

Abstract
Cissus quadrangularis (Vitaceae) is a perennial climber endemic to Africa and is characterized by succulent angular stems. The plant grows in arid and semi-arid regions of Africa especially in the African savanna. The stem of C. quadrangularis has a wide range of applications in both human and animal medicine, but there is limited cytogenetic information available for this species. In this study, the chromosome number, genome size, and genome composition for C. quadrangularis were determined. Flow cytometry results indicated that the genome size of C. quadrangularis is approximately 2C = 1.410 pg. Fluorescence microscopy combined with DAPI stain showed the chromosome numbers to be 2n = 48. It is likely that C. quadrangularis has a tetraploid genome after considering the basic chromosome numbers in Cissus genus (n = 10, 11, or 12). A combination of low-throughput genome sequencing and bioinformatics analysis allowed identification and quantification of repetitive elements that make up about 52% of the C. quadrangularis genome, which was dominated by LTR-retrotransposons. Two LTR superfamilies were identified as Copia and Gypsy, with 24% and 15% of the annotated clusters, respectively. The comparison of repeat elements for C. quadrangularis, Vitis vinifera, and four other selected members in the Cissus genus revealed a high diversity in the repetitive element components, which could suggest recent amplification events in the Cissus genus. Our data provides a platform for further studies on the phylogeny and karyotype evolution in this genus and in the family Vitaceae.
Keywords: Cissus quadrangularis, Genome size, Copia, Repeat elements, Chromosome counts, Gypsy, Flow cytometry, C value
Introduction
Cissus is the largest genus in the grape family Vitaceae with about 300 species (Wen, 2007). The species in this genus show pan-tropical intercontinental disjunction, occurring in Asia, the Americas, Australia, and Africa (Jackes, 1988; Lombardi, 2015; Wen, 2007; Rodrigues, Lombardi & Lovato, 2014; Latiff, 2001). The greatest concentration of Cissus species is found in Africa with approximately 135 species; this is considered the ancestral area for this genus (Liu et al., 2013; Adams et al., 2016). The majority of research focuses on phylogenetic relationships within the genus and the grape family (Liu et al., 2013; Rodrigues, Lombardi & Lovato, 2014; Wen et al., 2018). The tribe Cisseae contains only Cissus, which is based on the new phylogenetic tribal classification of Vitaceae (Wen et al., 2018).
Cissus quadrangularis is one of the perennial succulent plants within Cissus that is widely distributed in Africa, the Arabian Peninsula, northern India, and Southeast Asia (GBIF Backbone Taxonomy). Ting, Sternberg & Deniro (1983) identified a facultative crassulacean acid metabolism (CAM) pathway in C. quadrangularis that could contribute to its excellent tolerance to drought conditions. Scientific interest in this species has increased recently due to its value in veterinary and human medicine (Latiff, 2001; Mishra, Srivastava & Nagori, 2010; Stohs & Ray, 2012; Ganguly, Ganguly & Banerjee, 2018; Vedha, Bn & Devi, 2013; Indran & Raj, 2015).
Previously reported chromosome numbers for Cissus quadrangularis have been inconsistent. Raghavan (1957) examined the chromosome number for Indian medicinal plants and determined the chromosome numbers for C. quadrangularis to be 2n = 24. He suggested that earlier reports of a diploid with 45 chromosomes could have been incorrect or that the specimen was possibly a tetraploid. Further, Robert et al. (2001) examined C. quadrangularis from Kenya and its two variants (A & B) and determined their chromosome numbers to be 24 and 28, respectively. Variant A was identified with smooth stem angles and was proposed to be the type variety for C. quadrangularis while variant B had rough stem angles and was considered to be a new variety of C. quadrangularis. Karkamkar, Patil & Misra (2010) reported the chromosome numbers of seven Cissus species (2n = 24, 48). In addition, Chu et al. (2018) reported the chromosome numbers for seven other Cissus species (Table 1, 2n = 24, 40, 48, or 66) and suggested a linear relationship between the chromosome numbers and genome size (1C = 0.37–1.03 pg). These results implicate polyploidization and repetitive element modifications in the expanded genome size in this genus. However, considering the large number of species in the Cissus genus, much is unknown about the genome size, chromosome numbers, and genome characteristics of the members comprising this genus.
