

Abstract
Pseudomonas aeruginosa secretes multiple proteases that are implicated in its pathogenesis, and most of them are regulated by quorum sensing (QS). In this study, we found that the activities of three major extracellular proteases, protease IV (PIV), elastase A (LasA), and elastase B (LasB), are reduced considerably when expressed in a QS mutant (MW1). PIV and LasA expressed in MW1 exhibited little activity, even when purified, and their activities were inhibited by noncleavage or binding of their propeptides. LasB was activated by a QS-dependent factor, indicating that, unlike what has been proposed previously, LasB is not autoactivated. When LasB was relieved from inhibition, it activated PIV, which then sequentially processed pro-LasA to mature LasA. When activated, LasB was not inhibited by exogenous addition of its propeptide, but LasA and PIV were inhibited by their propeptides, even after prior activation. These differences may be explained by the fact that LasB can degrade its own propeptide but PIV and LasA cannot. We also found that, although PIV is the preferred LasA-activating factor, LasB can also partially activate LasA. Overall, LasB, PIV, and LasA were activated postsecretionally in a cascading manner in which the initial activation of LasB was controlled tightly by QS at the protein level in addition to the well-known transcriptional control of these proteases by QS. Interestingly, human elastase also activated LasA, indicating that the activation cascade is triggered by host factors during infection. In summary, a QS-induced proteolytic cascade activates secreted proteases from P. aeruginosa.
Keywords: quorum sensing, protease, metalloprotease, protein processing, protein secretion, protein–protein interaction, inhibition mechanism, microbial pathogenesis
Introduction
Pseudomonas aeruginosa is a Gram-negative opportunistic human pathogen that causes a variety of infections, such as burn wound infections, keratitis, and pneumonia, and often infects cystic fibrosis patients (1, 2). During these infections, P. aeruginosa secretes many virulence factors, including proteases, toxins, phenazines, and pyocyanin, that neutralize the host immune defense and cause damage to organs (3–5). In particular, three extracellular proteases of P. aeruginosa, elastase B (LasB,2 Pseudolysin), elastase A (LasA, Staphylolysin), and protease IV (PIV) are very important virulence factors that degrade host tissues and immune components (6, 7).
Interestingly, PIV, LasB, and LasA have a common domain organization without similarity in the amino acid sequence. They have a signal peptide (SP) at the N terminus, a propeptide (PP) domain in the middle, and a mature protease domain at the C terminus (Fig. S1). These proteases are expressed initially in their prepro-form with all three domains and processed twice from the N terminus during secretion to generate their mature forms (8). In P. aeruginosa, these proteases are known to be secreted in two steps: Sec system–mediated translocation across the inner membrane and transportation across the outer membrane, probably through the terminal branch of the type II secretion system (T2SS) (8, 9). Their SPs are first cleaved during translocation over the cytoplasmic membrane by the Sec system to generate their pro-forms, and their PPs are cleaved in the periplasm to generate mature proteases (5). When this processing is incomplete, these proteases have been proposed to be inactive (5), but this has not been clearly demonstrated for all of these three proteases.
Regarding LasB, the most studied one, it has been reported that the inactive pro-form of LasB (pro-LasB) is initially exported into the periplasmic space (Fig. S1A) and undergoes PP-mediated folding to become activated (10). Because the PP of LasB (LasBpp) is required for proper folding and secretion of LasB, LasBpp has been proposed as an intramolecular chaperone (11). The current model regarding the processing and activation of LasB is that, after PP-mediated folding in the periplasm, pro-LasB is cleaved by autoproteolysis and further secreted to the extracellular space via the Xcp machinery, which is a T2SS of P. aeruginosa (8–10, 12). Interestingly, the LasBpp released during processing has been proposed to bind to and inhibit mature LasB (13). Although this model has greatly expanded our understanding of LasB, our new findings in this study show that this model needs to be somewhat modified.
LasA was discovered originally as a second protease with elastolytic activity, but it was later characterized as a protease with high staphylolytic activity, causing rapid lysis of Staphylococcus aureus (14, 15). Similar to PIV and LasB, LasA is encoded as a 45.5-kDa protease precursor by the lasA gene (Fig. S1B), and mature LasA (20 kDa) is produced by two successive cleavages from the N terminus during secretion (8). Although LasB has been found to be secreted as a noncovalent complex with its PP, LasA has been detected to be exported in its incompletely processed pro-form (pro-LasA), which has been suggested to be processed by other proteases, such as LasB or a serine protease, rather than by autoprocessing (8, 16). Similar to LasB, LasA has also been suggested to be secreted extracellularly via the Xcp system (8), but there is little experimental evidence of its maturation and secretion.
Despite the importance of PIV in P. aeruginosa infections (2, 17, 18), the mechanism of secretion or activation of PIV has received less attention. A recent study elucidated the extracellular activation mechanism of PIV; PIV is inhibited by its own PP (PIVpp) and activated postsecretionally through degradation of PIVpp by LasB (19). This mechanism was revealed during the process of identifying the cause of PIV inactivation in a quorum-sensing (QS) mutant (19). LasB, LasA, and PIV are under transcriptional control of QS in P. aeruginosa (17, 20). QS is a bacterial cell-to-cell communication system that regulates a large number of genes encoding these virulence factors in a cell density–dependent manner (21). P. aeruginosa has multiple QS systems, and acyl homoserine lactone (acyl-HSL)–based QS systems, las and rhl, trigger the entire QS regulation. LasR, QscR, and RhlR receive N-(3-oxododecanoyl)-l-HSL (3oxo-C12-HSL) and N-butanoyl-l-HSL (C4-HSL) as the signal molecules that activate their regulon genes (21, 22).
Regulation by QS is believed to occur at the transcription level, but a previous study reported that artificial expression of PIV under control of a non-QS promoter (arabinose-inducible promoter) failed to complement the PIV-deficient phenotype of the QS mutant. This suggests that, even when PIV is overexpressed to a sufficient level, the activity of PIV was barely detected in the QS mutant (19). In addition, PIV overexpressed in the QS mutant was processed normally, but released PIVpp released has been found to bind to and inhibit PIV, indicating that the cleaved PIVpp is degraded rapidly in the WT, whereas it is not degraded in the QS mutant (19). These results showed not only how PIV was activated but also that the QS mutants could be useful for studying the mechanism of protease activation because many proteases are not expressed in the QS mutant, so the proteases that should be processed proteolytically are not or incompletely processed. Indeed, this study found that, in the QS mutant, even when overexpressing LasB and LasA artificially, the activities of LasB and LasA do not appear. Therefore, this study attempted to identify how LasB, LasA, and PIV interact with each other for their activation using this QS mutant system.
Results
LasA expressed in the QS mutant is fully secreted but inactive
The mechanism of LasA activation was examined first. LasA activity is usually measured using staphylolytic activity (14, 23), but there have been controversial results regarding a second staphylolytic enzyme in P. aeruginosa (23, 24). To confirm that LasA is the only enzyme with significant staphylolytic activity in the WT strain (PAO1) used in this study, a lasA mutant (ΔlasA) was constructed, and staphylolytic activity was measured (Fig. S2 and Table S1). Staphylolytic activity was fully abolished in the culture supernatants of the lasA mutant (CSΔlasA), showing that LasA is the only factor for staphylolytic activity in PAO1 (Fig. 1 and Fig. S2). The LasA activity in the QS mutant (MW1; lasI−, rhlI−) was also measured, and there was little LasA activity in the culture supernatants of MW1 (CSMW1), to a similar extent as that observed in culture supernatants of the lasA mutant (Fig. 1). This result might be explained by the current model because lasA expression is regulated tightly by QS at the transcription level (20). However, when LasA was overexpressed artificially in MW1 using pSP101, which has a QS-independent and arabinose-inducible promoter (Table S1), the LasA activity in the culture supernatants of the LasA-overexpressing MW1 (CSMW1-LasA) was still very low (Fig. 1). In the lasA mutant, however, LasA activity was restored completely by complementation with pSP101 (CSΔlasA-LasA) to WT levels (Fig. 1), confirming that pSP101 is working properly. This suggests that, like PIV, LasA also requires the QS system for activation, even after transcription.
