

Abstract
The catalytic use of a small peptide scaffold for the biosynthesis of amino acid-derived natural products is a recently discovered new biosynthetic strategy. During this process, a peptide-amino acyl tRNA ligase (PEARL), adds amino acids to the C-terminus of a small peptide scaffold in an ATP and tRNA-dependent process. The mechanism of this unusual transformation is currently not known. In this study, we present a detailed biochemical and mechanistic study of TglB (UniProtKB - F3HQJ1), a PEARL that catalyzes the addition of Cys to the C-terminus of the peptide TglA in the biosynthesis of 3-thiaglutamate in the plant pathogen Pseudomonas syringae. TglB recognizes several important residues close to the C-terminus of TglA to perform its activity, and is tolerant with respect to the last amino acid of its substrate peptide. The enzyme recognizes the acceptor stem of tRNACys as micro- and minihelices, truncated versions of full length tRNACys that contain the acceptor stem, were also accepted. Mutagenesis of conserved residues in TglB identified several key residues for catalysis and did not support the possibility of TglB adopting various ping-pong mechanisms to catalyze the amino acid addition reaction. Using isotopic labeling studies, we demonstrate that ATP is used to directly phosphorylate the C-terminal carboxylate of TglA. Collectively, the data support a general mechanism for the amino acid addition reaction catalyzed by this class of enzyme.
Graphical Abstract
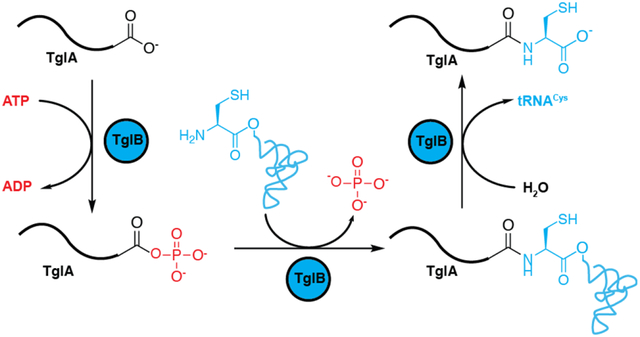
Introduction
Oligopeptide extension of a genetically encoded peptide using aminoacyl-tRNA was until recently limited to ribosome catalysis. During this process, the ribosome catalyzes amide bond formation by attack of the amino group of an aminoacyl-tRNA onto the ester by which the nascent peptide is linked to the tRNA of the most recently incorporated amino acid (Figure 1A). A recent study1 described an alternative route to peptide extension in which amino acid residues are added to the C-termini of ribosomally synthesized peptides in an ATP and tRNA dependent manner (Figure 1B). The enzymes catalyzing this transformation have been termed peptide aminoacyl tRNA ligases (PEARLs). This process is involved in the biosynthesis of a range of natural products in phylogenetically diverse bacteria including the ammosamides and 3-thiaglutamate.1 At present the details of how this process takes place in the absence of an RNA template that guides amide bond formation in the ribosome are unknown.
Figure 1 ∣. Different routes towards peptide extension and processes catalyzed by PEARL and LanB enzymes.

(A) Ribosome catalyzed amide bond formation. (B) Amide bond formation catalyzed by the PEARL TglB. (C) LanB enzymes glutamylate Ser/Thr residues in their substrate peptides and subsequently eliminate the glutamate to form dehydroamino acids. PEARL proteins have homology with the glutamylation domain of LanB proteins but lack the glutamate elimination domain. Dha, dehydroalanine; Dhb, dehydrobutyrine. (D) Proposed biosynthetic pathway towards 3–thiaglutamate.1 SAM, S-adenosyl-L-methionine. CysRS, cysteine tRNA synthetase. SAH, S-adenosyl-L-homocysteine.
PEARLs were discovered during our studies on ribosomally synthesized and post-translationally modified peptides (RiPPs), which are a large and diverse group of natural products.2,3 RiPPs are biosynthesized from a precursor peptide consisting of a leader peptide that serves as a recognition motif for the biosynthetic enzymes and a core peptide that is converted to the final product. Included in this group of natural products is the lanthipeptide nisin, which has been used for decades against food-borne pathogens with very little development of stable resistance.4 During the maturation of nisin, Ser and Thr residues are first glutamylated by the LanB dehydratase NisB in a glutamyl-tRNA dependent mechanism (Figure 1C).3,5 Subsequently, the glutamate is eliminated by the elimination domain of NisB to generate dehydroamino acids. Recent studies in thiopeptide biosynthesis6 and goadsporin maturation7 revealed that the glutamylation and elimination steps for dehydroamino acid formation can also be carried out by split LanB dehydratase enzymes composed of stand-alone glutamylation and elimination proteins. A survey of bacterial genomes also revealed more than 600 genes encoding LanB-like proteins lacking the elimination domain altogether in the genome.1 This observation suggested that these stand-alone glutamylation domains might add an amino acid in a tRNA-dependent fashion without subsequent elimination. Indeed, experiments on such an enzyme termed TglB from the plant pathogen Pseudomonas syringae transferred a cysteine residue from cysteinyl-tRNA to a ribosomally synthesized precursor peptide TglA. Surprisingly the Cys was not attached to a nucleophilic side chain of one the amino acids in TglA, but instead to the C-terminus of the peptide in an ATP-dependent manner (Figure 1B).1 After addition of the Cys, this amino acid is further modified to furnish 3-thiaglutamate by a series of enzymatic transformations that involve β-carbon excision, carboxymethylation, and proteolysis to yield an amino acid derived final product (Figure 1D). PEARL enzymes are not limited to the use of Cys-tRNACys as other gene clusters encode up to seven different PEARL enzymes and one family member attaches Trp in a Trp-tRNATrp dependent fashion in the ammosamide biosynthetic pathway.1 Thus, these studies revealed an interesting new paradigm for the generation of small, amino acid derived natural products by a posttranslational modification process. These pathways provide a variant of canonical RiPP biosynthesis because the final product is not encoded on a core peptide but added after ribosomal synthesis. Thus, unlike other RiPPs, the possible structure of the final product is not predicted in the gene cluster. Here, we describe our studies of the PEARL TglB that focused on its substrate specificity and mechanism of peptide extension.
