

Abstract
Objectives:
The goal of this study was to investigate genes associated with essential hypertension from a system perspective, making use of bioinformatic tools to gain insights that are not evident when focusing at a detail-based resolution.
Methods:
Using various databases (pathways, Genome Wide Association Studies, knockouts etc.), we compiled a set of about 200 genes that play a major role in hypertension and identified the interactions between them. This enabled us to create a protein–protein interaction network graph, from which we identified key elements, based on graph centrality analysis. Enriched gene regulatory elements (transcription factors and microRNAs) were extracted by motif finding techniques and knowledge-based tools.
Results:
We found that the network is composed of modules associated with functions such as water retention, endothelial vasoconstriction, sympathetic activity and others. We identified the transcription factor SP1 and the two microRNAs miR27 (a and b) and miR548c-3p that seem to play a major role in regulating the network as they exert their control over several modules and are not restricted to specific functions. We also noticed that genes involved in metabolic diseases (e.g. insulin) are central to the network.
Conclusion:
We view the blood-pressure regulation mechanism as a system-of-systems, composed of several contributing subsystems and pathways rather than a single module. The system is regulated by distributed elements. Understanding this mode of action can lead to a more precise treatment and drug target discovery. Our analysis suggests that insulin plays a primary role in hypertension, highlighting the tight link between essential hypertension and diseases associated with the metabolic syndrome.
Keywords: bioinformatics, blood pressure, essential hypertension, hypertension, networks, systems biology
INTRODUCTION
progress toward defining the genetic basis of hypertension has been slow, mainly as arterial blood pressure (BP) is a complex, polygenic trait that is influenced by multiple variants, gene–gene interactions and the environment, all contributing to the likelihood of this condition [1,2].
Arterial pressure is determined by a range of physical factors such as blood volume, cardiac output and vascular resistance and compliance. These elements are regulated by a range of neural, endocrine and paracrine factors [2]. Thus, understanding the causes of hypertension requires consideration of the many systems contributing to BP homeostasis [3,4].
Systems biology deals with the organization and control of large and complex biological systems composed of a large number of genes, proteins and metabolites. It aims to explain biological processes in terms of the complex networks of interactions between these elements. The logic behind this approach is that such a broad analysis can find novel insights and address deeper questions that are not amendable to a reductionist approach of analyzing each component separately.
Current knowledge of the mechanisms of BP control and the pathogenesis of hypertension is incomplete. However, it seems that essential hypertension is a perturbation of the system and the effect of any single gene is likely to be small and inconsistent across different genetic and environmental backgrounds [1,5,6].
The goal of this study was to conduct an initial investigation of essential hypertension from a system perspective, making use of bioinformatic tools and to thereby provide a road map for additional studies.
METHODS
The analysis was conducted in four stages: first, identification of the genes and proteins that play a role in the system; second, identification of the interactions between these genes, thereby describing a network; third, investigation of the topological properties of the network to identify those genes that play major roles in the network; and fourth, characterization of the genes according to their associations with the modules (or subsystems) that comprise the BP regulation system and the search for regulators [either transcription factors or microRNAs (miRNAs)] that control a large number of BP genes (Fig. 1).
FIGURE 1.

Flowchart describing the methodology used in this study.
Selection of hypertension genes
The hypertension gene list (HTNGL) was compiled from multiple sources. The initial effort was focused on identifying the major players that participate in the four major pathways related to hypertension: renin–angiotensin–aldosterone system (RAAS), endothelin, ANP/cGMP and bradykinin. The list was expanded with additional genes related to the sympathetic nervous system and proteins that are hypertension drug targets. In addition, we used Genome Wide Association Study (GWAS) results and various publications dealing with essential hypertension research. Additional cross references for the candidate list were the set of hypertension-related genes extracted from human disease databases. The data were also corroborated with mouse phenotypes databases. A detailed list of sources, including links and references, can be found in Supplementary Digital Content and in Table S1, http://links.lww.com/HJH/A894.
Protein–protein interactions
We used the search tool for the retrieval of interacting genes/proteins (STRING) [7] (http://string-db.org/) database to search for interactions among the genes in the HTNGL. STRING is a meta-resource that aggregates most of the available information on protein–protein associations and includes both direct physical interactions and literature-based interactions. We also used PrePPI [8] (predicting protein–protein interactions), (https://bhapp.c2b2.columbia.edu/PrePPI/) – which focuses more on physical interactions, derived using a prediction algorithm which combines structural and nonstructural information. The interaction data were visualized with Cytoscape [9] (http://www.cytoscape.org/), an open-source bioinformatics software platform for visualizing molecular interaction networks (Supplemental Digital Content Fig S1, http://links.lww.com/HJH/A894). BioLayout [10] (http://www.biolayout.org/) was used within Cytoscape to calculate the Closeness and Betweenness centrality measures for each node. Betweenness centrality quantifies the number of times a node acts as a bridge along the shortest path between two other nodes, and Closeness centrality measures how close a node is to all other nodes in the graph. We also utilized Genomatix Pathay System, a literature-based Genomatix tool (https://www.genomatix.de/), to enable visualization of the complex graph of interactions as a tree-like graph, highlighting the most significant interactions (more details are provided in the Supplemental Digital Content, http://links.lww.com/HJH/A894).
Identification of hub genes
Hubs are defined by high ‘degree’ (the number of edges that the node touches) as well as high ‘centrality’ [centrality of a node http://en.wikipedia.org/wiki/Vertex_(graph_theory) measures its relative importance within a graph]. The more central a node is, the lower its total distance to all other nodes [11]. Hub genes are considered major players in a network [12,13].