Table 1. Chromosome numbers, genome size and ploidy types in Cissus.
Cissus quadrangularis data was obtained in the present study. Data for other members have been documented by Chu et al. (2018), Robert et al. (2001) & Raghavan (1957).
| Cissus species | Chromosome numbers (2n) | Ploidy type | Genome size (2C-Value) |
|---|---|---|---|
| C. rotundifolia Vahl | 24 | 2 × | 0.76 pg. |
| C. discolor Blume | 24 | 2 × | 0.86 pg. |
| C. tuberosa Moc. & Sesse ex DC. | 24 | 2 × | 0.9 pg. |
| C. javana DC. | – | – | 0.74 pg. |
| C. antarctica Vent | 40 | 4 × | 1.34 pg. |
| C. trifoliata (L.) L | 48 | 4 × | 1.58 pg. |
| C. microcarpa Vahl | 66 | 6 × | 2.06 pg. |
| C. quadrangularis-Raghavan (1957) | 24 | 2 × | – |
| C. quadrangularis-Robert et al. (2001) | 24, 28 | – | – |
| C, quadrangularis- This study | 48 | – | 1.410 pg |
Repetitive sequences form up to 90% of the plant genome and are dominated by long terminal repeats (LTR) in plants (Du et al., 2010). The disparity in the plant genome size is attributed to polyploidisation events and the variation in the amount of repetitive DNA, which is characterized by transposable elements and tandem repeats (Kidwell, 2002; Sessegolo, Burlet & Haudry, 2016). Mis-annotation of LTR has led to the characterization of some of LTR as genes, especially the low copy number fragments (Bennetzen et al., 2004). This makes the complete and accurate annotation of transposable elements in whole genome sequencing projects of plants necessary. Macas, Neumann & Navrátilová (2007) utilized short read sequencing technology to identify repeat elements without using a reference genome based on the similarity of their reads. Sequencing reads can be classified into clusters based on their similarities representing the repetitive elements.
The advent of robust sequencing technologies that can generate huge sequence data at a reduced cost, coupled with advanced assembling methods, has improved genome sequencing for both model and non-model plants (Michael & VanBuren, 2015). Whole genome sequencing projects are challenging due to a limited amount of data. Additionally, proportions of repetitive DNA components in the genome can impede sequencing. In maize, repetitive elements form about 80% (SanMiguel et al., 1996) of the genome with a complex organization that created difficulty when sequencing its whole genome (Chandler & Brendel, 2002). Challenges in incorporating repetitive DNA sequence data have been one of the limiting factors in the available draft genomes (Feuillet et al., 2011).
Kidwell (2002) reported that there is a closer relationship between repetitive DNA sequences and genome size. Li et al. (2017) confirmed a positive relationship between genome size and repetitive sequences. Their analysis revealed a stronger positive correlation between retrotransposons and genome size than with transposons. Among the retrotransposons, LTR-retrotransposons were shown to have the highest positive correlation. However, the contributions of LTR lineages (Ty1-Copia and Ty3-Gypsy) to the genome size were similar. Understanding the components of the repetitive elements will therefore undoubtedly provide clues about the factors that may have influenced the expanded genome in C. quadrangularis and may include multiplication of its repetitive elements.
In the present study, the chromosome numbers and genome size of C. quadrangularis were evaluated. Short-read sequencing data were generated from the genomic DNA of C. quadrangularis to characterize its major genomic components and the fractions of repetitive elements. Comparisons were made between the repetitive element components for C. quadrangularis, Vitis vinifera, and four other Cissus species (Table 1). The findings reported here increase our understanding of the genome variations in the Cissus genus and provide basic genomic and cytogenetic information for C. quadrangularis that forms a foundation for whole genome sequence studies.
Materials and Methods
Plant materials
Stem cuttings of C. quadrangularis were collected from the roadside in Namango, Kenya, (S02°32′, E36°49′). Duplicate voucher specimens (SAJIT 002306) were deposited in the Wuhan Botanical Garden herbarium (HIB) and the Herbarium of Jomo Kenyatta University of Agriculture and Technology (JKUAT). Young leaves and petioles were collected for genome size evaluation while root tips were collected for chromosome number determination. All materials for the genome sequencing and karyotyping of C.quadrangularis were obtained from a single individual.