Results
Characterization of the substrate specificity of TglB
Previous truncation studies on TglA demonstrated a 12-mer peptide corresponding to the C-terminus of TglA is sufficient for peptide extension.1 To investigate which residues within this 12-mer peptide are important for TglB binding, we generated a panel of TglA variants by replacing each residue with an alanine and performing competition fluorescence polarization (FP) assays with a fluorescein-labeled peptide corresponding to the C-terminal 20 amino acids of TglA to determine relative IC50 values (Table 1, Figure S1). These experiments revealed a set of TglA residues that are important for TglB binding (Ile41, Val43, Ile44, Glu45, Lys47 and Val48). In addition to several hydrophobic residues that appear to mediate protein-peptide interactions, the two charged residues (Glu45, Lys47) contribute most to the affinity between TglA and TglB.
Table 1 ∣. Competition binding of TglA-Ala variants to TglB.
A fluorescein labeled peptide corresponding to the C-terminal 20 amino acids of TglA was competed off with full length TglA and its mutants. FP traces are shown in Figure S1.
| TglA variant |
IC50 (μM) | TglA variant |
IC50 (μM) |
|---|---|---|---|
| WT TglA | 8 ± 1 | E45A | > 300 |
| D40A | 15 ± 3 | S46A | 11 ± 2 |
| I41A | 32 ± 4 | K47A | 255 ± 63 |
| E42A | 10.5 ± 0.3 | V48A | 113 ± 36 |
| V43A | 98 ± 36 | F49A | 4.2 ± 0.2 |
| I44A | 24 ± 2 |
We next evaluated the substrate specificity of TglB with respect to the last amino acid of TglA. A series of TglA variants were prepared in which the last Ala residue (Ala50, Figure 1B) was substituted with seven other amino acids ranging in size and polarity (Glu, Lys, Gln, Ser, Pro, Phe, and Gly; Figure S2A). These TglA variants were individually co-expressed with TglB in Escherichia coli. All variants were accepted by TglB except for the charged amino acid residues Glu and Lys. Based on the acceptance of the A50Q variant, it appears that as long as the charge is blocked, the TglA variant can be converted into the cysteinylated product, although the efficiency of conversion varied (Figure S2A). These data indicate that TglB is relatively permissive towards changing the last amino acid of its peptide substrate, and that the active site of the protein can accommodate different sizes of amino acid side chains. However, when the last Ala residue was removed from the TglA peptide, resulting in Phe49 as the C-terminal amino acid, no product formation could be detected even though the A50F mutant was a substrate (Figure S2A and B). These observations suggest a well-defined distance between the binding site on TglA and the C-terminus of the peptide where catalysis takes place.
Based on the previously proposed tRNA-NisB binding model,5 nucleotides within the tRNAGlu acceptor stem are believed to be the major recognition elements. Previous studies on the lanthipeptide dehydratase MibB also suggest that the nucleotide pairs at positions 4 and 69 do not appear to be very important for efficient dehydration.8 To characterize the region of tRNACys that is important for TglB recognition, we performed in vitro cysteinylation reactions with mini- and micro-helices9 based on the P. syringae tRNACys (Figure S3). The cysteinylated product was clearly observed using either of these simplified co-substrates, although at a lower level compared with the use of full length tRNACys. This finding suggests that the interaction between TglB and the acceptor stem of the tRNACys is sufficient for the cysteinylation reaction to occur. The observed low overall conversion probably results from the anticipated poor catalytic efficiency of CysRS to load Cys onto these tRNACys mini- and micro-helices, because the use of these truncated substrates resulted in a decrease in kcat/Km of several orders of magnitude relative to full-length tRNA in studies of amino-acyl tRNA synthetases,10-12 including a study on E. coli CysRS showing a decrease in kcat/Km of more than 2 × 105.13
Reaction with 18O labeled TglA substrate
Previous 31P NMR analysis of the reaction, as well as NH2OH quenching assays, suggests the C-terminal carboxylate of TglA is activated by ATP-dependent phosphorylation.1 To provide direct evidence for this hypothesis, we performed the reaction with a substrate containing a C-terminal 18O-labeled carboxylate. If phosphorylation of the C-terminal carboxylate of TglA indeed occurs, the 18O should be incorporated into the released phosphate, which can be tested by 31P NMR spectroscopy. To prepare the labeled substrate, we purified a TglA-intein fusion protein expressed in E. coli, incubated the protein with 2-mercaptoethanesulfonate, and hydrolyzed the resulting C-terminal thioester in H218O (see experimental methods). Analysis by ESI-MS of the endo-proteinase GluC-digested 18O-labeled TglA indicated ~85% of the C-terminal carboxylate contained one 18O (Figure 2A). Using this peptide for a multiple turnover assay and subsequent 31P NMR analysis confirmed 18O incorporation into phosphate (18O-Pi) (Figure 2B), which could only originate from the C-terminal carboxylate of TglA. The assay was repeated twice and gave approximately the same result. The incorporation of 18O originating from TglA into phosphate is consistent with direct phosphorylation of the peptide. In that scenario, the relatively small 18O-Pi peak compared to that of 16O-Pi (Figure 2B) could result from uncoupled cleavage of ATP by TglB. Such uncoupling was indeed observed by 31P NMR analysis of TglB incubated with ATP in the absence of TglA (Figure S4). Alternatively, the relatively small amount of 18O in the phosphate could be indicative of a mechanism that does not involve direct phosphorylation of the carboxylate of TglA but instead involves phosphorylation of the enzyme. For instance, mechanism I in Figure 2C would predict that 18O from the substrate slowly accumulates in the enzyme in a multi-turnover assay, and then subsequently starts to be found in the phosphate produced after a few turnovers. According to this mechanism, no 18O would be incorporated into phosphate in the first turnover, and 25% in the second turnover (Figure 2C).
Figure 2 ∣. 18O incorporation into phosphate in multi-turnover reaction.

(A) ESI-MS analysis of GluC digested 18O labeled TglA. (B) 31P NMR analysis shows 18O incorporation into phosphate when using 50 μM 18O labeled TglA for a multiple turnover reaction catalyzed by 5 μM TglB in the presence of 50 μM Cys-tRNACys, 0.5 mM ATP, and 5 mM MgCl2. (C) Two possible mechanisms that could explain the 18O incorporation into phosphate in a multi-turnover reaction. In mechanism I, ATP-dependent phosphorylation occurs on a carboxylic acid residue on TglB, and subsequently involves the intermediacy of a TglA-TglB anhydride. This would result in 18O-labeled TglB, which could deposit the label into phosphate in subsequent turnovers. In mechanism II, it is the C-terminal carboxylate of TglA that is directly activated during reaction.