Figure 1.
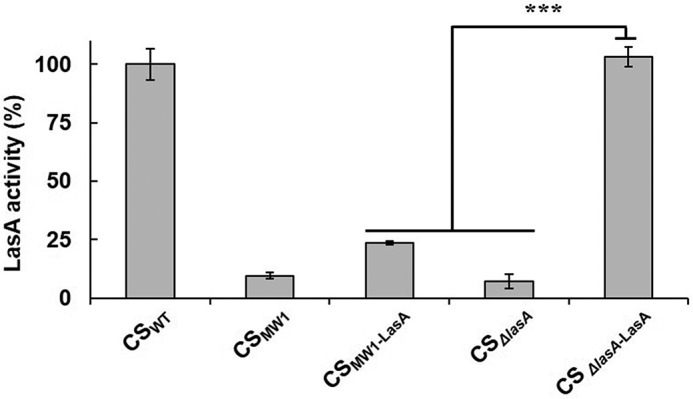
LasA activity in the culture supernatants of QS and ΔlasA mutants. The CSs were prepared from PAO1 (CSWT), MW1 (CSMW1), or ΔlasA (CSΔlasA) and their isotype strains harboring a LasA-overexpressing plasmid (pSP101 in Table S1; CSMW1-LasA and CSΔlasA-LasA). For fair comparison, the strains that did not harbor pSP101 were transformed by an empty plasmid (pJN105) as a vector control. LasA activity was measured in culture supernatants containing the same amount of proteins (2 μg). ***, p < 0.005.
Previous studies have suggested that LasB and PIV play key roles in extracellular processing of pro-LasA in P. aeruginosa (8, 16), and it was confirmed that pro-PIV is activated by LasB-mediated PP degradation (19). This study examined whether LasB or PIV could restore the activity of overexpressed LasA in CSMW1-LasA. When purified LasB and PIV were added to CSMW1-LasA, LasA activity was fully restored by both LasB and PIV in a dose-dependent manner (Fig. 2, A and B). This shows that LasA activity does not appear in the QS mutant despite sufficient expression of LasA. Therefore, LasA is inactive without the QS system, even though it is transcribed independently of QS, and both LasB and PIV can activate this inactivated LasA.
Figure 2.
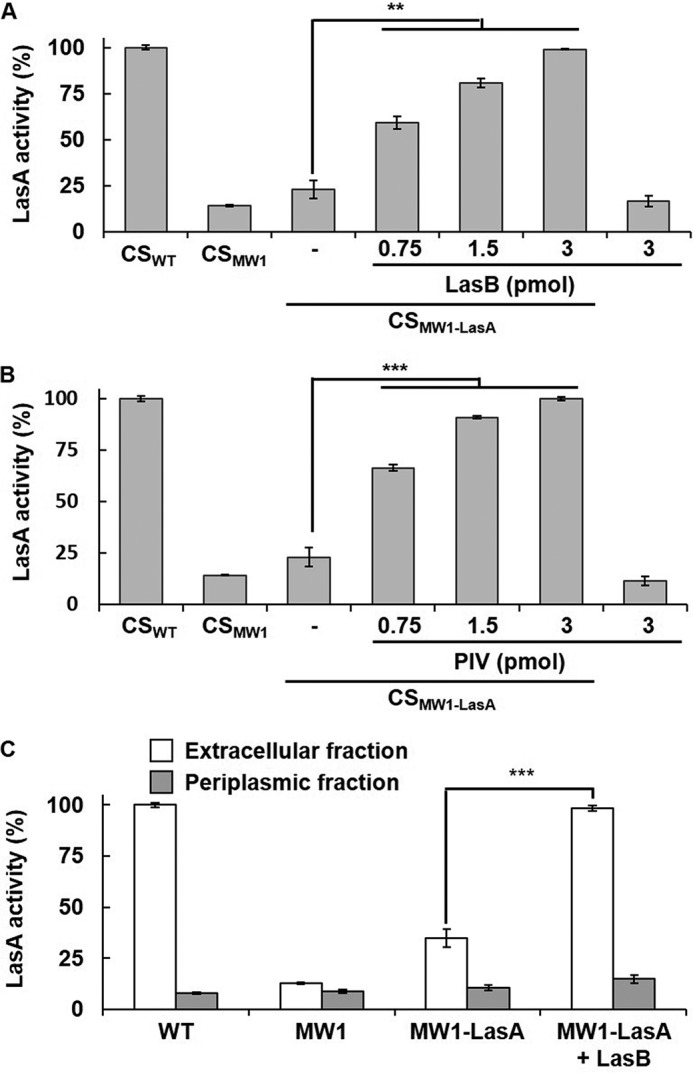
Restoration of the LasA activity in CSMW1-LasA by LasB and PIV. A and B, the culture supernatants of CSMW1-LasA (2 μg) were incubated with various amounts of purified LasB (A) or PIV (B) for 30 min, and LasA activity was measured. As a control, PAO1 and MW1 were transformed with pJN105, and their culture supernatants (CSWT and CSMW1) were prepared to be measured for LasA activity. C, LasA activity was measured in the periplasmic and extracellular fractions of PAO1 (WT), MW1, and LasA-overexpressing MW1 (MW1-LasA). Each fraction containing 1 μg of protein was used for the LasA activity assay. To restore the activity of LasA, the fraction of LasA-overexpressing MW1 was mixed with 0.6 pmol of purified LasB. **, p < 0.01; ***, p < 0.005.
Both LasA and LasB have been suggested to be secreted in a two-step manner and transported through T2SS in the second step (10). Because the expression of xcpP, the key component of the T2SS of P. aeruginosa, is QS-regulated (25), we examined whether LasA was secreted from the QS mutant. The results showed that LasB mostly restored the activity of LasA overexpressed in MW1 in the extracellular fraction, not in the periplasmic fraction, suggesting that overexpressed LasA was normally secreted from the QS mutant (Fig. 2C).
LasA is secreted in its pro-form and activated primarily by PIV
Previous studies have shown that LasA is initially secreted in the form of pro-LasA (8). In contrast, PIV is secreted in the processed state, but in the QS mutant, the cleaved PIVpp is not degraded and inhibits PIV by specific binding (19). To determine the precise processing status of LasA in the QS mutant, LasA was purified from PAO-T7 and PAO-T7-MW1 harboring a His-tagged, LasA-overexpressing plasmid (pQF21c-LasA, Table S1), called P-LasA and M-LasA, respectively (Fig. 3, A and B). In the case of PIV, the PIV from PAO-T7-MW1 (M-PIV) was copurified with cleaved PIVpp in a noncovalently bound complex (19). Unlike PIV, most of the M-LasA was in its pro-LasA form (Fig. 3, A and B), suggesting that processing of LasA did not occur in the QS mutant. Therefore, purified M-LasA showed significantly lower activity than P-LasA (Fig. S3), explaining the result shown in Fig. 1.