Functional classification
The functional annotation tool in Database for Annotation Visualization and Integrated Discovery (DAVID) [14,15] (http://david.abcc.ncifcrf.gov/summary.jsp) was used to obtain information on biological processes and pathways as well as relevance to other diseases that are most significant (P<< 0.05) for the gene list. DAVID results were compared with two additional tools – the Genomatix gene ranker of the Genomatix Software Suite and the ingenuity pathway analysis (IPA) tool, a web-delivered application that enables biologists to discover, visualize and explore networks that may be therapeutically relevant to their experimental results, such as gene expression array data sets (http://www.ingenuity.com/products/ipa) [16]. For schematic representation of the phenotypic abnormality results, we used the WebGestalt tool [17] (http://bioinfo.vanderbilt.edu/webgestalt/).
Identification of enriched transcription factor binding sites for hypertension genes
For the identification of enriched transcription factors involved in the regulation of the HTNGL, we used two approaches. The first approach is prediction of transcription factor binding sites (TFBS) in regions adjacent to relevant genes, based on the DNA sequence motif preference of the transcription factors. Relevant transcription factors were identified by examining the TFBS of each HTNGL gene. Binding sites were searched in the region from 1000 base pairs upstream to 500-base pairs downstream of the transcriptional start site using the University of California Santa Cruz Genome Browser [18] Genome Browser version 19 (GRCh37/hg19) (https://genome.ucsc.edu/). Only transcription factors that appear in all three of the following resources were considered: cis-regulatory elements in mammalian genome (cREMaG) [19] (http://www.cremag.org/), transcription element listening system (TELiS) [20] (http://www.telis.ucla.edu/index.php?cmd=background) and transcription factor matrix explorer (TFM-Explorer) [21] (http://bioinfo.lifl.fr/TFM/TFME/). In the second approach, the sites were identified using databases containing the results of experimental high-throughput binding assays results. This analysis was performed based on literature and gene expression data using the IPA software [16].
Prediction of transcription factor regulation is not trivial as some transcription factors may control hundreds of genes, and many genes are controlled by several transcription factors. Also, much of the experimental data measure binding of a transcription factor to a particular segment of DNA, which may or may not be related to regulation. Thus, to obtain reliable candidates and reduce false positives, we cross-referenced several resources.
Identification of enriched microRNA’s in hypertension genes
Identification of miRNA’s that interact with genes in the HTNGL was carried out with the Partek Genomic Suite (http://www.partek.com/pgs), using TargetScan 6.0 as the miRNA database. In addition, we used the IPA software mentioned above. Only miRNA’s that appeared significant (P < 0.05) in both tools were considered.
Statistics
In this work, we have used a variety of bioinformatic tools. Each of these tools has its own procedure to evaluate the statistical significance of the results provided by the tool. Generally, Fisher’s exact test P values are calculated to show the overrepresentation of the genes of interest for each regulatory element or annotation term in the database, whereas multiple testing correction is performed using the Benjamini–Hochberg False Discovery Rate method. More detailed Information about these statistical procedures can be found via the links provided for each tool.
RESULTS
Hypertension gene list generation
Candidate genes related to essential hypertension were extracted from literature and databases based on text-mining tools and then corroborated against annotation repositories. Supplemental Digital Content Table S1, http://links.lww.com/HJH/A894 lists the publication sources from which the selected data set was generated. The final list contains 198 genes, referred to as the HTNGL (HyperTensioN Gene List). This work deals only with essential hypertension and not with secondary hypertension [22] such as hyperaldosteronism (Conn’s Syndrome) [23], renovascular hypertension [24], hypertension of pregnancy [25].
Protein–protein interactions
To identify proteins that interact with each other in the pathogenesis of hypertension, we used a bioinformatics approach based on literature mining of protein cooccurrence in scientific articles. This method used advanced natural language processing tools and statistical methods to extract real interactions. Here, we identified protein interactions within the HTNGL from the STRING and Genomatix databases (see METHODS section) and constructed an interaction network, visualizing the result in a Cytoscape graph comprising 195 nodes and 1491 edges (Supplemental Digital Content Fig. S1, http://links.lww.com/HJH/A894); in this depiction, genes are represented as nodes and interactions as edges. This graphical representation makes it possible to conduct a mathematical network analysis of attributes that can suggest the role of the genes within the network, using the centrality measures described in METHODS section. An analysis of the distribution of nodes and edges of the HTNGL network shows the network exhibits a scale-free behavior (Supplemental Digital Content Fig. S2, http://links.lww.com/HJH/A894), meaning that most nodes have few connections to other genes, whereas a small number of nodes have a large number of connections, a common property of many biological networks [26].
Figure 2 shows a tree-like representation of the protein–protein interaction map, with subsystems identified by gene-level functional annotation. As shown, the topologic organization of the tree is modular, separating some of the many functions of the BP regulation system. Genes annotated for a specific function appear in close proximity to each other within the tree, reinforcing the subsystem organization emerging from the network representation. It is interesting to note the involvement of genes related to the sympathetic system in several different regions of the map, suggesting that these genes may play a role in coordinating the different arms of the BP system [27].
FIGURE 2.