To test the feasibility of phosphorylation of the enzyme or the peptide substrate (Figure 2C, mechanisms I or II), 31P NMR analysis of a single turnover assay was performed. In mechanism I, ATP is used to phosphorylate a carboxylate side chain of Glu/Asp or the C-terminal carboxylate in TglB, then the C-terminal carboxylate of TglA attacks this phosphorylated intermediate to form a substrate-protein conjugate via an anhydride linkage, and attack by Cys-tRNACys followed by hydrolysis would provide the product. Phosphorylation of an enzymatic carboxylate would be analogous to the ATP-grasp domain-containing enzymes from the ADP-forming carboxylate-amine/thiol ligase superfamily.14-17 Previous mechanistic studies on enzymes such as phosphotransferases18,19 and P-type ATPases,20 revealed a mechanism with a covalent aspartyl-phosphate intermediate. If such a mechanism were to be used by TglB, then under multiple turnover conditions, the side chain carboxylate of TglB that is used for catalysis would gradually be enriched with 18O (Figure 2C) resulting in partial incorporation of the label into phosphate consistent with the observations in Figure 2B. Conversely, if TglB catalyzes the cysteinylation reaction through mechanism I, no 18O-Pi would be detected in the first turnover (Figure 2C, mechanism I). Thus, a single turnover reaction was performed, and 18O-Pi accounted for ~33% of the total phosphate (Figure 3), thereby ruling out the possibility of the intermediacy of a TglA-TglB anhydride. Theoretically, with one of the carboxylate oxygens of the C-terminus of TglA labeled with 85% 18O (Figure 2A), 42% label would have been expected (for discussion of the expected 18O content, see Figure S4). The explanation for this apparent discrepancy (33% measured vs 42% expected) comes from integration of the peaks corresponding to ADP and phosphate (16O-Pi + 18O-Pi) in the 31P NMR spectrum. The peak area of labeled and unlabeled phosphate combined is larger than that of ADP produced. Thus, either during the assay or during the prolonged time of the NMR experiment, a portion of ADP was hydrolyzed to generate 16O-Pi (also supported by the detection of AMP). When corrected for this additional amount of unlabeled phosphate that is not the result of enzymatic turnover (see legend Fig S4), the ratio of 18O-Pi to 16O-Pi would be ~ 1 : 1.4, which is very close to the theoretical ratio of the single turnover reaction for mechanism II when starting with a peptide that is 85% labeled with one 18O atom (1: 1.35, see legend Figure S4). The single turnover assay was repeated resulting in the same 18O-Pi to 16O-Pi ratio, ruling out the possibility that the discrepancy between observed and theoretical ratio originates from the variability of experimental conditions. These data are not compatible with mechanism I and support mechanism II, in which the C-terminal carboxylate of TglA is directly activated by phosphorylation. Attempts to detect the phosphorylated TglA by NMR spectroscopy or MS were unsuccessful, but as mentioned previously evidence for an activated substrate peptide was obtained by trapping experiments with hydroxylamine that resulted in detection of the hydroxamate of TglA.1
Figure 3 ∣. 31P NMR analysis of a single turnover TglB reaction using 18O labeled TglA.

The 18O incorporation in the product phosphate strongly supports mechanism II in Figure 2C.
Mutagenesis experiments argue against a ping-pong mechanism for the cysteinylation reaction
TglB catalyzes the conjugation of two highly dissimilar macromolecules (peptide and aminoacyl-tRNA) without the templating by an RNA molecule that the ribosome uses. A possibility to achieve such a reaction is to first covalently link either reactant to the enzyme. One such ping-pong mechanism could involve the formation of an acyl-enzyme intermediate between a hydroxyl group on TglB and the carboxyl group of Cys (from Cys-tRNACys) similar to the mechanism of the cyclodipeptide synthases (Figure S5).21 We therefore performed a sequence alignment of TglB with its orthologs from various PEARL-containing gene clusters and identified four conserved Ser or Thr residues (Ser11, Thr180, Ser540, and Ser780; Figure S6). TglB variants were generated in which these Ser or Thr residues were replaced by Ala and these variants were co-expressed with TglA in E. coli. All of the TglB mutants retained the ability to add Cys to the C-terminus of TglA (Figure S7A), suggesting that these conserved Ser or Thr residues are not involved in formation of the corresponding acyl-enzyme intermediates. Further mutagenesis of a single fully conserved Tyr residue in TglB to Phe do not support the possibility of a conserved Tyr to form an acyl-enzyme intermediate during catalysis (Figure S7B); no conserved Cys residue was identified in the alignment. These data suggest that TglB does not utilize a ping-pong mechanism to generate a covalent cysteinyl-enzyme intermediate for catalysis. In addition, these experiments also argue against mechanisms in which TglA is covalently linked to a hydroxyl group of a conserved amino acid residue on TglB after phosphorylation of the TglA C-terminus (Figure S8).