Genome size estimation
Flow cytometry was used to determine nuclear DNA content with minor modifications using hand chopped material as originally described by Galbraith et al. (1983). In order to isolate the nuclei, a woody plant buffer (WPB) was used, which contained 0.2 M Tris-HCl, 4 mM MgCl2⋅6H2O, 2 mM EDTA Na2⋅2H2O, 86 mM NaCl, 10 mM sodium metabisulfite, 1% PVP-10, 1% (v/v) Triton X-100, with a pH 7.5 (Loureiro et al., 2007a; Loureiro et al., 2007b). Raphanus sativus cv. Saxa (radish) seeds provided by the Institute of Experimental Botany, Czech Republic, were germinated and the plantlets were used as reference standards. The plantlets of C. quadrangularis were pre-treated for 4 days in the dark. Petioles from the young leaves of C. quadrangularis and young leaves from radishes were collected (approximately 50 mg for each sample) and hand chopped using a sharp razor on ice in 1.5 mL WPB as described by Pfosser et al. (1995). The suspension was filtered through a 40-µm-nylon mesh (Cat. 352340, Falcon, USA) to eliminate the excess debris. RNase A was added to 100 ng/mL in the nuclei homogenate solution. Propidium Iodide (50 mg/mL) was used to stain the nuclei for at least 2 min on ice before the samples were run in the flow cytometer (BD Accuri C6). Three independent samples were run on the cytometer and the genome size was calculated using the following formula: sample 2C DNA content = [(sample G1 peak mean)/(internal standard peak mean)] × Internal standard DNA content.
Chromosome count
Root tips were obtained from C. quadrangularis plantlets propagated from the same individual and collected from the field. Samples were treated with a saturated solution of 1-Bromonaphthalene for 3 h at room temperature to halt cell division (Mirzaghaderi, 2010). Microscopic slides were then prepared from the treated root tips using the protocol as developed by Kirov et al. (2014) with minor modifications to obtain the chromosomes at metaphase stage. To digest the cell wall, the root tips were incubated for 60 min at 37 °C in a 1% enzyme mix (1% pectinase and 1% cellulase in freshly prepared 0.1 M citrate buffer). Relative humidity was maintained between 40 and 60% during the dropping step. The prepared microscopic slides were stained with 4′, 6-diamidino-2-phenylindole (DAPI) and chromosome fluorescent images were captured using a fluorescence microscope (Leica DMi8) fitted with a camera (Leica DFC 550).
DNA isolation, sequencing, and data analysis
Genomic DNA was extracted from the leaves of C. quadrangularis using the plant genomic DNA kit (Tiangen, China) following the manufacturer’s protocol. DNA libraries were constructed the using NEBNext® Ultra™ DNA Library Prep Kit for Illumina. Paired-end sequences (2 × 150 bp and 400–450 bp insert size) were generated on the Illumina HiSeq X Ten platform produced by Novogene (Tianjing, China). The sequencing data were deposited in the SRA database under accession number SRR8573652.
To analyze the repetitive components of this species, graph-based clustering was performed using the RepeatExplorer platform (Novák, Neumann & Macas, 2010). Short-read sequencing data was subjected to the Galaxy-based RepeatExplorer platform as described by Novak et al. (2013). The quality of the reads was determined by the FastQC tool and poor-quality reads were discarded. Clean reads were converted into the FASTA format Using the FASTQ to FASTA converter. It was possible to infer the repeat composition for a species with typically 0.1–0.5× genome coverage using the RepeatExplorer (Macas et al., 2015). A total of 700,000 paired-end reads (150 bp) were randomly selected, which were equal to approximately 6.56% of the predicted genome for clustering. An all-to-all comparison was carried out, grouping similar sequences into respective clusters in RepeatExplorer. The genome proportions for each cluster were calculated based on the read percentages.