Hypertension gene list tree-like map indicating subsystem functional annotations. Shaded areas are regions with a high representation of various pathways and systems affecting blood-pressure regulation (the sympathetic activity components, marked by a yellow border, appear in five regions of the map, the other modules are unique). Annotation was performed manually based on gene ontology annotations and uniprot gene descriptions (see Functional Classification section and Table S2 in Supplemental Digital Content for functional assignments, http://links.lww.com/HJH/A894).
We then focused on the 10 genes with the highest degree (i.e. the number of immediate neighbors) in the graph representation of the HTNGL (Table 1). These genes are marked in the protein network visualization as largest red circles (Supplemental Digital Content Fig. S1, http://links.lww.com/HJH/A894) and have corresponding highest centrality measures (Closeness centrality >0.5, Table 1). Among these genes are INS, AGT, REN, EDN1, NOS3 and VEGFA, which are well known effectors of BP regulation. Out of the 10 genes, insulin (INS) stood out with 78 interactions, highlighting the importance of glucose metabolic regulation within the hypertension phenotype. This finding was somewhat unexpected, as angiotensinogen, renin and angiotensin-I-converting enzyme are usually considered the most prominent effectors of hypertension [28] but were ranked lower in centrality measures within our network.
TABLE 1.
Hub genes of the hypertension gene list
| Gene name |
Gene description |
Degree | Betweenness centralitya |
Closeness centralitya |
|---|---|---|---|---|
| INS | Insulin | 78 | 0.16 | 0.60 |
| REN | Renin | 70 | 0.14 | 0.57 |
| KNG1 | Kininogen 1 | 70 | 0.11 | 0.57 |
| ACE | Angiotensin–I– converting enzyme | 62 | 0.05 | 0.55 |
| GCG | Glucagon | 58 | 0.04 | 0.54 |
| AGT | Angiotensinogen | 52 | 0.05 | 0.53 |
| EDN1 | Endothelin 1 | 45 | 0.02 | 0.51 |
| NOS3 | Nitric oxide synthase 3 | 44 | 0.01 | 0.51 |
| VEGFA | Vascular endothelial growth factor A | 42 | 0.02 | 0.51 |
| NPY | Neuropeptide Y | 40 | 0.03 | 0.51 |
HTNGL, hypertension gene list.
Betweeness and Closeness centralities for the most central HTNGL genes (degree ≥40). Larger numbers indicate more hub-like properties.
In addition to the STRING database, which is mainly based on literature-mining of protein cooccurrence, we also used the PrePPI database which predicts protein–protein interactions using information on the structure of the proteins. This analysis resulted in a connected graph of 138 nodes and 481 edges. An additional 30 nodes and 11 edges were disconnected from the main graph (occurring as singletons, doubletons or a tripleton) and thus, were not included in the centrality calculations, and there were 30 missing nodes for which no information was available in the database. Although the main graph was substantially smaller in size relative to the network created based on STRING data (195 nodes and 1461 edges), INS still plays a major role with the INS receptor as the most central node and the INS gene itself as the fifth most central.
Functional clustering and classification
Once a list of genes was established, the next task was to identify which biological processes, pathways and conditions are associated with members of the list. To ensure the validity of our results, we used several tools and searched for consensus [29,30].
Table 2 lists the gene ontology terms for biological processes that show significant enrichment scores for BP-related processes using the DAVID functional annotation tool. Supplemental Digital Content Table S3, http://links.lww.com/HJH/A894 shows enrichment scores for diseases and disorders, physiological system functions and canonical pathways obtained with the IPA tool. Equivalent Genomatix results are given in the Supplemental Digital Content Table S2, http://links.lww.com/HJH/A894. The four principal BP-related processes (circulatory system, blood circulation, regulation of BP and vascular process in the circulatory system) appear in all gene ontology terms and Genomatix analyses, whereas IPA suggests cardiovascular and hematological system functions as the most closely related processes.
TABLE 2.
Enriched biological process gene ontology terms
| Biological process GO terma | Genes | P value | P value corrected for multiple testing |
|---|---|---|---|
| Regulation of blood pressure | 47 | 1.20E – 59 | 2.70E – 56 |
| Blood circulation | 55 | 3.50E – 57 | 3.90E – 54 |
| Circulatory system process | 55 | 3.50E – 57 | 3.90E – 54 |
| Regulation of systemic arterial blood pressure | 27 | 9.90E – 42 | 7.30E – 39 |
| Regulation of system process | 52 | 2.10E – 40 | 1.20E – 37 |
| Chemical homeostasis | 61 | 6.20E – 39 | 2.70E – 36 |
| Regulation of systemic arterial blood pressure mediated by a chemical signal | 23 | 4.90E – 37 | 1.80E – 34 |
| Homeostatic process | 66 | 3.00E – 34 | 9.40E – 32 |
| Vascular process in the circulatory system | 25 | 8.50E – 30 | 2.10E – 27 |
| Regulation of systemic arterial blood pressure by hormones | 18 | 2.10E – 29 | 4.60E – 27 |
| Regulation of vasoconstriction | 21 | 4.70E – 29 | 8.60E – 27 |
DAVID, Database for Annotation Visualization and Integrated Discovery; GO, gene ontology; HTNGL, hypertension gene list.
The most enriched GO biological processes in the HTNGL gene list as derived using DAVID.