Although the 18O-labeling experiments described above argue against mechanism I (Figure 2C), they do not rule out a mechanism in which ATP is used to first activate the TglA C-terminus, followed by attack by a carboxylate residue on TglB to form an anhydride and subsequent attack by the cysteinyl-tRNA (Figure S9). We therefore performed a series of experiments designed to trap any activated carboxylate intermediates. As noted previously, performing the reaction in the presence of hydroxylamine resulted in trapping of an activated TglA intermediate as the corresponding hydroxamate,1 but those experiments did not look for any trapping of an activated intermediate on the enzyme. To probe whether TglB forms either an acyl-phosphate intermediate during catalysis (Figure S10) or an TglA-TglB anhydride (Figure S9), chemical trapping with hydroxylamine or sodium borohydride (NaBH4) was attempted using protocols reported for the study of phosphotransferases.22 However, we were not able to detect any modified peptide fragments in the LC-ESI-MS analysis of proteinase-digested TglB, which disfavors the generation of any intermediate in which either phosphate or TglA is covalently linked to TglB during catalysis. Collectively, mechanism II (Figure 2C) is the most likely mechanism used by the enzyme even though it is peculiar that in this mechanism the activated carboxyl group of Cys-tRNACys is not used and is merely hydrolyzed. Such hydrolysis is consistent with experiments conducted in H218O which produced the product peptide with one 18O label.1
Identification of TglB residues important for catalysis
To further identify residues important for catalysis by TglB, we took a closer look at the multiple sequence alignment and identified eighteen additional conserved residues with side chains that could be involved in catalysis (Figure S6). Each of these residues was replaced with Ala and co-expressed with TglA in E. coli. The activity of these mutant enzymes was qualitatively assessed by MALDI-TOF-MS analysis of the co-expression product (Table S1). A set of ten residues (Arg10, Arg173, Lys177, Asp179, Asp542, Asp564, His566, Lys752, Glu801 and Arg803) was identified as possibly involved in substrate binding or catalysis. To confirm that any diminished activity in E. coli did not result from low production of the enzyme, the ten TglB variants were also individually expressed and purified from E. coli (Figure S11) and tested for in vitro activity (Figure S12). There was good consistency between the in vivo and in vitro experimental data which demonstrated that of the ten residues, Arg10, Arg173, Arg177, Asp542, Glu801, and Arg803 are indispensable, whereas Asp179, Glu564, and His566 are not important. Mutation of Lys752 led to a variant with reduced activity.
Repeated attempts to crystallize TglB proved recalcitrant. Thus, a predicted model of TglB was generated using the Phyre2 web portal23 based on the crystal structure of the glutamylation protein TbtB involved in thiomuracin biosynthesis.24 In this homology model (Figure 4A), an Arg/Lys-rich region suitable for interacting with the negatively charged phosphodiester groups of the amino acyl-tRNA was observed. The model also showed a helix-turn-helix structural motif, which is also present in the lanthipeptide dehydratases NisB5 and MibB8 and is predicted to be the leader peptide binding element (or RiPP recognition element, RRE)25 engaged in TglA recognition. Moreover, the TglB residues identified to have diminished activity through Ala substitution assays discussed above are all clustered in a putative active site, which further corroborates the role of these residues during catalysis and the reliability of the model.
Figure 4 ∣. Phyre2 homology model of TglB.

(A) The overall predicted structure of TglB is shown in grey. The Arg/Lys-rich region putatively involved in tRNA binding is colored in navy. The RRE motif engaged in precursor peptide recognition is colored in yellow. Residues identified to have diminished cysteinylation activity in TglB are clustered in the predicted structure as shown in the insert. (B) Calculated electrostatic potential mapped onto the TglB surface showing the basic patch (blue) that probably engages in ATP and/or Cys-tRNACys binding. The sites of putative ATP and tRNA binding are based on mutagenesis data in this study.
We reasoned that the variants that no longer conjugated Cys with TglA could still potentially carry out half-reactions and hence might be good candidates for trapping any intermediates. We searched for potential intermediates in which the substrate peptide is covalently linked to the tRNA as predicted by the various mechanisms, but we were not able to detect any such species. This finding suggests that none of the residues mutated is solely responsible for hydrolysis of the peptide-tRNA intermediate. Next, we again performed NH2OH quenching assays with the TglB mutants that displayed no detectable conjugation enzyme activity. The resulting data suggest that Arg10, Arg173 and Lys177 do not participate in the activation of TglA during catalysis, as mutation of these residues did not affect the formation of a C-terminal hydroxamate of TglA. In contrast, residues Asp542, Glu801 and Arg803 could potentially be involved in the phosphorylation step of the reaction, either through ATP/peptide binding or catalyzing acyl-phosphate formation on TglA, because mutation of these residues to Ala completely abolished the NH2OH addition to TglA (Figure S13). Calculation of the electrostatic potential of the Phyre2 homology model also reveals the basic nature of the active site by virtue of a Arg/Lys-rich region (Figure 4B), which is consistent with the expected interaction with ATP, the C-terminal carboxylate of the peptide substrate and aminoacyl-tRNA. This highly basic active site may also explains why replacing the last Ala of TglA with charged residues did not result in product formation, as the electrostatic interaction could cause misorientation of the C-terminal carboxylate of TglA in the active site.
Discussion
PEARLs are intriguing enzymes that perform C-terminal peptide extension reactions in an ATP and tRNA dependent manner. They are not the first examples of ribosome-independent amino acid-peptide ligases that use amino acyl-tRNA as such activities are also found in cell wall and natural product biosynthesis,26-29 but to the best of our knowledge they are the first examples of enzymes that catalyze C-terminal extension of ribosomally synthesized peptides, which usually is performed by the ribosome. In our previous work we suggested that the reason why the amino acids that are added by PEARL enzymes to the C-termini of their peptide substrates are not genetically encoded is that the independent addition and subsequent modification of these amino acids to the mature natural products allows catalytic use of the substrate peptide as a scaffold. In other words, rather than making a single molecule of 3-thiaglutamate from a 51-amino acid precursor, the organism can make many molecules of 3-thiaglutamate from a 50-amino acid precursor by re-addition of Cys from Cys-tRNACys after each cycle of making 3-thiaglutamate. This biosynthetic paradigm has resemblances with both non-ribosomal peptides synthesis, which uses the peptide carrier protein as scaffold, and RiPP biosynthesis by using the same mechanism of binding of the scaffold peptide to an RRE domain.
In this study, we performed detailed biochemical and mechanistic studies of TglB involved in the biosynthesis of 3-thiaglutamate to characterize its substrate specificity and investigate the general mechanism of this class of enzyme. We found that two charged residues of TglA are the most critical for binding to the RRE containing TglB protein, in addition to an array of hydrophobic residues that also appear to contribute to protein recognition. With the exception of charged residues, TglB is surprisingly tolerant with respect to the last amino acid of the substrate peptide suggesting that the C-terminal amino acid is not an important part of substrate recognition. However shortening the distance from the putative binding site on TglA to the C-terminus by just one amino acid abolished activity suggesting that binding to the RRE sets the substrate in the right register for peptide extension. With respect to recognition of the other co-substrate, Cys-tRNACys, use of truncated tRNACys analogs illustrates the importance of the acceptor stem of the tRNACys for interaction.