Figure 3.
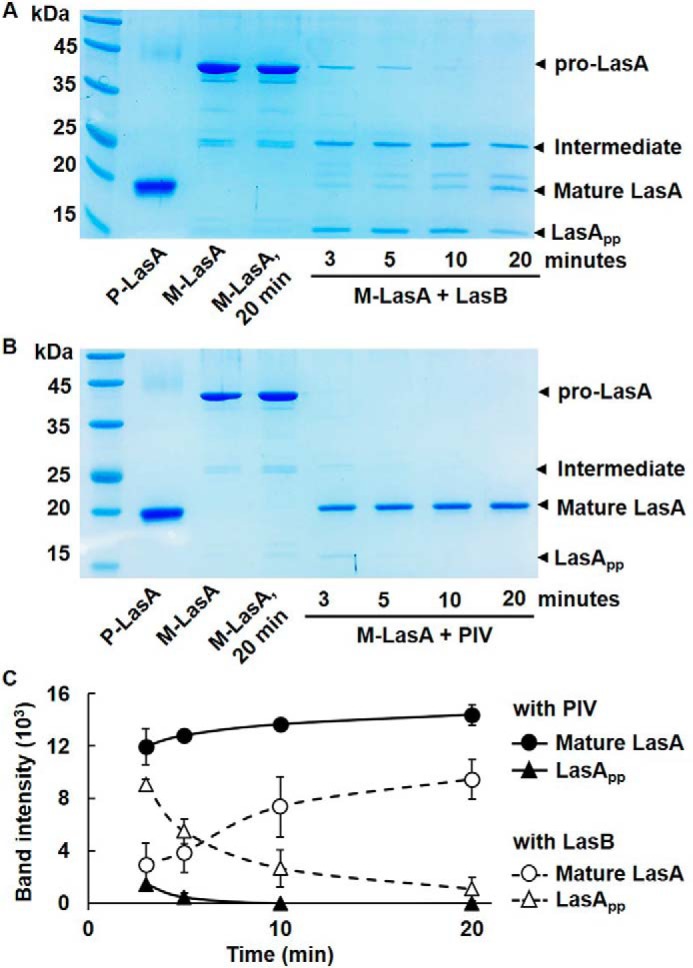
Processing of pro-LasA by LasB and PIV. A and B, LasA was purified from CSWT-LasA (P-LasA) and CSMW1-LasA (M-LasA), respectively, and M-LasA (1 μg) was mixed with 3 pmol of LasB (A) or PIV (B) in a 20-μl reaction volume. After incubation for the indicated time, the reaction mixture was applied to 15% SDS-PAGE. As a control, the same amounts of P-LasA, M-LasA, and M-LasA that were incubated for 20 min without LasB or PIV were loaded. The arrowheads indicate the bands for pro-LasA, the intermediate form, LasApp, and mature LasA. C, the processing rates of M-LasA by LasB and PIV were compared by quantifying the band intensities of LasA and LasApp.
Our results showed that both LasB and PIV could activate LasA, and an attempt was made to determine which of them was the primary factor to activate LasA. Because the staphylolysis method requires long incubation time with S. aureus cells (∼1.5 h), it was inappropriate to accurately compare the efficiency of LasA activation. Instead, purified pro-LasA (M-LasA) was mixed with LasB or PIV, and processing of M-LasA was monitored directly by SDS-PAGE. The results showed that PIV converted M-LasA to P-LasA (mature LasA) more quickly by cutting off and degrading LasApp than LasB (Fig. 3, A and B). During this conversion, both LasB and PIV generated the intermediate form of LasA, which has been observed by other groups (8). With PIV, the intermediate form and PP of LasA disappeared rapidly, within 3 min, quickly generating mature LasA, whereas they persisted after 10 min, and the mature LasA was generated slowly with LasB (Fig. 3C). Overall, inactivation of LasA in the QS mutant is due to incomplete processing, and PIV is a primary LasA-activating factor that preferentially processes LasA.
Three proteases are activated sequentially in the order LasB, PIV, and LasA
A previous study showed that LasB activates PIV by degrading PIVpp (19), and this study also showed that activated PIV, in turn, activates LasA by processing pro-LasA. To confirm these results in vivo, a lasB mutant (ΔlasB) was constructed (Table S1 and Fig. S4), and the activities of LasB, PIV, and LasA in the lasB mutant were measured. The mutation on lasB could fully abolish the activities of both LasA and PIV, confirming that LasB activates both LasA and PIV (Fig. 4, A and B). In particular, more than 60% of total LasA activity was abolished in Δpiv (Fig. 4A), suggesting that PIV is more important in LasA activation and that LasB alone cannot sufficiently activate LasA. However, this result shows that LasB also contributes to LasA activation to some extent. This result is consistent with the in vitro results showing that PIV converted the pro-LasA preferentially to mature LasA (Fig. 3). The mutation on lasA did not affect PIV activity (Fig. 4B), and the mutation on piv did not affect LasB activity (Fig. 4C), suggesting that LasA is not involved in PIV activation and PIV is not involved in LasB activation. This means that three proteases are activated sequentially in the order LasB, PIV, and LasA. In addition, LasB triggers this cascade activation of PIV and LasA.
Figure 4.
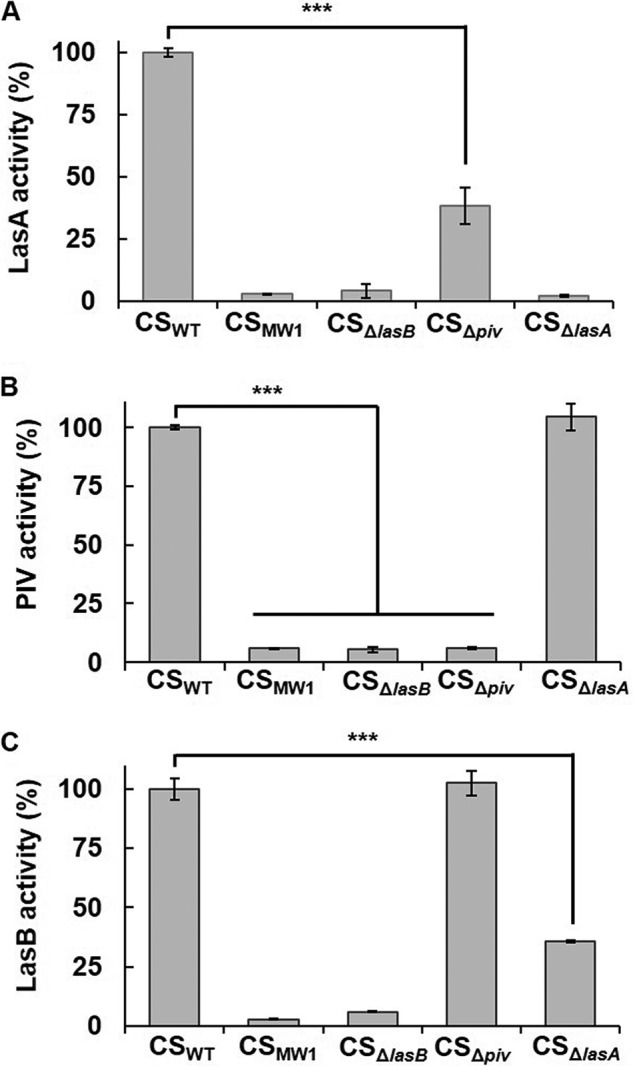
Activity of LasB, PIV, and LasA in WT and protease mutants. A–C, culture supernatants of the WT and each protease mutant were prepared, and the activities of LasA (A), PIV (B), and LasB (C) were measured. ***, p < 0.005.