Based on these results, essential hypertension seems to be most associated with cardiovascular disease, endocrine system disorders, and metabolic, renal and urological diseases. The most prominent functional category of the HTNGL is signaling and signal transduction (Supplemental Digital Content Table S3, http://links.lww.com/HJH/A894). Specific signaling annotations include G-Protein Coupled Receptor signaling, cAMP-mediated signaling, Gas/Gai signaling and AMPK signaling and components of the sympathetic nervous system involved in essential hypertension. The angiotensin, nitric oxide, cGMP, endothelin, adrenergic, insulin and protein kinase A pathways were found to be enriched, suggesting a connection to background disease manifested by the metabolic syndrome. A detailed list of annotations and genes can be found in Supplemental Digital Content Tables S2 and S4, http://links.lww.com/HJH/A894.
Figure 3 shows human phenotypic abnormalities that are enriched for HTNGL as generated using Webgestalt (see details in Supplemental Digital Content, http://links.lww.com/HJH/A894). The leftmost branch relates to the urinary system and associated abnormalities of renal excretion. The expected annotations regarding the renin–angiotensin system and the cardiovascular systems are directly related to essential hypertension. The rightmost branch represents the homeostasis of metabolic processes, specifically abnormalities associated with potassium and sodium ion transport. These systems play a crucial role in regulation of arterial pressure, as well as in other diseases related to the metabolic syndrome – for example obesity and sdiabetes [31].
FIGURE 3.

Phenotypic abnormalities associated with hypertension gene list. Chart of phenotypic abnormalities generated by Webgestalt. Branches represent relevant major human systems and diseases; enriched annotations are marked in red. Phenotypes are taken from the Human Phenotype Ontology Website (http://human-phenotype-ontology.github.io/).
Regulation of hypertension gene expression
The regulatory elements we considered were transcription factors for transcription control and miRNAs for post-transcription mRNA modulation:
Transcription factor regulation
For TFBS identification, based on sequence motifs (see METHODS section), we used three web-based tools: cREMaG, TELiS and TFM-Explorer (the first two with two different transcription factor databases), allowing us to perform a meta-analysis (Supplemental Digital Content Table S5, http://links.lww.com/HJH/A894). When we searched for transcription factors identified using more than one tool with P < 0.05, SP1 transcription factor was the only one found in four out of the five searches.
For literature and gene expression data analysis, we queried the ingenuity tool (IPA), which annotates transcription factors and their binding sites. This tool also found SP1 as the most significant transcription factor related to the HTNGL (P = 1.23E –12). Taken together, these two approaches indicate that SP1 is a significant transcription factor regulator for the HTNGL (the 19 genes regulated by SP1 are listed in Supplemental Digital Content Table S6, http://links.lww.com/HJH/A894).
Posttranscriptional regulation
Utilization of both the Partek and IPA tools (see METHODS section) in the identification of enriched miRNAs for the HTNGL produced similar significant results. Both tools identified hsa-mir27a-3p and its close homolog hsa-mir27b-3p with P values of 6.69E – 04 and 6.7E – 04 (for Partek and IPA analyses, respectively) and hsa-mir548c-3p with P values of 4.07E – 03 and 4.4E – 0.3 (for Partek and IPA analyses, respectively) associated with targets for 22 and 27 HTNGL genes, respectively.
Figure 4 shows the genes in the HTNGL network predicted to interact with these miRNAs, and Supplementary Table S6, http://links.lww.com/HJH/A894 lists all targets. Nine HTNGL genes (CACNA1A, ENPEP, GFPT1, GOSR2, IGF1, INSR, NLN, PTGER3 and VEGFC) are targets of both miRNAs. The miRNA predicted interactions span many branches of the hypertension gene graph, indicating a system-wide regulation pattern rather than a subsystem-specific (branch-specific) regulation mechanism.
FIGURE 4.
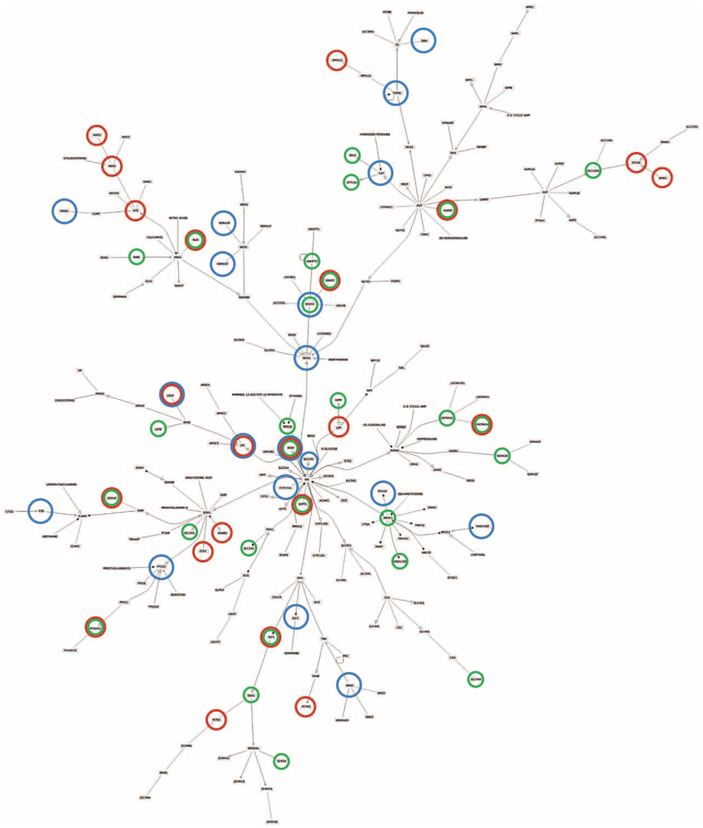
Hypertension network regulation. Tree-like representation of the hypertension protein interaction map, showing genes controlled by specific regulators. Circled genes are regulated by enriched elements: Blue – SP1 transcription factor, Green – microRNA548c, Red – microRNA27 (detailed target list in Supplemental Digital Content Table S6, http://links.lww.com/HJH/A894).