Repeat clusters contributing no less than 0.01% of the genome proportions were considered for further annotation while those with smaller contributions were ignored. Repeat clusters with known protein domains were annotated directly on the RepeatExplorer platform, while similarity searches against GenBank databases (nt and nr) using BLASTn and BLASTx (Altschul et al., 1990) were carried out manually with the E-value at 1e−5 to classify other repeat clusters.
Comparison of repeat content in C. quadrangularis, grape, and 4 other Cissus species
To investigate variations in repeat elements among the species and their roles during the evolution of the Cissus genus, we collected short-read sequencing data for other Cissus species, which is publicly available at NCBI. Four Cissus species whose genome size and sequencing data are available (C. tuberosa, C. trifoliata, C. discolor, and C. microcarpa), were selected for co-clustering (last accessed May 20, 2019). Sequencing and genome size data are essential in determining the proper number of reads representing similar genome proportions. Co-clustering analysis was performed using RepeatExplorer (Novak et al., 2013). Reads from C. quadrangularis, four other Cissus species and grape were simultaneously clustered. The species were randomly grouped and considered to have equal probabilities for common repeats and frequencies among the 6 species. Co-clustering allowed the grouping of similar reads from individual species suggesting similar ancestral origin. In order to avoid a sensitivity bias, the number of reads that were analyzed for all species are proportional to the genome sizes of the corresponding species. Randomly selected paired-end reads from C. quadrangularis, grape, Cissus microcarpa, Cissus trifoliata, Cissus discolor, and Cissus tuberosa, (ca. 0.06× coverage for each genome) were combined for RepeatExplorer co-clustering (Novak et al., 2013). The sources for clustering data are provided in Table 2. The genome size data used for annotation of repeats for Cissus species has been reported by Chu et al. (2018). Plastid repeats are phylogenetically uninformative due to their abundance, which is linked to the photosynthetic dynamics in plant tissues (Dodsworth et al., 2016) and were manually excluded in further analysis.
Table 2. Genomic and co-clustering data for Cissus quadrangularis, Grape (Pinot noir) (Vitis vinifera) and four other Cissus species.
| Species | Data source | Genome size (1C) | Length of reads | 6% in reads number | Reads used |
|---|---|---|---|---|---|
| Cissus quadrangularis | This study | 689 | 130 | 318000 | 318000 |
| Vitis vinifera | SRR5627797 | 475 | 130 | 219230.7692 | 219000 |
| Cissus discolor | SRX1323033 | 420.54 | 130 | 194096.3846 | 194000 |
| Cissus microcarpa | SRX1322892 | 1007.34 | 130 | 464926.1538 | 465000 |
| Cissus trifoliata | SRX1322890 | 772.62 | 130 | 356593.8462 | 357000 |
| Cissus tuberosa | SRX1322889 | 440.1 | 130 | 203123.0769 | 203000 |
Results and Discussion
C-value determination in Cissus quadrangularis
The nuclear DNA content of C. quadrangularis was evaluated by flow cytometry using radish (2C = 1.11 pg) as the reference standard. The genome size for the C. quadrangularis individual considered in this study was estimated as 2C = 1.410 pg. (Fig. 1) representing 689 Mbp/1C (1 pg. = 978 Mbps) (Dolezel et al., 2003). According to the classification by Soltis et al. (2003) the C. quadrangularis genome falls within the group of plants with very small genome. The size of the Cissus quadrangularis genome is approximately in the same range as Cissus antarctica and Cissus trifoliata which are tetraploid species (Chu et al., 2018). In addition, its genome is roughly double that of Cissus javana and Cissus rotundifolia which have diploid genomes (Chu et al., 2018).
Figure 1. Fluorescence histograms for genome size assessments in Cissus quadrangularis by flow cytometry.

(A) Radish (2C = 1.1 pg.) with peaks at about 585000 and 1146000, (B) Cissus quadrangularis with a peak at about 719000, (C) Cissus quadrangularis combined with Radish; G1—radish 2C peak, G2—Cissus quadrangularis 2C peak, G3—radish 4C peak.