Remarkably, LasB activity was reduced partly by a lasA mutation (Fig. 4C), indicating that LasA may contribute to LasB activity to some extent. This result is probably related to the previous finding that LasA can increase the activity of some elastases (26). That study suggested that LasA interacts with the elastin substrate rather than modifying elastase because LasA enhances the elastolytic activity of several elastases, including LasB, thermolysin, proteinase K, and human neutrophil elastase (NE) in vitro, and does not enhance the proteolytic activity of LasB (26).
LasB is also inactive when expressed in the QS mutant: LasB cannot autoactivate alone and requires a QS-dependent factor for activation
This study examined how LasB is activated initially. LasB is believed to be activated by itself (8, 27, 28). If it is true, then artificial expression of LasB by an arabinose-inducible promoter should complement LasB activity in the QS mutant. However, although artificial expression of LasB complemented the lasB mutant well, it failed to complement the QS mutant, and LasB overexpressed in MW1 showed very low activity (Fig. 5A). Interestingly, when the culture supernatants of the QS mutant overexpressing LasB (CSMW1-LasB) were mixed with the culture supernatants of the lasB mutant (CSΔlasB), it dramatically restored LasB activity (Fig. 5B), suggesting that LasB activity does not appear in the QS mutant despite sufficient expression. Because PIV or LasA was unable to activate LasB (Fig. 5B), this clearly shows that LasB is activated extracellularly by other QS-dependent factor(s), not by its own activity.
Figure 5.
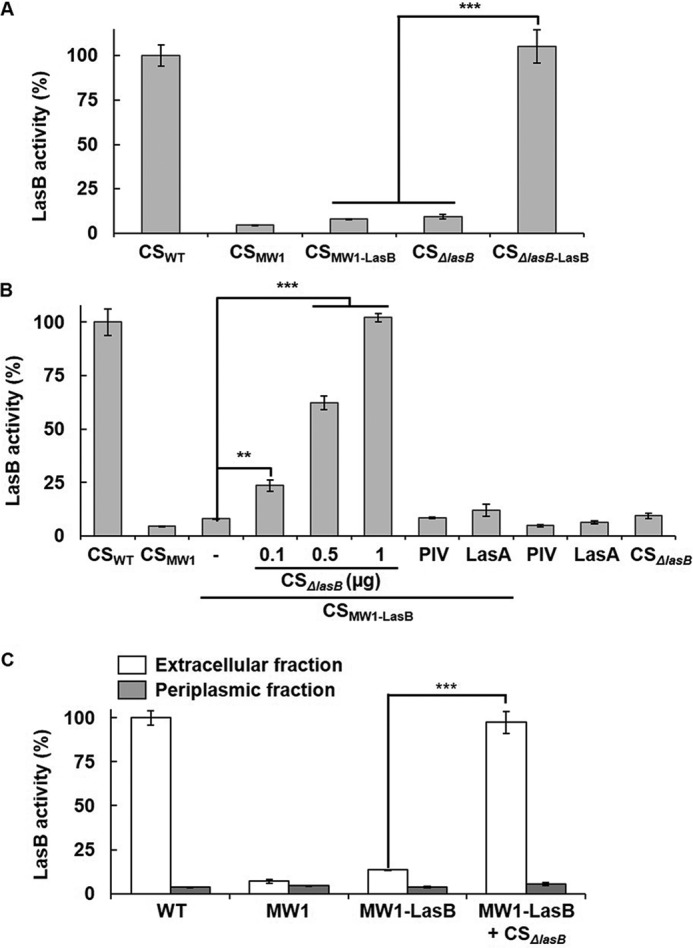
LasB activity in the QS mutant and restoration of the LasB activity by CSΔlasB. A, LasB activity in culture supernatants (2 μg) prepared from PAO1 (CSWT), MW1 (CSMW1), or ΔlasB (CSΔlasB) and their isotype strains harboring the LasB-overexpressing plasmid (pSP201, CSMW1-LasB and CSΔlasB-LasB) were measured. The strains that did not harbor pSP201 were transformed by pJN105 as a vector control. B, CSMW1-LasB was mixed with various amounts of CSΔlasB (containing 0.3, 0.5, and 1 μg total proteins, respectively) or 100 ng of purified PIV or LasA and incubated for 30 min, and LasB activity was measured. As a control, the same amount of PIV, LasA, or CSΔlasB containing 1 μg of total proteins were incubated for 30 min without CSMW1-LasB, and LasB activity was measured. C, LasB activity was measured in the periplasmic and extracellular fractions of PAO1 (WT), MW1, and MW1-LasB. Each fraction containing 1 μg of protein was used for the LasB activity assay. To restore the activity of LasB, the fraction of LasB-overexpressing MW1 was mixed with CSΔlasB containing 1 μg of total proteins. **, p < 0.01; ***, p < 0.005.
As mentioned above, LasB has been suggested to be secreted via T2SS (10), and QS induces xcp in P. aeruginosa (25). We therefore examined whether LasB is fully secreted from the QS mutant. The results showed that the activity of LasB overexpressed in MW1 was restored in the extracellular fraction by CSΔlasB, not in the periplasmic fraction (Fig. 5C), suggesting that overexpressed LasB had been fully secreted, similar to LasA.
LasA can be inhibited by exogenous addition of its propeptide but LasB cannot
A previous study showed that mature PIV can be inhibited by exogenous addition of PIVpp (19). Similarly, LasApp (amino acids 1–236 of LasA) and LasBpp (amino acids 1–197 of LasB) was overexpressed in Escherichia coli and purified (Fig. S1). When mature LasA (P-LasA) was incubated with purified LasApp, activity was inhibited significantly in a dose-dependent manner (Fig. 6A), suggesting that, similar to PIV, LasA can be inhibited by its PP, even when already activated. In contrast, LasB was inhibited partially by exogenous LasBpp (Fig. 6A), indicating that, when activated, LasB is no longer subject to inhibition by its PP. To determine why, LasApp was mixed with mature LasB, PIV, or LasA (P-LasA), and the state of the PPs was checked. Although LasApp was well-degraded by both LasB and PIV, it was not degraded by LasA (Fig. 6B), similar to PIVpp, which was not degraded by PIV (19). In contrast, when LasBpp was mixed with mature proteases, LasBpp was degraded by LasB (Fig. 6C). PIV also degraded LasBpp, but LasA could not degrade LasBpp (Fig. 6B). These results explain why LasBpp was unable to inhibit mature LasB. When LasB is activated, it can degrade its PP, making it resistant to its PP. On the other hand, LasA cannot degrade its PP and is still subject to inhibition by its own PP. Finally, inhibition of mature LasA by LasApp was relieved by addition of LasB and PIV (Fig. 6D). These results consistently demonstrate that LasApp acts as an inhibitor for mature LasA, but LasBpp only acts as an inhibitor until LasB is activated and does not act for the already activated LasB. Both LasB and PIV relieve inhibition of LasA by its PP, whereas inhibition of LasB is relieved by other QS-dependent factor(s).
Figure 6.