DISCUSSION
In this study, we set out to construct an initial molecular map of genes involved in the BP system. The work identified the major genes involved in BP regulation, mapped the potential interactions of the corresponding proteins and identified a transcription factor and two miRNAs that appear to regulate many elements of the system. This map should be useful for follow-up studies that further explore the molecular basis of BP regulation.
The choice of genes for the analysis plays an important role in this study. Hence, our reliance on a wide array of sources that contributes to the overall confidence of the selection. Our approach led to a large set of genes (≈200) that are sufficient to reveal the properties of the network. We note that we used a zero/one approach in construction of the protein interactions map, that is if the calculated score of an interaction was above a threshold, it was included in the network. In future analysis, the network can be refined to include the strength of each interaction within the network.
In analyzing our results, we first discuss the observation that stood out most strongly – the centrality of INS in the BP system.
Although the relevance of INS resistance as a precursor to diabetes is widely appreciated, the pathogenesis of hypertension in INS-resistant patients is less obvious. The association between essential hypertension, INS resistance and hyperinsulinemia is well known [32–34]. Hyperinsulinemia stimulates the sympathetic nervous system and the RAAS [35], both of which result in sodium retention and volume expansion. In addition, abnormalities in INS signaling may lead to elevated BP through their effect on endothelial cells and on calcium influx into vascular smooth muscle cells [36–39].
In an INS-resistant milieu, the angiotensin II type 1 receptor is upregulated [40], which enhances the physiologic actions of angiotensin II. Angiotensin II, in addition to being a potent peripheral vasoconstrictor, counteracts many of the effects of INS and IGF1 [37,41]. Angiotensin II also inhibits INS-induced glutamine-4 uptake into skeletal muscle cells, thus decreasing cellular glucose uptake which attenuates INS-stimulated vasodilatation [42]. Finally, angiotensin II has mitogenic effects on vascular smooth muscle cells that result in chronic vascular hypertrophy and a resultant rise in peripheral vascular resistance [43]. These pathogenic mechanisms, which are reflected in our map, may explain the link between primary hypertension and INS resistance.
We view the BP regulation mechanism as a system-of-systems, composed of several contributing subsystems and pathways, rather than a single module. Our objective was to describe the molecular components that are the main players of the BP regulation system. This was accomplished by compiling a comprehensive list of the involved genes from various databases (Supplemental Digital Content Table S1, http://links.lww.com/HJH/A894) and utilizing a variety of bioinformatic tools to create gene network maps and identify regulatory elements. It is clear that these data are not complete, and additional components should be added to the system as more computational and experimental data are created and analyzed.
In recent years, several system-wide studies have been performed, both computationally and experimentally, to analyze the BP system: Huan et al. [44] integrated transcriptional profiling, GWAS and network modeling to search for key driver genes that play a role in BP regulation. Significantly, the list of key genes that was found in this study is very different from our list. This could be explained by the fact that the list in Huan et al. is mainly derived from a GWAS study, whereas our study incorporates a variety of complementary sources. Moreover, as these authors mentioned, their work is based on coexpression network analysis which creates a challenge in finding the ‘key’ regulatory genes and dissecting gene–gene interactions. To address this limitation, they employed graphical molecular networks (Bayesian and PPI) which are partial to the STRING/PrePPI/Genomatix databases that we utilized for network analysis.
Several studies have been conducted to explore hypertension using various experimental approaches;
Gajjala et al. [45] performed plasma analysis that identified a series of molecules that could be linked to hypertension. Although a list of such proteins has been detected, the authors mention that it is difficult to associate them to the pathogenesis of hypertension and to decide whether these proteins are related to the cause or the consequences of the disease.
Araki et al. [46] studied the plasma peptidome from hypertensive pregnant women serum. In this study, three out of the seven peptides were identified as fragments derived from kininogen-1, a finding that supports the centrality of this gene as reported by our analysis.
Myers et al. [47] similarly performed mass spectrometric studies to identify proteins as markers of preeclampsia. They identified that biomarker combinations, centered on INS-like growth factor acid labile subunit, have a potential to predict this condition in healthy women. This finding comes in alignment with our findings of the role of INS as well as INS-like growth factor genes (IGF1, IGF2) in hypertension. Our work complements these studies by providing a map that is based on a wide perspective that takes into consideration a multitude of sources and databases.
We further identified transcription factors and noncoding RNA molecules that may act as master regulators of hypertension: SP1 is a transcription factor that regulates the expression of a large number of genes involved in a variety of processes, specifically the expression of the vasodilator genes NRP1 [48] and NPR2 [49] and a number of genes expressed in the heart and vessel wall [50,51]. SP1 has been known to increase in response to promitogenic stimuli in vascular smooth muscle cells and may play a role in controlling the growth response that is activated by these stimuli [52]. A recent study has shown that SP1 is also tied to SCNN1A transcription, thus contributing to maximal aldosterone transactivation of SCNN1A in kidney collecting duct cells [53].
