick R., Smith J. L., Chaipan C., Friew Y., Chen J., Venkatachari N. J., Delviks-Frankenberry K. A., Hu W. S., and Pathak V. K. (2010) P body-associated protein Mov10 inhibits HIV-1 replication at multiple stages. J. Virol. 84, 10241–10253 10.1128/JVI.00585-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu C., Luo Z., Jäger S., Krogan N. J., and Peterlin B. M. (2012) Moloney leukemia virus type 10 inhibits reverse transcription and retrotransposition of intracisternal a particles. J. Virol. 86, 10517–10523 10.1128/JVI.00868-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Singleton M. R., Dillingham M. S., and Wigley D. B. (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 76, 23–50 10.1146/annurev.biochem.76.052305.115300 [DOI] [PubMed] [Google Scholar]
- 24. Izumi T., Burdick R., Shigemi M., Plisov S., Hu W. S., and Pathak V. K. (2013) Mov10 and APOBEC3G localization to processing bodies is not required for virion incorporation and antiviral activity. J. Virol. 87, 11047–11062 10.1128/JVI.02070-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nassal M. (2015) HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64, 1972–1984 10.1136/gutjnl-2015-309809 [DOI] [PubMed] [Google Scholar]
- 26. Nguyen D. H., and Hu J. (2008) Reverse transcriptase– and RNA packaging signal–dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J. Virol. 82, 6852–6861 10.1128/JVI.00465-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ko C., Lee S., Windisch M. P., and Ryu W. S. (2014) DDX3 DEAD-box RNA helicase is a host factor that restricts hepatitis B virus replication at the transcriptional level. J. Virol. 88, 13689–13698 10.1128/JVI.02035-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang H., Xing Z., Mani S. K., Bancel B., Durantel D., Zoulim F., Tran E. J., Merle P., and Andrisani O. (2016) RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology 64, 1033–1048 10.1002/hep.28698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato S., Li K., Kameyama T., Hayashi T., Ishida Y., Murakami S., Watanabe T., Iijima S., Sakurai Y., Watashi K., Tsutsumi S., Sato Y., Akita H., Wakita T., Rice C. M., et al. (2015) The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132 10.1016/j.immuni.2014.12.016 [DOI] [PubMed] [Google Scholar]
- 30. Lu H. L., and Liao F. (2013) Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J. Immunol. 191, 3264–3276 10.4049/jimmunol.1300512 [DOI] [PubMed] [Google Scholar]
- 31. Ma Y. X., Li D., Fu L. J., Fu B. Q., Chen S. J., Xu W. Z., Teng X., Song Z. W., and Gu H. X. (2015) The role of Moloney leukemia virus 10 in hepatitis B virus expression in hepatoma cells. Virus. Res. 197, 85–91 10.1016/j.virusres.2014.12.011 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Q., Chen J., Pan M., Liu J., Liu T., and Zhou Y. H. (2018) Comparison of replication competence of wild-type and lamivudine-resistant hepatitis B virus isolates from a chronic hepatitis B patient. Virus. Res. 255, 165–170 10.1016/j.virusres.2018.07.021 [DOI] [PubMed] [Google Scholar]
- 33. Fallows D. A., and Goff S. P. (1995) Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 69, 3067–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y., Li J., Chen J., Li Y., Wang W., Du X., Song W., Zhang W., Lin L., and Yuan Z. (2015) Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol. 89, 2287–2300 10.1128/JVI.02760-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu S., Zhao Q., Zhang P., Kulp J., Hu L., Hwang N., Zhang J., Block T. M., Xu X., Du Y., Chang J., and Guo J. T. (2017) Discovery and mechanistic study of benzamide derivatives that modulate hepatitis B virus capsid assembly. J. Virol. 91, e00519–17 10.1128/JVI.00519-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amini-Bavil-Olyaee S., Herbers U., Sheldon J., Luedde T., Trautwein C., and Tacke F. (2009) The rtA194T polymerase mutation impacts viral replication and susceptibility to tenofovir in hepatitis B e antigen-positive and hepatitis B e antigen-negative hepatitis B virus strains. Hepatology 49, 1158–1165 10.1002/hep.22790 [DOI] [PubMed] [Google Scholar]
- 37. Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H., Fu L., Song M., Chen P., Gao W., Ren B., et al. (2012) Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1, e00049 10.7554/eLife.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schultz U., Summers J., Staeheli P., and Chisari F. V. (1999) Elimination of duck hepatitis B virus RNA-containing capsids in duck interferon-α-treated hepatocytes. J. Virol. 73, 5459–5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guo J. T., Pryce M., Wang X., Barrasa M. I., Hu J., and Seeger C. (2003) Conditional replication of duck hepatitis B virus in hepatoma cells. J. Virol. 77, 1885–1893 10.1128/JVI.77.3.1885-1893.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Okuyama-Dobashi K., Kasai H., Tanaka T., Yamashita A., Yasumoto J., Chen W., Okamoto T., Maekawa S., Watashi K., Wakita T., Ryo A., Suzuki T., Matsuura Y., Enomoto N., and Moriishi K. (2015) Hepatitis B virus efficiently infects non-adherent hepatoma cells via human sodium taurocholate cotransporting polypeptide. Sci. Rep. 5, 17047 10.1038/srep17047 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.