Inhibition of LasA and LasB by their propeptides. A, active LasA and LasB were mixed with purified LasApp and LasBpp at various molar ratios. After 30-min incubation, the activities of LasA and LasB were measured. B and C, mature forms of LasB (100 ng), PIV (100 ng), and LasA (500 ng) were mixed with 1 μg of LasApp (B) or 1 μg of LasBpp (C), incubated for 30 min, and electrophoresed in 15% SDS-PAGE. D, P-LasA (mature LasA, 500 ng) was mixed with LasApp (850 ng), and mature LasB (100 ng) or PIV (100 ng) was then added to the mixture. LasA activity was measured after 30-min incubation. ***, p < 0.005
Human NE can activate LasA
NE has elastolytic activity similar to LasB but a different amino acid sequence. In humans and mice, as an important antibacterial protease, NE is involved in inflammation and innate immunity during an infection. This study investigated whether human NE can activate LasA or PIV like LasB. When human NE was added to M-LasA, activity was fully restored to that when LasB or PIV was added (Fig. 7A). This result is intriguing because the nascent prepro-LasA in the cytoplasm can be activated by human NE when it somehow comes out during an infection, and the virulence of P. aeruginosa can be exerted even after bacterial cells are lysed by antimicrobial peptides or host immunity. Unlike LasA, M-PIV, which is a PP-bound PIV purified from MW1 (19), was not activated by human NE (Fig. 7B). This indicates different substrate specificity of human NE and P. aeruginosa LasB.
Figure 7.
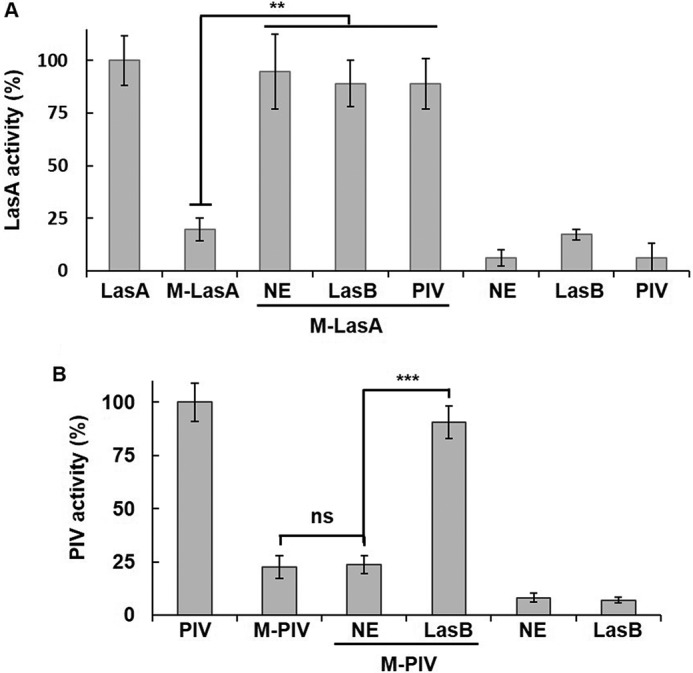
Activation of pro-LasA by human elastase. A, 500 ng of pro-LasA (M-LasA) was mixed with 50 ng of human NE, LasB, or PIV and incubated for 30 min, and LasA activity was measured. B, 100 ng of M-PIV (PIV purified from MW1) was mixed with 50 ng of NE or LasB and incubated for 30 min, and PIV activity was measured. **, p < 0.01; ***, p < 0.005; ns, no significance.
Discussion
Extracellular proteases, including LasB, PIV, and LasA, are believed to play key roles in P. aeruginosa infection (5). Coordinated expression of these proteases is very important for successful infection with P. aeruginosa, and QS is considered a mechanism to enable this. Because QS is a transcriptional regulatory system, this might be achieved by transcriptional regulation of the genes encoding these proteases. Indeed, all three protease genes are under transcriptional control of QS in P. aeruginosa (20), which must be an important regulation. Nevertheless, this study showed that this is not the case in controlling these protease activities, and QS is critically involved in the activation mechanism at the protein level. A previous study of PIV activation showed that PIV expressed in a QS mutant is inactive, even when purified, because of the lack of degradation of its PP in the QS mutant (19). In this study, a similar phenomenon was also observed with LasA; extracellular processing of LasA does not occur in the QS mutant. In both cases, artificial overexpression using the QS-independent promoter failed to elevate the activity of the proteases. This clearly shows that the QS system does not regulate the activity of these proteases through transcriptional regulation only and still controls their activity even after these proteases are secreted from the cell. This is a postsecretional activation mechanism by QS. Fig. 8 summarizes the regulatory mechanism revealed by this study. The PPs of these proteases act as a “safety pin” to prevent premature activation of these proteases and protects the bacterial cells from undesired degradation of the periplasmic proteins. LasB is involved in extracellular activation of PIV and LasA by cleaving and degrading their PPs, and PIV activates LasA in a similar manner, giving priority over LasB (Fig. 8).
Figure 8.
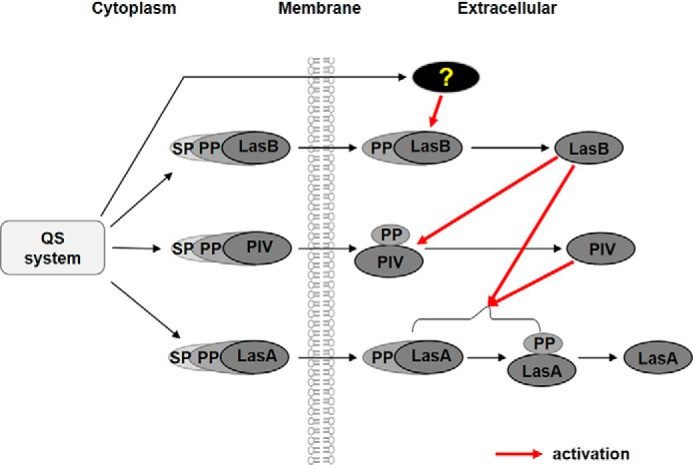
Proposed model of the postsecretional activation cascade of the three proteases in P. aeruginosa. In the cytoplasm, the prepro-forms of the proteases are expressed under transcriptional control of QS, and their SPs are removed during translocation over the cytoplasmic membrane. In the extracellular space, the pro-form of LasB is first processed to its mature active form by unknown QS-dependent factors. The activated LasB, in turn, activates PIV by degrading PIVpp. In the case of PIV, cleavage of PP occurs, even in the QS mutant. PIV relieved from inhibition by PP processes pro-LasA into mature and active forms. LasB also plays an auxiliary role in activation of LasA.
In this study, some previous perceptions were revised. The first is regarding the factor that activates LasA, and the second is about the activation mechanism of LasB. Although it has been suggested that pro-LasA is processed into active LasA by LasB and lysyl endopeptidase (8), another study has shown that LasB was unable to cleave pro-LasA (29). This study clearly showed that purified LasB could cleave and activate pro-LasA (Figs. 2A and 3A). Furthermore, direct evidence showing that both LasB and PIV are involved in extracellular processing of pro-LasA to activate LasA by removing LasApp was provided. In addition, PIV plays a primary role in degradation of LasApp. LasB is fully responsible for PIV activation but is only partially responsible for LasA activation. If LasB can activate LasA alone, then LasA should be activated normally in the piv mutant, but the results showed that this was not the case (Fig. 4A), indicating that PIV is critically involved in LasA activation; that is, it is not the simultaneous activation of all by LasB but sequential activation from LasB to PIV to LasA.
The autoactivation model through self-cleavage of LasB was revisited in this study. When sequential activation of LasB, PIV, and LasA was confirmed, LasA and PIV were first thought to be inactivated in the QS mutant because transcription of LasB was not induced without QS. However, we found that LasB is also inactive in the QS mutant, even when overexpressed to a sufficient level, and should be activated at the protein level. Our results showed that exogenous addition of culture supernatants from the QS-positive but lasB-deficient strain restored LasB activity in the QS mutant (Fig. 5B), which clearly shows that some QS-dependent factors are required for LasB activation. Apparently, regardless of whether its own activity is critical, LasB cannot activate itself alone and is still subject to tight regulation by QS. Therefore, LasB is not the first initiator in this activation cascade.