A recent review on the role of noncoding RNA in essential hypertension and BP regulation [54] emphasized the emerging key role of miRNAs in these conditions. Such findings emphasize the importance of our search for regulatory elements. Our system-wide approach enabled us to identify two miRNA molecules, miRNA27a/b and miRNA548C (miRNA27a was previously identified in Marques et al. [54]), that may play a more global role by affecting several components of the system.
miRNA27a and miRNA27b are members of the miR23~27~24 cluster, with miR27a and miR27b being highly expressed in endothelial cells. It has been shown recently that both miR27 members are positive regulators of angiogenesis [55]. These miRNAs are also involved in the regulation of repulsion of endothelial cells [56]. Both attraction and repulsion are essential elements in the establishment of a functional vascular network [55,56]. It has been shown that miR27 is highly expressed in the heart after myocardial infraction and influences capillary formation and fibrosis [57]. Other studies identified miR27 as a central regulatory hub in lipid metabolism [58] in addition to playing a role in pathological development of obesity [59–62] and the metabolic syndrome [54,63].
miRNA548 is a recently discovered human miRNA gene family [64]. It’s targets have shown the enrichment of pathways involving cardiac pathophysiology [65]; however, there is no prior clinical evidence linking this miRNA to hypertension. Thus, this finding should encourage more research into the possible role of miRNA548 in BP regulation.
From the analysis here, we can suggest that miRNA27 and miRNA548C are linked to arterial pressure via physiological mechanisms related to angiogenesis and cardiac functions.
Two observations can be made from the mapping of all three regulators (SP1, miRNA27 and miRNA548) to the protein interaction network in Fig. 4: first, some genes are regulated by two of the regulatory elements, and one gene (INS receptor) is controlled by all three regulators; second, the targets are spread over the entire system, rather than each regulator controlling a specific arm of the system. This observation strengthens the notion that BP is controlled by a multitude of interacting subsystems.
In summary, our investigation utilizing a systems biology approach has shown that BP regulation is a distributed system, which is affected by various control elements. We identified three regulatory elements (SP1, miRNA27a/b, miRNA548C) that may have a global effect on the network. The work also suggests that INS plays a primary role in hypertension, highlighting the tight link between essential hypertension and the metabolic syndrome diseases.
Future studies should attempt to understand in a more mechanistic way how these molecular components exert their influence on BP regulation in the normotensive and hypertensive populations. Furthermore, such a map has the potential to lead to a more precise treatment and to identify new drug targets as well as suggest attractive combinations of drugs that will have a wide effect and control BP more effectively than current therapies.
Supplementary Material
Reviewers’ Summary Evaluation.
Reviewer 1
This novel study uses a system biology approach to gain insight into the genesis and progression of essential hypertension that otherwise would be lost using a reductionist approach. Strengths include the use of the latest bioinformatics tools and the discovery of three regulatory elements globally affecting the network using an unbiased method.
ACKNOWLEDGEMENTS
We would like to thank Dr Ariel Azia and Dr Tirza Doniger for valuable assistance throughout this work.
J.M.’s contribution was partly supported by NIH R01 GM104436.
Previous presentations: 16th Israel Bioinformatics Symposium, Ramat Gan, Israel – Poster competition; The First International Multidisciplinary Symposium on Artificial Intelligence in Medicine, Dana Point, CA, USA – Abstract competition.
Support for authors: none.
Funding received: none.
Abbreviations:
- BP
blood pressure
- cREMaG
cis-regulatory elements in mammalian genome
- DAVID
database for Annotation Visualization and Integrated Discovery
- HTNGL
hypertension gene list
- IPA
ingenuity pathway analysis
- PrePPI
predicting protein–protein interactions
- RAAS
renin–angiotensin–aldosterone system
- STRING
search tool for the retrieval of interacting genes/proteins
- TELiS
transcription element listening system
- TFBS
transcription factor binding site
- TFM-Explorer
transcription factor matrix explorer
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Garcia EA, Newhouse S, Caulfield MJ, Munroe PB. Genes and hypertension. Curr Pharm Des 2003; 9:1679–1689. [DOI] [PubMed] [Google Scholar]
- 2.Cowley AW Jr. The genetic dissection of essential hypertension. Nat Rev Genet 2006; 7:829–840. [DOI] [PubMed] [Google Scholar]
- 3.Padmanabhan S, Caulfield M, Dominiczak AF. Genetic and molecular aspects of hypertension. Circ Res 2015; 116:937–959. [DOI] [PubMed] [Google Scholar]
- 4.Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med 2011; 17:1402–1409. [DOI] [PubMed] [Google Scholar]
- 5.Zheng J Rao DC, Shi G. An update on genome-wide association studies of hypertension. Appl Inform 2015; 2:10. [Google Scholar]
- 6.Saavedra JM The challenge of genetic studies in hypertension. Circ Res 2007; 100:1389–1393. [DOI] [PubMed] [Google Scholar]