The mucilage of the chopped plant tissues had a viscous texture and is composed of complex polysaccharides of various concentrations, which include galacturonic acids, rhamnose, galactose and others (Ovodov, 1998). Their sticky nature causes the aggregation of the nuclei making it difficult to isolate them for cytometric analysis and in majority of species, leaves are commonly used for flow cytometry analysis. However, in our experiment, young leaves yielded unsatisfying results, which may have been caused by high amounts of polysaccharides. Following the suggestion by Suda (2004), petioles were used yielding acceptable peaks. Nuclei isolation buffers, including the Tris⋅MgCl2 buffer and Galbraiths’ buffer (Dolezel, Greilhuber & Suda, 2007), yielded unsatisfactory results (data not shown). Loureiro et al. (2006) modified the constituents of the Tris⋅MgCl2 buffer to develop WPB, which counters the negative effects of tannic acid. The inclusion of sodium metabisulfite and PVP-10 in the buffer enhanced its efficacy by reducing the impact of phenol and other secondary metabolites. Dolezel, Greilhuber & Suda (2007), Loureiro et al. (2007a) and Loureiro et al. (2007b) used higher concentrations of a detergent (Triton-X), which had the effect of reducing the mucilage viscosity levels and minimizing their negative impacts. WPB has been employed in genome size determination for other members in genus the Cissus genus (Chu et al., 2018) and other stubborn woody plants (Loureiro et al., 2007a; Loureiro et al., 2007b). Dark-treatment of the samples improved our results and is a method that can be applied in genome size determination for species with higher levels of polysaccharides and other secondary metabolites.
Chromosome counts
The slides prepared with root tips were stained with a fluorescent dye, DAPI, to visualize the chromosomes. C. quadrangularis chromosomes are tiny and their numbers were quantified as 2n = 48 (Fig. 2). Additional chromosomal images have been supplied in Data S1. Observations revealed small chromosomes, making it difficult to identify the centromeres except for two pairs. To confirm the chromosome numbers, fluorescent in situ hybridization (FISH) using telomere labeled DNA probes was conducted. We used nick translation method to label DNA telomere probes (Ma et al., 2010). The sensitivity of FISH is to detect 3.5 kb target sequence on chromosome (Ma et al., 2010). Additionally, we used barley as the control for this experiment. We could not obtain clear telomere signals on all chromosome ends simultaneously in Cissus quadrangularis, while the telomere signals were clear in all barley chromosomes (data not shown). During FISH procedure, the DNA on the chromosomes could be lost/degraded if the fixation step is not optimized (Schwarzacher & Heslop-Harrison, 2000). In addition, high efficiencies FISH was achieved from mitotic metaphase chromosomes prepared from floral tissues of Arabidopsis thaliana, while almost no signals were detected on the chromosomes of root meristematic tissues with the same clones as probes. This is possibly due to the differences in chromatin structure between root meristematic tissues and floral tissues (Murata & Motoyoshi, 1995). In future, optimization of FISH protocol and use of different tissues to prepare mitotic metaphase chromosomes is therefore essential.
Figure 2. Cissus quadrangularis chromosome image.

Mitotic metaphase chromosome complements from Cissus quandrangularis. The image has been resized for easy counting. Bar = 3 um.
The basic chromosome number for members in the Cissus genus can be inferred to be 10, 11, or 12 (Chu et al., 2018). Cissus quadrangularis chromosomes have previously been reported (2n = 24, 28) for diploids and 2n = 45 for a specimen from India which was thought to be a tetraploid (Raghavan, 1957). Variations in chromosome numbers observed in individuals from different localities could be due to epigenetic factors such as DNA methylation and histone modification, which are thought to contribute to the silencing of transposable elements triggering chromosomal evolution in different biogeographical regions (Li et al., 2017). The limited sampling size prohibits a conclusive determination of the chromosome numbers for C. quadrangularis. In future studies, the sample size could be expanded to include more samples from different biogeographic locations.