The evidence of the autoprocessing of LasB is that, when lasB was expressed in E. coli, processing was independent of any other P. aeruginosa gene product(s) because cleavage of PP occurred (27). This experiment cannot perfectly exclude involvement of the E. coli factor that can act as a functional counterpart of the P. aeruginosa factor. Further evidence is that inactive mutant LasB was not processed (28), but this result is unclear because the same result can be obtained even when the LasB activity is inhibited by other factors. A previous study suggested that LasB is secreted as a noncovalent complex with its PP and that the PP is then degraded extracellularly, most probably by elastase itself (8). Our results also showed that LasB itself can degrade LasBpp extracellularly, but this degradation can occur after LasB is activated, before LasB is inhibited. This inhibition might be relieved by factors other than LasB. Although it is unclear what the factor is, it is believed to be a QS-dependent protein because the heat-inactivated culture supernatants of the QS-positive but lasB-deficient strain were unable to activate LasB in the same experiment as shown Fig. 5B (data not shown).
Interestingly, human NE can activate LasA (Fig. 7A). Because LasA has been reported to enhance the elastolytic activity of several proteases, including LasB and human NE, by interacting with the elastin substrate (26), LasA activity can mutually rise with human NE during P. aeruginosa infection. So far, NE has been known to play a role in preventing infection by degrading the outer membrane proteins of Gram-negative bacteria (30), but our results suggest a harmful role of NE that may assist P. aeruginosa with invasion.
From the results of this study, three new conclusions are proposed. First, LasB is not activated by itself. Instead, it requires a QS-dependent factor for activation. Second, PIV is activated by LasB, which, in turn, activates LasA. Both LasB and PIV can activate LasA, but PIV is a primary activator for LasA. Third, among these three proteases, the activities of LasA and PIV can be inhibited by adding their PPs exogenously, but exogenous LasBpp does not inhibit LasB. This is because when activated, LasB can degrade its own PP (but LasB is not autoactivated), so it is no longer subject to LasBpp.
Experimental procedures
Bacterial strains and culture conditions
Bacterial strains and plasmids used in this study are listed in Table S1. The P. aeruginosa and E. coli strains were generally cultivated in Luria-Bertani medium (LB; 1% bactotryptone, 0.5% yeast extract, and 0.5% NaCl) at 37 °C with vigorous shaking. For solid medium, agar was added at 1.5% (w/v). Antibiotics were used at the following concentrations: ampicillin, 100 μg/ml; tetracycline, 15 μg/ml for E. coli and 50 μg/ml for P. aeruginosa; and gentamicin, 10 μg/ml for E. coli and 50 μg/ml for P. aeruginosa. l-arabinose (0.02%) or IPTG (isopropyl 1-thio-β-d-galactopyranoside, 0.1–0.5 mm) was used to induce protein expression.
Preparation of culture supernatants
The culture supernatants were prepared as described elsewhere (19). Briefly, bacterial cells were grown overnight in 5 ml of LB and diluted 1:100 into 5 ml of fresh LB broth for main culture. Arabinose was added when protein induction was required. After further 16-h cultivation, the cells were removed by centrifugation at 4 °C, and the supernatants were filtered through a 0.22-μm filter (Sartorius). When necessary, the culture supernatants were concentrated by 10-kDa cutoff Centricon (Vivaspin®, Sartorius).
Construction of protein overexpression plasmids
To construct the plasmids that overexpress LasA and LasB, the DNA sequences for lasA (PA1871) and lasB (PA3724) were obtained from the Pseudomonas database (www.pseudomonas.com),3 and the DNA fragments were amplified by PCR reaction using the specific primers containing NdeI and XhoI sites (Table S2). The PCR products were digested with NdeI and XhoI and ligated into the NdeI- and XhoI-digested pQF21c to make pQF21c-LasA and pQF21c-LasB (Table S1), respectively. pQF21c is a plasmid that overexpresses a protein with a C-terminal histidine tag and has a broad host range replication origin, Ori1600, for replication in P. aeruginosa. To construct pET3a-Apro and pET3a-Bpro (Table S1), plasmids that overexpress the PPs of LasA (LasApp) and LasB (LasBpp), respectively, the internal regions of the lasA and lasB genes encoding the PP domains (from amino acids 1–236 of LasA and from amino acids 1–197 of LasB (Fig. S1)) were amplified by PCR with specific primers containing NdeI and BamHI sites (Table S2) and ligated into the NdeI- and BamHI-digested pET3a to generate pET3a-Apro and pET3a-Bpro, respectively. pET3a is a plasmid that overexpresses a native protein.
Preparation of periplasmic proteins
The periplasmic proteins were prepared as described previously (31). Briefly, 2 ml of bacterial cells cultured overnight were harvested by centrifugation for 10 min at 1100 × g. After thoroughly removing the supernatant, the cell pellet was resuspended in 100 μl of 10 mm Tris-HCl (pH 8.0), mixed with an equal volume of chloroform, vortexed briefly, and incubated at room temperature for 15 min. The aqueous fraction containing the periplasmic proteins was separated from chloroform by centrifugation at 6000 × g for 20 min and withdrawn carefully for use in the following experiments.
Overexpression and purification of histidine-tagged proteins
PIV, LasA, and LasB were overexpressed with the C-terminal histidine tag from the pQF21c-based plasmids and purified by affinity chromatography using nickel–agarose resin. The overexpression plasmids for PIV, LasA, and LasB (pQF21c-PIV, pQF21c-LasA, and pQF21c-LasB, respectively (Table S1)) were introduced to P. aeruginosa PAO-T7 or PAO-T7-MW1 by transformation. Overexpression and purification of these histidine-tagged proteases were carried out as described previously for PIV (17, 19). Briefly, cells harboring the overexpression plasmids were inoculated from the colony to 100 ml of fresh LB broth containing 150 μg/ml carbenicillin and cultivated at 37 °C for 6 h with vigorous shaking (180 rpm). The culture was transferred to 1 liter of LB broth with 150 μg/ml carbenicillin and cultivated further up to A600 = 0.5. The culture was chilled to 16 °C, and IPTG was added at 0.1 mm. After further cultivation at 16 °C for 16 h, the cells were removed by centrifugation at 4 °C, and the supernatant was filtered through a 0.22-μm filter. The resulting cell-free supernatant was applied to binding buffer (20 mm Tris-HCl (pH 8.0))–equilibrated nickel-nitrilotriacetic acid–agarose affinity resin (Novagen) for column chromatography. After washing the protein-bound resin with binding buffer, the histidine-tagged proteins were eluted by imidazole-containing elution buffer (20 mm Tris-HCl and 5–500 mm imidazole (pH 8.0)). The fractions containing the target protein were pooled and dialyzed in storage buffer (20 mm Tris-HCl and 5% glycerol (pH 8.0) for LasB and LasA; 50 mm Tris-HCl, 50 mm NaCl, 50 mm KCl, and 30% glycerol (pH 7.5) for PIV). The purified proteins were aliquoted and stored at −80 °C.