ATP is required for the peptide extension catalyzed by TglB, which is an important difference from LanB enzymes that catalyze dehydration of Ser/Thr, which do not require ATP.5 Regarding the role of ATP during the reaction, 18O labeling studies provide strong evidence for a mechanism in which ATP is used to directly activate the C-terminal carboxylate of TglA. Mutagenesis of TglB revealed several residues important for catalysis, and their potential function in catalysis was probed by NH2OH quenching assays and Phyre2 model prediction. Along with our previous study on the mechanism of TglB,1 the data presented herein support a general mechanism that may be adopted by PEARL enzymes for peptide extension reactions (Figure 2C, mechanism II). ATP is used to activate the C-terminal carboxylate of the peptide through phosphorylation, then the phosphorylated peptide is attacked by the amino group on the amino acyl-tRNA, and finally hydrolysis on the amino acid-tRNA ester bond resolves the peptide-tRNA complex and releases the final product. The use of Cys-tRNACys instead of just Cys appears wasteful as the activated ester in Cys-tRNACys is never used. Presumably, Cys-tRNACys is used as substrate as a vestige of a predecessor enzyme that uses aminoacyl tRNA, which could possibly be LanB. Remaining questions include how ATP is activated since PEARLs do not contain canonical ATP-binding sequences, and how LanBs and PEARLs are evolutionary related given that they catalyze fundamentally different chemical transformations.
Experimental Section
General materials and methods
Reagents used for molecular biology experiments were purchased from New England BioLabs (Ipswich, MA), Thermo Fisher Scientific (Waltham, MA), or Gold Biotechnology Inc. (St. Louis, MO). Other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Escherichia coli DH5α and BL21 (DE3) strains were used for plasmid maintenance and protein overexpression, respectively. Co-expression vector pRSFDuet-1 was obtained from Novagen. MALDI-TOF-MS analysis was performed using a Bruker UltrafleXtreme MALDI TOF-TOF mass spectrometer (Bruker Daltonics) in reflector positive mode at the University of Illinois School of Chemical Sciences Mass Spectrometry Laboratory. ESI-MS/MS analyses were performed using a SYNAPT ESI quadrupole TOF Mass Spectrometry System (Waters) equipped with an ACQUITY Ultra Performance Liquid Chromatography (UPLC) system (Waters). 31P NMR spectra were recorded on an Agilent 600 MHz spectrometer in 10% D2O. HiTrap columns for Ni-NTA affinity chromatography were purchased from GE Healthcare.
Molecular biology techniques
DNA was prepared using MiniPrep kits (Qiagen) using the manufacturer’s instructions from E. coli DH10b cells (New England Biolabs) made chemically competent by the KCM method.30 Plasmids were constructed with type-II restriction enzymes using the New England Biolabs Golden Gate Assembly Tool (http://goldengate.neb.com/editor) or manually designed and prepared by Gibson assembly. For plasmids assembled by Golden Gate Assembly, two primer pairs were designed for PCR to prepare both the gene and the vector with complementary sticky ends for type IIs restriction digest. BsaI and T4 DNA Ligase were used for single step DNA ligation. For plasmids assembled by Gibson Assembly, one primer pair was designed for PCR amplification of the gene with an overhang complementary to the vector. The vector was linearized by restriction digest with NdeI and XhoI and ligated with the gene of interest using NEBuilder® HiFi DNA Assembly Master Mix. Restriction enzymes and PCR polymerases were obtained from New England Biolabs. Genes for each of the Tgl gene cluster products and the P. syringae cysteine tRNA synthetase (CysRS) were identified using the Joint Genome Initiative Integrated Microbial Genomes and Microbiomes webtool (http://img.jgi.doe.gov). Primers for cloning and site-directed mutagenesis were obtained from Integrated DNA Technologies. Primers for P. syringae tRNACys were designed and the dsDNA template for in vitro transcription was prepared as previously described.31 Mutagenesis was accomplished using the QuikChange II Site-Directed Mutagenesis Kit according to the manufacturer’s instructions. Mutagenesis primers were designed using the QuikChange Primer Design webtool. Primers are listed in Table S2.
In vitro transcription of P. syringae tRNACys
Primers for P. syringae tRNACys were designed according to a previously described method.31 The tRNAGlu dsDNA template was generated from two overlapping synthetic deoxyoligonucleotides (Table S2). To prepare the dsDNA template for in vitro transcription, 5′ overhangs were filled in using the following conditions: NEB Buffer 2 (1×), primers (4 μM each), dNTP (100 μM each), and DNA polymerase I large (Klenow) fragment (1 U μg−1 DNA) in a final volume of 50 μL. The reaction was incubated at 25 °C for 15 min, quenched with EDTA (10 mM) at 75 °C for 25 min, and dsDNA tRNACys template was precipitated with cold EtOH overnight. In vitro transcription was performed using a previously described method.32 The transcribed tRNACys was then purified by acidic phenol extraction using a previously described method.33
Peptide expression and purification
For peptide expression, E. coli BL21(DE3) cells (New England Biolabs) expressing N-terminal His6-tagged TglA from His6-TglA-pRSF-Duet-1 were grown with 50 μg/mL kanamycin in autoinduction (AI) media (for 1 L total: 10 g bacto tryptone, 5 g yeast extract, 5 g NaCl, 3 g KH2PO4, 6 g Na2HPO4) with 1X AI sugar solution containing 0.5% (vol/vol) glycerol, 0.05% (w/vol) glucose, and 0.2% (w/vol) lactose (20 mL of 50× stock)). Cultures were shaken at 37 °C for 6-7 h following inoculation with 1 mL of saturated culture. Cells were harvested and lysed by sonication. Peptides were purified by immobilized metal affinity chromatography (IMAC). The lysate was clarified by centrifugation at 29,000 rcf and applied to a 5 mL NiNTA-agarose column (GE Healthcare) using a peristaltic pump. The immobilized peptide was washed with 5 column volumes (CV) of 90% lysis buffer (50 mM HEPES, 100 mM NaCl, pH 7.5), 10% elution buffer (50 mM HEPES, 100 mM NaCl, 500 mM imidazole, pH 7.5; final imidazole concentration in wash: 50 mM) and eluted with 100% elution buffer. The elution fraction was concentrated using a 3 kDa MWCO Amicon spin filter and washed with 10-20 CV of deionized water to remove imidazole. Crude peptide was desalted with a VYDAC® Bioselect C4 cartridge. Peptide elution fractions were lyophilized.