Based on the deduced chromosome duplication model in the Cissus genus, previous reports for C. quadrangularis and other members in the genus (Karkamkar, Patil & Misra, 2010; http://www.tropicos.org/Project/IPCN; Chu et al., 2018), C. quadrangularis individual considered in our study, with 2n = 48 can be said to be a tetraploid (Table 1). However, more information, which may include molecular data, will be required to confirm this assumption. This would include carrying out both meiotic and mitotic studies to test the possibility of having accessory chromosomes, the existence of aneusomatic division, or the presence of accessory chromosomes in roots (Gibbs & Semir, 1988). Obtaining such information will improve our understanding of chromosomal evolution in the Cissus genus. This will further assist in the interpretation of phylogenetic and biogeographic models such as Cladistic, Migrationist and Panbiogeography models (Wilson, 1991), rather than just relying on potentially out-of-date information. Differences in diploid somatic chromosomes may be indicative of intraspecific polyploidization (Husband, Baldwin & Suda, 2013). Additionally, the occurrence of species complexes with variable karyotypes have been reported in other species and it has been suggested that they are indicative of a series of dysploidy in diploid species followed by hybridization and polyploidization (Choi et al., 2008; Haga & Noda, 1976). Such species complexes could suggest the presence of cryptic lineages in this species and therefore more tests should be carried out to include morphological traits examinations and molecular phylogenetics.
The genome composition of C. quadrangularis
The complexity and tediousness of traditional molecular techniques have made it difficult to analyze genome components, especially in non-model organisms. In this study, short-read sequencing in combination with bioinformatics tools were used to analyze the C. quadrangularis genome. Genome analysis is efficient and economical using these techniques, despite the plant not being well-studied.
50,767 clusters were obtained from the RepeatExplorer results indicating that about 52% of the C. quadrangularis genome is composed of repetitive elements (Fig. 3). The output of clustering analysis forms a foundation for comprehensive study in the future to determine the repeat family structures and their variations. The raw RepeatExplorer output data has been provided in Data S2. The repeat composition was within the range of the estimates for plants with small genome sizes, which range between 25.04–66.42% in the Oryza genus (Zuccolo et al., 2007), 41.4% in grapes (Jaillon et al., 2007) and 61% in sorghum (Paterson et al., 2009).
Figure 3. Repeat composition of clusters generated in RepeatExplorer (similarity-based partitioning) of 700000 reads (7.61% of genome size).

X-axis: cumulative proportion of clusters of the genome. Y-axis: numbers of reads.
The top 324 clusters constituting no less than 0.01% of the genome proportions were considered for further annotation. The relatively higher proportions of the single or low-copy repeat families were not considered. However, the variation of these elements may impact the phenotypic characteristics of plants (Barghini et al., 2015). To analyze the variations of the single copy retroelements, a reference genome in combination with genotype re-sequencing is needed, as in many model species. The singletons in this study represent the low-copy fraction of the genome that could not be assembled into clusters using RepeatExplorer (Fig. 3).
Characterization of LTR-retrotransposons of Cissus quadrangularis
Repeat cluster annotation and characterization were performed by a sequence similarities search against the NCBI database (Altschul et al., 1990) and on the RepeatExplorer platform for clusters with known protein domains. LTR-retrotransposons formed the major part of the identified repeats with two subfamilies: Gypsy and Copia. The Copia family comprised 24% of the LTR components while Gypsy made up of 15% of the LTR components (Fig. 4A). According to Wicker et al. (2007), the two main superfamilies, Gypsy and Copia, can further be classified into 6 and 7 lineages, respectively. The majority of the Copia lineages are of Angela type (Fig. 4B) (28% of the Copia elements) while the majority of the Gypsy lineages are of the chromovirus type (Fig. 4C) (29% of the Gypsy elements). A higher percentage of the analyzed clusters had unidentified components that represented clusters whose components could not be annotated (Fig. 4A). This is attributed to absence of protein domains in clusters and/or the limited repeat sequences of closely related species in public databases. Considerably higher numbers of unidentified clusters have been observed in the sunflower and Eragrostis tef (Gebre et al., 2016; Mascagni et al., 2015), while in sea grass (Posidonia oceanica) (Barghini et al., 2015), the proportions of unidentified clusters were considerably lower. Small clusters that were not analyzed (<0.01% genome proportion) also represented a significant proportion of the genome that was not well-characterized. Other components that were identified included LINEs, DNA transposons, organellar DNA, satellite repeats, and rDNA among others (Fig. 4A).