Purification of LasApp and LasBpp
pET3a-Apro and pET3a-Bpro (Table S1) were introduced to E. coli BL21 (DE3) to overexpress LasApp and LasBpp, respectively, and cells harboring each plasmid were inoculated into 1 liter of LB broth with 100 μg/ml ampicillin and cultivated at 37 °C up to A600 = 0.5 with shaking at 180 rpm. The cultures were then chilled to 16 °C, and 0.5 mm of IPTG was added to induce proteins. After further cultivation at 16 °C for 16 h, the cells were harvested by centrifugation and washed with buffer (20 mm Tris-HCl (pH 8.0)) at 4 °C to remove the spent medium. The cells were resuspended in a binding buffer (20 mm Tris-HCl (pH 8.0)) and lysed by sonication. The resulting cell debris and insoluble fractions were removed by centrifugation at 15,000 rpm for 20 min. The soluble fractions were loaded onto a Q Sepharose column (GE Healthcare) and separated with NaCl gradient elution in 20 mm Tris-HCl buffer (pH 8.0) by fast FPLC. Fractions containing pure LasApp or LasBpp were pooled, dialyzed in a storage buffer (20 mm Tris-HCl (pH 8.0)), and stored at −80 °C.
LasA activity assay
LasA activity was determined by a staphylolysis assay as described elsewhere (14, 23). Briefly, S. aureus cells were grown overnight, harvested by centrifugation, and washed with water. The cells were resuspended in 25 mm diethanolamine (pH 9.5) and heat-killed at 100 °C for 10 min. The culture supernatants or purified proteases were preincubated in the same diethanolamine buffer for 10 min and mixed with the heat-killed S. aureus cells (final A595 = 1.0). After 1.5-h incubation at 37 °C, the extent of S. aureus cell lysis was determined by measuring the A595 using a spectrophotometer (Optizen 3220UV). With this method, higher LasA activity indicates a lower value. For easier and more intuitive understanding, the activity of LasA was converted to the relative value of the LasA activity in the WT (100%) using the following equation: LasA activity (%) = [(1 − A595 of the staphylolytic reaction with sample)/(1 − A595 of staphylolytic reaction with the WT)] × 100.
LasB activity assay
The activity of LasB was determined from its elastolytic activity using the elastin–Congo red method as described elsewhere with a slight modification (14, 32). Briefly, the culture supernatants prepared from the indicated strains or purified LasB were mixed with 10 mg/ml elastin–Congo red in 10 mm Tris-HCl (pH 8.0) and incubated at 37 °C for 16 h. After centrifugation at 15,000 × g for 10 min to remove any precipitation, A490 was measured using an iMarkTM microplate reader (Bio-Rad). Like LasA activity, LasB activity was also converted to the relative LasB activity in the WT (100%).
PIV activity assay
PIV activity was determined using a chromogenic substrate (N-(p-tosyl)-Gly-Pro-Lys-4-nitroanilide acetate salt, Sigma) as described elsewhere (2). The indicated amount of purified proteins or culture supernatants prepared from the various strains was added to 100 μl of reaction buffer (50 mm Tris-HCl (pH 8.0)) containing 200 μm of chromogenic substrate. The reaction was incubated at 37 °C for 10 min, and A410 was measured using a spectrophotometer (Optizen 3220UV). The PIV activity is also presented as the relative PIV activity in the WT (100%). No significant cross-reactivity was observed in the assays for LasA, LasB, and PIV.
Inhibition of proteases by exogenous PPs
A 500-ng sample of purified mature LasA or LasB was mixed with purified LasApp or LasBpp at the indicated molar ratio and incubated at room temperature for 30 min, and the activity was then measured as described above.
Mutant construction
The mutants used in this work were obtained by gene replacement using the suicide plasmid pEX19Ap (Table S1) and a sacB-based selection strategy (33). Briefly, the up- and downstream regions of the target gene (longer than 500 bp) were amplified by PCR using specific primers (Table S2) and inserted into pEX-19Ap using specific enzyme sites (PstI and XbaI for lasA upstream, KpnI and EcoRI for lasA downstream, SacI and XbaI for lasB upstream, and KpnI and SacI for lasB downstream). The tetracycline resistance (TcR) cassette obtained from the original transposon (Table S1) by PCR was inserted into the XbaI and KpnI sites between the upstream and downstream regions. As a result, the regions covering the full ORFs of lasA and lasB were replaced with the TcR cassette on pEX19Ap. To generate the lasA and lasB mutants, the recombinant plasmids were introduced into P. aeruginosa PAO1 by conjugation (through E. coli SM10 (Table S1)), and the strain with chromosomal integration of the plasmid as a merodiploid was selected on VBMM medium (0.3% trisodium citrate, 0.2% citric acid, 1% K2HPO4, 0.35% NaNH4PO4, 1 mm MgSO4, 100 μm CaCl2, pH 7.0) containing 50 μg/ml tetracycline. The merodiploid was then resolved by plating the strain on VBMM medium containing 5% sucrose. Because the strain that harbors the SacB-containing plasmid moiety cannot survive on sucrose, only the strain that removes the plasmid moiety, leaving the TcR cassette during the resolution of merodiploid, can survive on the plate containing sucrose and tetracycline. The mutants were obtained by successive selection on a plate containing sucrose and tetracycline and confirmed by PCR with specific primers (Table S2) and loss of protease activity.
Statistical analysis
A Student's t test (two-sample assuming equal variances) was used to determine the significance of the differences using Microsoft Office Excel. p < 0.05 was considered significant. All experiments were carried out in triplicate and repeated at least twice independently.
Author contributions
X.-H. L. formal analysis; X.-H. L. writing-original draft; J.-H. L. supervision; J.-H. L. project administration; J.-H. L. writing-review and editing.
Supplementary Material
This work was supported by a National Research Foundation of Korea grant funded by the Korean government (NRF-2019R1A2C1010087). This work was also supported by a grant from the National Research Foundation of Korea (2017M3A9E4078553). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4, Tables S1 and S2, and references.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- LasB
- elastase B
- LasA
- elastase A
- PIV
- protease IV
- SP
- signal peptide
- PP
- propeptide
- T2SS
- type II secretion system
- CS
- culture supernatant
- QS
- quorum sensing
- HSL
- homoserine lactone
- NE
- neutrophil elastase
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside.