Protein expression and purification for TglB and CysRS.
The general expression protocol for TglB was as follows: E. coli BL21 (DE3) cells (50 μL) were electroporated with His6-TglB-pET28a (50 ng), and cells were plated on LB agar plates supplemented with kanamycin (50 μg/mL) and grown at 37 °C for 12–15 h. A single colony was used to inoculate 20 mL of LB broth supplemented with kanamycin, the culture was grown for 12–15 h at 37 °C, and used to inoculate 2 L of TB media, supplemented with kanamycin, to an OD600 of 0.025. Cultures were grown at 37 °C to a final OD600 of 0.6–0.8. Protein overexpression was induced by the addition of IPTG to a final concentration of 0.2 mM, and cultures were grown at 18 °C for 18 h. Cells were harvested by centrifugation, collected in 50 mL tubes and frozen in liquid nitrogen. For purification of His6-TglB and His6-CysRS, cells were thawed, resuspended in lysis buffer (50 mM HEPES, 100 mM NaCl, pH 7.5; 30 mL per 10 g wet cell paste) and lysed by treatment with lysozyme (100 μg/mL) and sonication (3 min active time; 1 s pulse, 2 s rest at 60 % max amplitude using a 1 cm tip). Proteins were purified by IMAC. Phenylmethylsulfonyl fluoride (PMSF, 0.1 mM) was added to prevent degradation by serine proteases and 1 mM TCEP was included to maintain reduced thiols. The eluate was concentrated to 2.5 mL in a 30 kDa MWCO centrifuge filter and desalted on a PD-10 size-exclusion column (GE Healthcare Life Sciences) with storage buffer (50 mM HEPES, 500 mM NaCl, 10% glycerol, pH 7.5). Protein was separated into aliquots and stored at −70 °C. Protein purity was judged by SDS-PAGE.
Peptide purification and LC-MS
Following IMAC and C4 desalting, TglA was purified using an Agilent 1200 HPLC. HPLC buffers were 0.1% TFA in water (HPLC buffer A) and 0.1% TFA in acetonitrile (HPLC buffer B). The desalted material was lyophilized, resuspended in buffer A, and applied to a Phenomenenx Luna C18 column equilibrated with 95% HPLC buffer A and 5% HPLC buffer B. A linear gradient to 100% HPLC buffer B was run over 20 min at 0.1 mL min−1. TglA eluted near the end of the gradient. TglA-Cys behaved similarly and eluted slightly later. Full-length and AspN- or trypsin-digested peptides were analyzed by LC-MS/MS on a Waters Synapt Q-TOF equipped with a Waters UPLC and Phenomenex C18 Luna or C4 Jupiter columns. Buffers for LC-MS were 0.1% formic acid in water (LC-MS buffer A) and 0.1% formic acid in LC-MS grade acetonitrile (LC-MS buffer B).
In vitro assay of TglB
In a 0.5 mL centrifuge tube, reaction components were combined to the following optimized final concentrations: TglA (50 μM), TglB (5 μM), CysRS (10 μM), ATP (5 mM), L-cysteine (5 mM), TCEP (1 mM), tRNACys (10 μM), P. syringae CysRS (10 μM), and HEPES assay buffer (100 mM HEPES pH 7.5; 5 mM MgCl2, 100 mM NaCl). The assay mixture was incubated at 30 °C for 30 min, desalted and concentrated by ZipTip, eluted in 50% acetonitrile, 50% water, 0.1% TFA, and analyzed by MALDI-TOF MS. For 31P NMR analysis, reaction components were combined to final concentrations as follows: TglA (50 μM), TglB (5 μM), Cys-tRNACys (50 μM), ATP (0.5 mM), Tris assay buffer (50 mM Tris pH 7.5, 5 mM MgCl2, 100 mM NaCl). Cys-tRNACys was obtained using a similar procedure for preparation of Glu-tRNAGlu reported previously.6 For single turnover reactions, the reaction components were combined to final concentrations as follows: TglA (10 μM), TglB (50 μM), Cys-tRNACys (50 μM), ATP (10 μM), Tris assay buffer (50 mM Tris pH 7.5, 5 mM MgCl2, 100 mM NaCl). The reaction was incubated at 30 °C for 15 min, then Chelex resin was added to remove Mg2+. After using an Amicon Ultra 3K MWCO (3,000 Da cut-off) ultrafiltration device to remove proteins and tRNA, the sample was added into an NMR tube for analysis.
Fluorescence polarization (FP) binding assay
The interaction between fluorescently labeled peptides and TglB was quantified using an FP assay. Protein was serially diluted into binding buffer (50 mM HEPES, pH 7.5, 300 mM NaCl, 2.5% (v/v) glycerol, 0.5 mM TCEP) and mixed with 25 nM of the appropriate FITC labeled peptides. Binding assays were carried out in nonbinding-surface, 384-black-well polystyrene microplates (Corning) and measured using a Synergy H4 Hybrid plate reader (BioTek) with λex = 485 nm and λem = 538 nm and data were recorded with Gen5 software. Prior to measurement, the dilutions were equilibrated with shaking for 30 min at 25 °C. Dissociation constant (Kd) values were calculated from the 50% saturation point using a dose-response curve fit in Origin Pro 9.1 (OriginLab) with three independent titrations. Background fluorescence from the proteins alone was subtracted from the fluorescence polarization signal obtained with the fluorophore.