- 7.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011; 39:D561–D568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang QC, Petrey D, Garzón JI, Deng L, Honig B. PrePPI: a structure-informed database of protein-protein interactions. Nucleic Acids Res 2013; 41:D828–D833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theocharidis A, van Dongen S, Enright AJ, Freeman TC. Network visualization and analysis of gene expression data using BioLayout Express3D. Nat Protoc 2009; 4:1535–1550. [DOI] [PubMed] [Google Scholar]
- 11.Valente TW, Coronges K, Lakon C, Costenbader E. How correlated are network centrality measures? Connect 2008; 28:16–26. [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, Gerstein M, Snyder M. Getting connected: analysis and principles of biological networks. Genes Dev 2007; 21:1010–1024. [DOI] [PubMed] [Google Scholar]
- 13.Seo CH, Kim JR, Kim MS, Cho KH Hub genes with positive feedbacks function as master switches in developmental gene regulatory networks. Bioinformatics 2009; 25:1898–1904. [DOI] [PubMed] [Google Scholar]
- 14.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 15.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol 2007; 8:R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krämer A, Green J, Pollard J Jr, Tugendreich S Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014; 30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 2013; 41:W77–W83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.James Kent W, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piechota M, Korostynski M, Przewlocki R. Identification of cis-regulatory elements in the mammalian genome: the cREMaG database. PLoS One 2010; 5:e12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole SW, Yan W, Galic Z, Arevalo J, Zack JA. Expression-based monitoring of transcription factor activity: the TELiS database. Bioinformatics 2005; 21:803–810. [DOI] [PubMed] [Google Scholar]
- 21.Tonon L, Touzet H, Varré JS. TFM-Explorer: mining cis-regulatory regions in genomes. Nucleic Acids Res 2010; 38:W286–W292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rimoldi SF, Scherrer U, Messerli FH. Secondary arterial hypertension: when, who, and how to screen? Eur Heart J 2014; 35:1245–1254. [DOI] [PubMed] [Google Scholar]
- 23.Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 2006; 48:2293–2300. [DOI] [PubMed] [Google Scholar]
- 24.Textor SC. Secondary hypertension: renovascular hypertension. J Am Soc Hypertens 2014; 8:943–945. [DOI] [PubMed] [Google Scholar]
- 25.Shopen N, Schiff E, Koren-Morag N, Grossman E. Factors that predict the development of hypertension in women with pregnancy-induced hypertension. Am J Hypertens 2016; 29:141–146. [DOI] [PubMed] [Google Scholar]
- 26.Barabasi AL, Albert R. Emergence of scaling in random networks. Science 1999; 286:509–512. [DOI] [PubMed] [Google Scholar]
- 27.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci 2006; 7:335–346. [DOI] [PubMed] [Google Scholar]
- 28.Lima SG, Hatagima A, Silva NL. Renin-angiotensin system: is it possible to identify hypertension susceptibility genes? Arq Bras Cardiol 2007; 89:427–433. [DOI] [PubMed] [Google Scholar]
- 29.Jesmin J, Rashid MS, Jamil H, Hontecillas R, Bassaganya-Riera J. Gene regulatory network reveals oxidative stress as the underlying molecular mechanism of type 2 diabetes and hypertension. BMC Med Genomics 2010; 3:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satoh J Molecular network analysis of human microRNA targetome: from cancers to Alzheimer’s disease. BioData Min 2012; 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abate N, Chandalia M, Cabo-Chan AV Jr, Moe OW, Sakhaee K. The metabolic syndrome and uric acid nephrolithiasis: novel features of renal manifestation of insulin resistance. Kidney Int 2004; 65:386–392. [DOI] [PubMed] [Google Scholar]
- 32.Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 2001; 37:1053–1059. [DOI] [PubMed] [Google Scholar]
- 33.Saad MF, Rewers M, Selby J, Howard G, Jinagouda S, Fahmi S, et al. Insulin resistance and hypertension: the Insulin Resistance Atherosclerosis study. Hypertension 2004; 43:1324–1331. [DOI] [PubMed] [Google Scholar]
- 34.Grunfeld B, Balzareti M, Romo M, Gimenez M, Gutman R. Hyperinsulinemia in normotensive offspring of hypertensive parents. Hypertension 1994; 23 (1 Suppl):I12–I15. [DOI] [PubMed] [Google Scholar]
- 35.Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities – the role of insulin resistance and the sympathoadrenal system. N Engl J Med 1996; 334:374–381. [DOI] [PubMed] [Google Scholar]
- 36.Walsh MF, Barazi M, Pete G, Muniyappa R, Dunbar JC, Sowers JR. Insulin-like growth factor I diminishes in vivo and in vitro vascular contractility: role of vascular nitric oxide. Endocrinology 1996; 137:1798–1803. [DOI] [PubMed] [Google Scholar]
- 37.Sowers JR. Insulin and insulin-like growth factor in normal and pathological cardiovascular physiology. Hypertension 1997; 29:691–699. [DOI] [PubMed] [Google Scholar]
- 38.Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J 1998; 333 (Pt 3):471–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walsh MF, Ali SS, Sowers JR. Vascular insulin/insulin-like growth factor-1 resistance in female obese Zucker rats. Metabolism 2001; 50:607–612. [DOI] [PubMed] [Google Scholar]
- 40.Nickenig G, Röling J, Strehlow K, Schnabel P, Böhm M Insulin induces upregulation of vascular AT1 receptor gene expression by posttranscriptional mechanisms. Circulation 1998; 98:2453–2460. [DOI] [PubMed] [Google Scholar]