References
- 1. Hancock R. E., and Speert D. P. (2000) Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Resist. Updat. 3, 247–255 10.1054/drup.2000.0152 [DOI] [PubMed] [Google Scholar]
- 2. Engel L. S., Hill J. M., Caballero A. R., Green L. C., and O'Callaghan R. J. (1998) Protease IV, a unique extracellular protease and virulence factor from Pseudomonas aeruginosa. J. Biol. Chem. 273, 16792–16797 10.1074/jbc.273.27.16792 [DOI] [PubMed] [Google Scholar]
- 3. Coggan K. A., and Wolfgang M. C. (2012) Global regulatory pathways and cross-talk control pseudomonas aeruginosa environmental lifestyle and virulence phenotype. Curr. Issues Mol. Biol. 14, 47–70 [PubMed] [Google Scholar]
- 4. Rada B., and Leto T. L. (2013) Pyocyanin effects on respiratory epithelium: relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol. 21, 73–81 10.1016/j.tim.2012.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoge R., Pelzer A., Rosenau F., and Wilhelm S. (2010) Weapons of a pathogen: proteases and their role in virulence of Pseudomonas aeruginosa. Current Research, Technology and Education Topics in Applied Microbiology and Microbial Biotechnology 2, 383–395 [Google Scholar]
- 6. Young J. A., and Collier R. J. (2007) Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 76, 243–265 10.1146/annurev.biochem.75.103004.142728 [DOI] [PubMed] [Google Scholar]
- 7. Dolly J. O., and Aoki K. R. (2006) The structure and mode of action of different botulinum toxins. Eur. J. Neurol. 13, 1–9 10.1111/j.1468-1331.2006.01648.x [DOI] [PubMed] [Google Scholar]
- 8. Kessler E., Safrin M., Gustin J. K., and Ohman D. E. (1998) Elastase and the LasA protease of Pseudomonas aeruginosa are secreted with their propeptides. J. Biol. Chem. 273, 30225–30231 10.1074/jbc.273.46.30225 [DOI] [PubMed] [Google Scholar]
- 9. Filloux A., Michel G., and Bally M. (1998) GSP-dependent protein secretion in Gram-negative bacteria: the Xcp system of Pseudomonas aeruginosa. FEMS Microbiol. Rev. 22, 177–198 10.1111/j.1574-6976.1998.tb00366.x [DOI] [PubMed] [Google Scholar]
- 10. Braun P., Ockhuijsen C., Eppens E., Koster M., Bitter W., and Tommassen J. (2001) Maturation of Pseudomonas aeruginosa elastase: formation of the disulfide bonds. J. Biol. Chem. 276, 26030–26035 10.1074/jbc.M007122200 [DOI] [PubMed] [Google Scholar]
- 11. McIver K. S., Kessler E., Olson J. C., and Ohman D. E. (1995) The elastase propeptide functions as an intramolecular chaperone required for elastase activity and secretion in Pseudomonas aeruginosa. Mol. Microbiol. 18, 877–889 10.1111/j.1365-2958.1995.18050877.x [DOI] [PubMed] [Google Scholar]
- 12. Cianciotto N. P. (2005) Type II secretion: a protein secretion system for all seasons. Trends Microbiol. 13, 581–588 10.1016/j.tim.2005.09.005 [DOI] [PubMed] [Google Scholar]
- 13. Kessler E., and Safrin M. (1994) The propeptide of Pseudomonas aeruginosa elastase acts an elastase inhibitor. J. Biol. Chem. 269, 22726–22731 [PubMed] [Google Scholar]
- 14. Kessler E., Safrin M., Olson J. C., and Ohman D. E. (1993) Secreted LasA of Pseudomonas aeruginosa is a staphylolytic protease. J. Biol. Chem. 268, 7503–7508 [PubMed] [Google Scholar]
- 15. Kessler E., Safrin M., Abrams W. R., Rosenbloom J., and Ohman D. E. (1997) Inhibitors and specificity of Pseudomonas aeruginosa LasA. J. Biol. Chem. 272, 9884–9889 10.1074/jbc.272.15.9884 [DOI] [PubMed] [Google Scholar]
- 16. Gustin J. K., Kessler E., and Ohman D. E. (1996) A substitution at His-120 in the LasA protease of Pseudomonas aeruginosa blocks enzymatic activity without affecting propeptide processing or extracellular secretion. J. Bacteriol. 178, 6608–6617 10.1128/jb.178.22.6608-6617.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park S. J., Kim S. K., So Y. I., Park H. Y., Li X. H., Yeom D. H., Lee M. N., Lee B. L., and Lee J. H. (2014) Protease IV, a quorum sensing-dependent protease of Pseudomonas aeruginosa modulates insect innate immunity. Mol. Microbiol. 94, 1298–1314 10.1111/mmi.12830 [DOI] [PubMed] [Google Scholar]
- 18. Bradshaw J. L., Caballero A. R., Bierdeman M. A., Adams K. V., Pipkins H. R., Tang A., O'Callaghan R. J., and McDaniel L. S. (2018) Pseudomonas aeruginosa protease IV exacerbates pneumococcal pneumonia and systemic disease. mSphere 3, e00212–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oh J., Li X. H., Kim S. K., and Lee J. H. (2017) Post-secretional activation of protease IV by quorum sensing in Pseudomonas aeruginosa. Sci. Rep. 7, 4416 10.1038/s41598-017-03733-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schuster M., Lostroh C. P., Ogi T., and Greenberg E. P. (2003) Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 185, 2066–2079 10.1128/JB.185.7.2066-2079.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fuqua C., and Greenberg E. P. (2002) Listening in on bacteria: acyl-homoserine lactone signalling. Nat. Rev. Mol. Cell Biol. 3, 685–695 10.1038/nrm907 [DOI] [PubMed] [Google Scholar]
- 22. Lee J. H., Lequette Y., and Greenberg E. P. (2006) Activity of purified QscR, a Pseudomonas aeruginosa orphan quorum-sensing transcription factor. Mol. Microbiol. 59, 602–609 10.1111/j.1365-2958.2005.04960.x [DOI] [PubMed] [Google Scholar]
- 23. Folders J., Tommassen J., van Loon L. C., and Bitter W. (2000) Identification of a chitin-binding protein secreted by Pseudomonas aeruginosa. J. Bacteriol. 182, 1257–1263 10.1128/JB.182.5.1257-1263.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park S., and Galloway D. R. (1995) Purification and characterization of LasD: a second staphylolytic proteinase produced by Pseudomonas aeruginosa. Mol. Microbiol. 16, 263–270 10.1111/j.1365-2958.1995.tb02298.x [DOI] [PubMed] [Google Scholar]
- 25. Chapon-Hervé V., Akrim M., Latifi A., Williams P., Lazdunski A., and Bally M. (1997) Regulation of the xcp secretion pathway by multiple quorum-sensing modulons in Pseudomonas aeruginosa. Mol. Microbiol. 24, 1169–1178 10.1046/j.1365-2958.1997.4271794.x [DOI] [PubMed] [Google Scholar]
- 26. Peters J. E., and Galloway D. R. (1990) Purification and characterization of an active fragment of the LasA protein from Pseudomonas aeruginosa: enhancement of elastase activity. J. Bacteriol. 172, 2236–2240 10.1128/jb.172.5.2236-2240.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McIver K., Kessler E., and Ohman D. E. (1991) Substitution of active-site His-223 in Pseudomonas aeruginosa elastase and expression of the mutated lasB alleles in Escherichia coli show evidence for autoproteolytic processing of proelastase. J. Bacteriol 173, 7781–7789 10.1128/jb.173.24.7781-7789.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McIver K. S., Olson J. C., and Ohman D. E. (1993) Pseudomonas aeruginosa lasB1 mutants produce an elastase, substituted at active-site His-223, that is defective in activity, processing, and secretion. J. Bacteriol. 175, 4008–4015 10.1128/jb.175.13.4008-4015.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park S., and Galloway D. R. (1998) Pseudomonas aeruginosa LasD processes the inactive LasA precursor to the active protease form. Arch. Biochem. Biophys. 357, 8–12 10.1006/abbi.1998.0787 [DOI] [PubMed] [Google Scholar]
- 30. Belaaouaj A., Kim K. S., and Shapiro S. D. (2000) Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science 289, 1185–1188 10.1126/science.289.5482.1185 [DOI] [PubMed] [Google Scholar]
- 31. Ames G. F., Prody C., and Kustu S. (1984) Simple, rapid, and quantitative release of periplasmic proteins by chloroform. J. Bacteriol. 160, 1181–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rust L., Messing C. R., and Iglewski B. H. (1994) Elastase assays. Methods Enzymol. 235, 554–562 10.1016/0076-6879(94)35170-8 [DOI] [PubMed] [Google Scholar]
- 33. Hoang T. T., Karkhoff-Schweizer R. R., Kutchma A. J., and Schweizer H. P. (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86 10.1016/S0378-1119(98)00130-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.