Competition FP binding assay
In general, 3 μM TglB was mixed with 25 nM of the appropriate FITC-labeled peptide in binding buffer. The peptides were serially diluted into binding buffer. Binding assays were carried out on the same instrument and analyzed with the same software as above. IC50 values were calculated from the 50% saturation point using a dose-response curve fit in Origin Pro 9.1 (OriginLab) with three independent titrations. Kd values for unlabeled ligands were calculated using the following equation:
Where:
Kd = Dissociation constant for the unlabeled peptide
[L] = Concentration of labeled peptide
Kd,labeled = Dissociation constant for the labeled peptide
LC-MS analysis for a potential anhydride intermediate
Assays were performed by reacting 25 μM TglB in the presence or absence of ATP (5 mM) and TglA (25 μM) for 5 min at 30 °C. Then assays were quenched with 20% ice-cold trichloroacetic acid and kept on ice for 20 min to precipitate the protein. The precipitate was separated by centrifugation (10,000 × g for 10 min) at 4 °C, washed with 3 × 400 μL of 10 mM ice-cold HCl, dried under vacuum, and dissolved in 20 μL of DMSO. To this solution, 15 μL of 0.1 M NaBH4 in DMSO was added and incubated at 30 °C for 10 min, followed by addition of 1 mL of ice-cold perchloric acid (0.44 M). The mixture was incubated on ice for another 30 min, followed by centrifugation at 10,000 × g for 20 min at 4 °C. The resulting precipitate was washed with 3 × 400 μL of 10 mM ice-cold HCl, and dried under vacuum. The dried protein was re-dissolved in 6 M guanidine-HCl and diluted with H2O to 1 M guanidine HCl. The protein was digested with 0.02 mg min−1 chymotrypsin at 37 °C overnight and analyzed by LC-MS.
NH2OH quenching assay
Assays were performed by reacting 50 μM TglB and 50 μM TglA in the presence or absence of ATP (5 mM) for 5 min at 30 °C. Then assays were quenched with NH2OH to a final concentration of 1 M and incubated at 30 °C for another 20 min. The assay was then desalted by ZipTip and analyzed by MALDI-TOF-MS.
Preparation of plasmid pTXB1-TglA-intein-chitin
The gene coding for TglA was amplified by PCR using appropriate primers (Table S2), and purified by gel extraction from a 1% (w/v) agarose gel using the QIAquick Gel Extraction Kit. The vector pTXB1 was digested using NdeI and SapI (NEB) restriction endonucleases and purified by gel extraction as described previously. The DNA fragment was inserted by Gibson one-step isothermal DNA assembly, and an aliquot of 20 μL of the Gibson assembly reaction was used to transform E. coli DH5α cells using the heat shock method. The cells were plated on LB plates supplemented with ampicillin (100 μg/mL) and the plates were incubated at 37 °C for 12–15 h. Single colonies were picked and grown in LB supplemented with ampicillin (100 μg/mL) at 37 °C for 12–15 h and the plasmids were isolated using a QIA prep Spin Miniprep Kit. Insert integrity was verified by sequencing the plasmids with the appropriate primers (Table S2).
Preparation of C-terminal 18O-labeled TglA
E. coli BL21 (DE3) cells (50 μL) were electroporated with pTXB1-TglA-intein-chitin (50 ng), and cells were plated on LB agar plates supplemented with 50 μg/mL kanamycin and grown at 37 °C for 12–15 h. A single colony was used to inoculate 20 mL of LB broth supplemented with 100 μg/mL ampicillin, grown for 12–15 h at 37 °C, and the culture was used to inoculate 2 L of terrific broth (TB) media, supplemented with ampicillin, to an OD600 of 0.025. Cultures were grown at 37 °C to a final OD600 of 1.0. Protein overexpression was induced by the addition of IPTG to a final concentration of 1 mM, and cultures were grown at 37 °C for 3 h. The cells were harvested by centrifugation, and then frozen in liquid nitrogen and stored at −80 °C.
For the purification of the TglA peptide, the cell paste was placed into a metal sonication cup using a minimal amount of resuspension buffer (20 mM Tris-HCl, 500 mM NaCl, 1 mM EDTA, pH 7.5) to transfer all of the cell paste. The cell paste was lysed by sonication (65% amplitude with 3 s pulses and 9 s delays) for 16 min. The sonicated cells were centrifuged for 30 min at 13,000 × g. The supernatant was poured into a column containing 20 mL of chitin beads pre-equilibrated with resuspension buffer. The supernatant and beads were gently shaken/rocked at 4 °C for 2 h to allow binding of the TglA-intein-chitin binding domain fusion protein to the resin. The column was drained and washed with 600 mL of resuspension buffer to remove all impurities. To the chitin beads was then added 40 mL of cleavage buffer (50 mM HEPES, 200 mM NaCl, 1 mM EDTA, 20 mM 2-mercaptoethanesulfonate, pH 7.7) and the column was capped and sealed with parafilm. The column was rocked at 4 °C for 6 h, and the solution was drained from the column directly into an Amicon Ultra 3K MWCO (3,000 Da cut-off) tube. The Amicon was then centrifuged until the protein solution had been concentrated to ~10 mL. The concentrated peptide solution was frozen in liquid nitrogen and lyophilized.
The lyophilized peptide was re-dissolved in basic H218O buffer (100 mM Na2CO3, pH 10.0) and incubated at 30 °C for 1 h, and was purified on a Shimadzu Prominence Preparative Liquid Chromatography system equipped with a Phenomenex Luna C18 column (250 × 10 mm, 10 μm particle size, 100 Å pore size). Acetonitrile and 10 mM aq. NH4HCO3 were used as the mobile phases, and a gradient of 2-80% aq. MeCN over 45 min at 5 mL min−1 was used for purification.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM R01 058822).