- 41.Isenovic ER, Divald A, Milivojevic N, Grgurevic T, Fisher SE, Sowers JR Interactive effects of insulin-like growth factor-1 and beta-estradiol on endothelial nitric oxide synthase activity in rat aortic endothelial cells. Metabolism 2003; 52:482–487. [DOI] [PubMed] [Google Scholar]
- 42.Baron AD. Vascular reactivity. Am J Cardiol 1999; 84:J25–J27. [DOI] [PubMed] [Google Scholar]
- 43.Lever AF. Angiotensin II, angiotensin-converting enzyme inhibitors, and blood vessel structure. Am J Med 1992; 92:S35–S38. [DOI] [PubMed] [Google Scholar]
- 44.Huan T, Meng Q, Saleh MA, Norlander AE, Joehanes R, Zhu J, et al. Integrative network analysis reveals molecular mechanisms of blood pressure regulation. Mol Syst Biol 2015; 11:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gajjala PR, Jankowski V, Heinze G, Bilo G, Zanchetti A, Noels H, et al. Proteomic-biostatistic integrated approach for finding the underlying molecular determinants of hypertension in human plasma. Hypertension 2017; 70:412–419. [DOI] [PubMed] [Google Scholar]
- 46.Araki Y, Nonaka D, Tajima A, Maruyama M, Nitto T, Ishikawa H, et al. Quantitative peptidomic analysis by a newly developed one-step direct transfer technology without depletion of major blood proteins: Its potential utility for monitoring of pathophysiological status in pregnancy-induced hypertension. Proteomics 2011; 11:2727–2737. [DOI] [PubMed] [Google Scholar]
- 47.Myers JE, Tuytten R, Thomas G, Laroy W, Kas K, Vanpoucke G, et al. Integrated proteomics pipeline yields novel biomarkers for predicting preeclampsia. Hypertension 2013; 61:1281–1288. [DOI] [PubMed] [Google Scholar]
- 48.Rossignol M, Pouysségur J, Klagsbrun M. Characterization of the neuropilin-1 promoter; gene expression is mediated by the transcription factor Sp1. J Cell Biochem 2003; 88:744–757. [DOI] [PubMed] [Google Scholar]
- 49.Thompson IR, Chand AN, King PJ, Ansorge O, Karavitaki N, Jones CA, et al. Expression of guanylyl cyclase-B (GC-B/NPR2) receptors in normal human fetal pituitaries and human pituitary adenomas implicates a role for C-type natriuretic peptide. Endocr Relat Cancer 2012; 19:497–508. [DOI] [PubMed] [Google Scholar]
- 50.Flesch M On the trail of cardiac specific transcription factors. Cardiovasc Res 2001; 50:3–6. [DOI] [PubMed] [Google Scholar]
- 51.Xu Q, Ji YS, Schmedtje JF Jr. Sp1 increases expression of cyclooxygenase-2 in hypoxic vascular endothelium. Implications for the mechanisms of aortic aneurysm and heart failure. J Biol Chem 2000; 275:24583–24589. [DOI] [PubMed] [Google Scholar]
- 52.Rahmutula D, Cui J, Chen S, Gardner DG. Transcriptional regulation of type B human natriuretic peptide receptor gene promoter: dependence on Sp1. Hypertension 2004; 44:283–288. [DOI] [PubMed] [Google Scholar]
- 53.Yu Z, Kong Q, Kone BC. Sp1 trans-activates and is required for maximal aldosterone induction of the αENaC gene in collecting duct cells. Am J Physiol Renal Physiol 2013; 305:F653–F662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marques FZ, Booth SA, Charchar FJ. The emerging role of noncoding RNA in essential hypertension and blood pressure regulation. J Hum Hypertens 2015; 29:459–467. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Q, Gallagher R, Ufret-Vincenty R, Li X, Olson EN, Wang S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23~27~24 clusters. Proc Natl Acad Sci USA 2011; 108:8287–8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Urbich C, Kaluza D, Frömel T, Knau A, Bennewitz K, Boon RA, et al. MicroRNA-27a/b controls endothelial cell repulsion and angiogenesis by targeting semaphorin 6A. Blood 2012; 119:1607–1616. [DOI] [PubMed] [Google Scholar]
- 57.Dai Y, Khaidakov M, Wang X, Ding Z, Su W, Price E, et al. MicroRNAs involved in the regulation of postischemic cardiac fibrosis. Hypertension 2013; 61:751–756. [DOI] [PubMed] [Google Scholar]
- 58.Vickers KC, Shoucri BM, Levin MG, Wu H, Pearson DS, Osei-Hwedieh D, et al. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology 2013; 57:533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karbiener M, Fischer C, Nowitsch S, Opriessnig P, Papak C, Ailhaud G, et al. microRNA miR-27b impairs human adipocyte differentiation and targets PPARgamma. Biochem Biophys Res Commun 2009; 390:247–251. [DOI] [PubMed] [Google Scholar]
- 60.Zhu Y, Zhang X, Ding X, Wang H, Chen X, Zhao H, et al. miR-27 inhibits adipocyte differentiation via suppressing CREB expression. Acta Biochim Biophys Sin (Shanghai) 2014; 46:590–596. [DOI] [PubMed] [Google Scholar]
- 61.Kong X, Yu J, Bi J, Qi H, Di W, Wu L, et al. Glucocorticoids transcriptionally regulate miR-27b expression promoting body fat accumulation via suppressing the browning of white adipose tissue. Diabetes 2015; 64:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z. A role of miR-27 in the regulation of adipogenesis. FEBS J 2009; 276:2348–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karolina DS, Tavintharan S, Armugam A, Sepramaniam S, Pek SL, Wong MT, et al. Circulating miRNA profiles in patients with metabolic syndrome. J Clin Endocrinol Metab 2012; 97:E2271–E2276. [DOI] [PubMed] [Google Scholar]
- 64.Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2007; 2:e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta MK, Halley C, Duan ZH, Lappe J, Viterna J, Jana S, et al. miRNA-548c: a specific signature in circulating PBMCs from dilated cardiomyopathy patients. J Mol Cell Cardiol 2013; 62:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.