Figure 4. (A) The repeat class distribution of the 342 top clusters with no less than 0.01% of genome proportion from Illumina assemblies using RepeatExplorer, (B) & (C) lineage distributions of the main super-families, Copia and Gypsy, respectively.
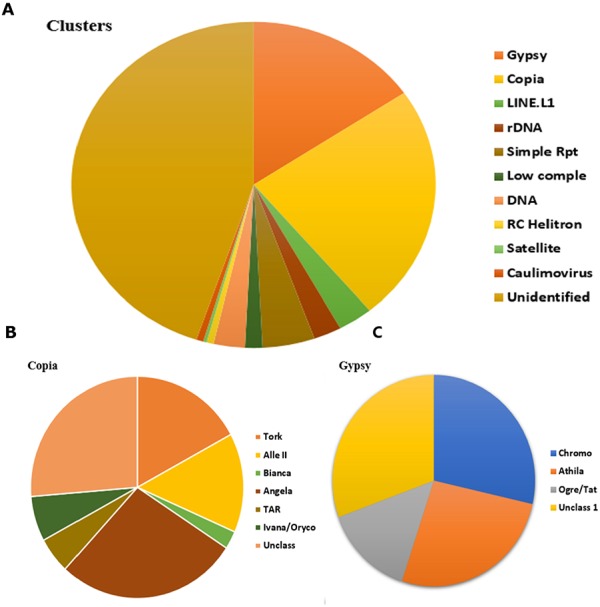
The values are percentages for each of the lineage composition.
The possibility of chloroplast DNA insertions into the nuclear genome has been infrequently reported (Kejnovsky et al., 2006). In maize, the presence of chloroplast DNA in the nuclear genome has been reported (Roark et al., 2010), which may explain the presence of some levels of organellar DNA in our sample, while the majority could have been as a result of contamination during DNA extraction. The presence of rDNA can be due to biases generated during sequencing (Macas et al., 2011). Low efficiency during sequencing has been identified in GC-enriched segments when using Illumina technology (Nakamura et al., 2011; Aird et al., 2011) and leads to vulnerability of GC-rich templates to biases during Illumina sequencing. It is likely that the quality of the sequencing sample caused the bias in the representation of rDNA.
In this study, only 6 of the Copia subfamilies were found, which may be due to the limited consideration of clusters whose genome compositions were no less than 0.01% each were considered and therefore a large number of elements with lower representations were not annotated. In addition, part of the plant genome is not yet annotated into the main subfamilies as inferred from the analysis by RepeatExplorer. Therefore, more extensive sequencing techniques and coverage are required to decisively annotate the whole genome. We carried out repeat composition comparison analysis for C. quadrangularis and other well-studied species. From our comparison, a linear relationship between the genome size and LTR contents was observed but with exceptions (Table 3). For example, potato (Solanum tuberosum) has a genome size smaller than C. quadrangularis but a higher LTR composition. Our observations are in agreement with Wang et al. (2014) and Li et al. (2017) who identified a positive correlation between genome size and quantity of repetitive sequences in plants.
Table 3. Comparison of Cissus quadrangularis repeat composition with other studied plant species.
| Species | Genome size | LTR (%) | References |
|---|---|---|---|
| Norway spruce | 20 Gbps | 60 | Schnabel et al. (2009) |
| Hordeum vulgare L | 4,289 Mbp | 75 | Nystedt et al. (2013) |
| Zea may | 2,300 Mbp | 70.10 | Mayer et al. (2012) |
| Solanum lycopersicum | 900 Mbp | 61.8 | Sato et al. (2012) |
| Solanum tuberosum | 844 Mbp | 29 | Xu et al. (2011) |
| Cissus quadrangularis | 689 Mbp | 40 | This study |
| Actinidia chinensis | 616.1 Mbp | 13.36 | Huang et al. (2013) |
| Vitis vinifera | 478 Mbp | 6.30 | Jaillon et al. (2007) |
Variation in genome sizes for angiosperms have been attributed to differences in transposable element content, especially the LTR (Feng et al., 2017). This has been demonstrated in Spirodela polyrhiza (158 Mbp) with chromosome numbers 2n = 40 and Lemma minor which has the same chromosome number but a 481 Mbp genome size (Feng et al., 2017). Genomic repeats serve as vital indices in phylogenetic studies (Dodsworth et al., 2015). Therefore, changes in the LTR composition and its impact on genome evolution could be applied in evolutionary studies for C. quadrangularis and its relatives as new technologies are developed.