Footnotes
Notes The authors declare no competing financial interest
Supporting Information
Experimental details and supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Ting CP; Funk MA; Halaby SL; Zhang Z; Gonen T; van der Donk WA Use of a scaffold peptide in the biosynthesis of amino acid-derived natural products Science 2019, 365, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian KD; Fischbach MA; Garavelli JS; Göransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Müller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJ; Rebuffat S; Ross RP; Sahl HG; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Süssmuth RD; Tagg JR; Tang GL; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature Nat. Prod. Rep 2013, 30, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ortega MA; van der Donk WA New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products Cell Chem. Biol 2016, 23, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lubelski J; Rink R; Khusainov R; Moll GN; Kuipers OP Biosynthesis, immunity, regulation, mode of action and engineering of the model lantibiotic nisin Cell. Mol. Life Sci 2008, 65, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ortega MA; Hao Y; Zhang Q; Walker MC; van der Donk WA; Nair SK Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB Nature 2015, 517, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hudson GA; Zhang Z; Tietz JI; Mitchell DA; van der Donk WA In vitro biosynthesis of the core scaffold of the thiopeptide thiomuracin J. Am. Chem. Soc 2015, 137, 16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ozaki T; Kurokawa Y; Hayashi S; Oku N; Asamizu S; Igarashi Y; Onaka H Insights into the Biosynthesis of Dehydroalanines in Goadsporin ChemBioChem 2016, 17, 218. [DOI] [PubMed] [Google Scholar]
- (8).Ortega MA; Hao Y; Walker MC; Donadio S; Sosio M; Nair SK; van der Donk WA Structure and tRNA specificity of MibB, a lantibiotic dehydratase from Actinobacteria involved in NAI-107 biosynthesis Cell Chem. Biol 2016, 23, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Francklyn C; Schimmel P Aminoacylation of RNA minihelices with alanine Nature 1989, 337, 478. [DOI] [PubMed] [Google Scholar]
- (10).Francklyn C; Schimmel P Enzymatic aminoacylation of an eight-base-pair microhelix with histidine Proc. Natl. Acad. Sci. U. S. A 1990, 87, 8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gale AJ; Shi JP; Schimmel P Evidence that specificity of microhelix charging by a class I tRNA synthetase occurs in the transition state of catalysis Biochemistry 1996, 35, 608. [DOI] [PubMed] [Google Scholar]
- (12).Lovato MA; Chihade JW; Schimmel P Translocation within the acceptor helix of a major tRNA identity determinant Embo J. 2001, 20, 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hamann CS; Hou YM Enzymatic aminoacylation of tRNA acceptor stem helices with cysteine is dependent on a single nucleotide Biochemistry 1995, 34, 6527. [DOI] [PubMed] [Google Scholar]
- (14).Noike M; Matsui T; Ooya K; Sasaki I; Ohtaki S; Hamano Y; Maruyama C; Ishikawa J; Satoh Y; Ito H; Morita H; Dairi T A peptide ligase and the ribosome cooperate to synthesize the peptide pheganomycin Nat. Chem. Biol 2014, 11, 71. [DOI] [PubMed] [Google Scholar]
- (15).Fawaz MV; Topper ME; Firestine SM The ATP-grasp enzymes Bioorg. Chem 2011, 39, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yamaguchi H; Kato H; Hata Y; Nishioka T; Kimura A; Oda J. i.; Katsube Y Three-dimensional structure of the glutathione synthetase from Escherichia coli B at 2·0 Å resolution J. Mol. Biol 1993, 229, 1083. [DOI] [PubMed] [Google Scholar]
- (17).Fan C; Moews PC; Shi Y; Walsh CT; Knox JR A common fold for peptide synthetases cleaving ATP to ADP: glutathione synthetase and D-alanine:d-alanine ligase of Escherichia coli Proc. Natl. Acad. Sci. U. S. A 1995, 92, 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Collet JF; Stroobant V; Pirard M; Delpierre G; Van Schaftingen E A new class of phosphotransferases phosphorylated on an aspartate residue in an amino-terminal DXDX(T/V) motif J. Biol. Chem 1998, 273, 14107. [DOI] [PubMed] [Google Scholar]
- (19).Collet JF; Stroobant V; Van Schaftingen E Evidence for phosphotransferases phosphorylated on aspartate residue in N-terminal DXDX(T/V) motif Methods Enzymol. 2002, 354, 177. [DOI] [PubMed] [Google Scholar]
- (20).Post RL; Kume S Evidence for an aspartyl phosphate residue at the active site of sodium and potassium ion transport adenosine triphosphatase J. Biol. Chem 1973, 248, 6993. [PubMed] [Google Scholar]
- (21).Moutiez M; Schmitt E; Seguin J; Thai R; Favry E; Belin P; Mechulam Y; Gondry M Unravelling the mechanism of non-ribosomal peptide synthesis by cyclodipeptide synthases Nat. Comm 2014, 5, 5141. [DOI] [PubMed] [Google Scholar]
- (22).Collet J-F; Stroobant V; Van Schaftingen E Evidence for phosphotransferases phosphorylated on aspartate residue in N-terminal DXDX(T/V) motif Methods Enzymol. 2002, 354, 177. [DOI] [PubMed] [Google Scholar]
- (23).Kelley LA; Mezulis S; Yates CM; Wass MN; Sternberg MJE The Phyre2 web portal for protein modeling, prediction and analysis Nat. Protoc 2015, 10, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bothwell IR; Cogan DP; Kim T; Reinhardt CJ; van der Donk WA; Nair SK Characterization of glutamyl-tRNA-dependent dehydratases using nonreactive substrate mimics Proc. Natl. Acad. Sci. U. S. A 2019, 116, 17245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Burkhart BJ; Hudson GA; Dunbar KL; Mitchell DA A prevalent peptide-binding domain guides ribosomal natural product biosynthesis Nat. Chem. Biol 2015, 11, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang W; Ntai I; Kelleher NL; Walsh CT tRNA-dependent peptide bond formation by the transferase PacB in biosynthesis of the pacidamycin group of pentapeptidyl nucleoside antibiotics Proc. Natl. Acad. Sci. U. S. A 2011, 108, 12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Moutiez M; Belin P; Gondry M Aminoacyl-tRNA-utilizing enzymes in natural product biosynthesis Chem. Rev 2017, 117, 5578. [DOI] [PubMed] [Google Scholar]
- (28).Bougioukou DJ; Mukherjee S; van der Donk WA Revisiting the biosynthesis of dehydrophos reveals a tRNA-dependent pathway Proc. Natl. Acad. Sci. U. S. A 2013, 110, 10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ulrich EC; van der Donk WA Cameo appearances of aminoacyl-tRNA in natural product biosynthesis Curr. Opin. Chem. Biol 2016, 35, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chung CT; Niemela SL; Miller RH One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution Proc. Natl. Acad. Sci. U. S. A 1989, 86, 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sherlin LD; Bullock TL; Nissan TA; Perona JJ; Lariviere FJ; Uhlenbeck OC; Scaringe SA Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization RNA 2001, 7, 1671. [PMC free article] [PubMed] [Google Scholar]
- (32).Rio DC; Ares MJ; Hannon GJ; Nilsen TW RNA: A Laboratory Manual; Cold Spring Harbor Laboratory Press, 2011. [Google Scholar]
- (33).Walker SE; Fredrick K Preparation and evaluation of acylated tRNAs Methods 2008, 44, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.