Comparing the repeat contents for Cissus quadrangularis, grape, and 4 other Cissus species
The alignment of homologous DNA sequences has been the basis for molecular systematics. The differences in the sequence alignment patterns are used for the construction of phylogenetic trees. Insufficiency in divergences for homologous protein domains for repetitive elements between taxa makes repetitive elements unsuitable for phylogenetic analysis. However, variations in the number of repeat types and specific retrotransposons can be used quantitatively for phylogeny reconstruction (Dodsworth et al., 2015). Distantly related species exhibit divergences in the structure of their repetitive elements, whereas there is a level of uniformity noted in closely related species (Dodsworth et al., 2015).
Based on our comparison, 45S rDNA was the major element present in all 6 species (Fig. 5A) which agreed with the high homology of 45S rDNA in angiosperm (Roa & Guerra, 2012). The application of repeat elements for phylogenetic inferences is defined up to the genus level due to a limited number of common repeats beyond genus level (Dodsworth et al., 2016). Therefore, phylogenetic inferences were restrained using repeats from the Cissus genus and grape. Other major elements represented in all of the six species studies include 5S rDNA, Ty_copia, Ty3_gypsy, and some elements whose protein domains could not be identified in the annotated clusters and which are identified as unknown in Figs. 5A & 5B. It was further noted that the most abundant repeats were shared by the 5 Cissus species (Fig. 5B). These elements include Ty3_gypsy, Ty1_copia, 45S rDNA, hAt:hAt, pararetrovirus:PARA and elements whose protein domains could not be identified (Fig. 5B). The noticeable conservation of the Copia elements may explain their homology for the species considered. Gypsy elements were shared among some species but the degree was lower compared to Copia elements. This indicates the less conservative nature of these elements as observed by Barghini et al. (2015). Other elements such as satellites and LINE: LINE were shared by some Cissus species, which may indicate that they are newly formed. These elements display a lower uniformity in the considered species and reflect variations in proliferation rates among different and related species (Hawkins et al., 2006).
Figure 5. Comparison of transposable elements (TEs) for different Cissus species and grape.

The numbers in X-axis represent the number of species combinations randomly selected. Different colors in Y-axis display types of TEs while the heights of the bars represent the number of reads. (A) The number of reads with high homology between Cissus species and grape. For example, the column ‘5’ represent the reads of different TEs in five Cissus species and grape, and the column ‘0’ represent the reads of different TEs unique to grape. (B) The number of reads with high homology across Cissus species without grape. For example, the column ‘5’ represent the reads of different TEs in five Cissus species (all Cissus), and column ‘1’ represent the reads of different TEs in one Cissus.
Comparative analysis conducted on Musaceae family indicated quantitative differences in the repeat elements and the classified elements at different taxonomic levels (Novak et al., 2014). In our comparison, as a result of limited homology for majority of the repeat elements in Cissus, it was not possible to infer phylogenetic relationships. Dodsworth et al. (2015) noted a similar problem in constructing bifurcating phylogenetic trees while evaluating relationships in legume tribe Fabeae with homoploid and polyploid hybridization. In addition, comparative analysis of repetitive elements involving several species have unveiled variation in the sequences for probed repeat families and their abundances (Kelly et al., 2015; Macas et al., 2015). Novak et al. (2014) deduced that these differences may be due to the incomplete assembly and the unclear constituents that are encountered in the assembly of the genome. Considering the huge number of species in the Cissus genus, our sample size for co-clustering could contribute to the limited phylogenetic information in this genus. However, the information from this study is a foundation for future work in this genus.
Conclusion
In this study, the genomic characteristics have been identified in the widely used species of the Cissus genus, C. quadrangularis. The high proportions of repetitive elements observed in C. quadrangularis could suggests that the expanded genome arose through the amplification of the repetitive elements coupled with polyploidization. Transposable element activities such as silencing and proliferation may have facilitated the observed karyotypic variations in this genus.
The probability of C. quadrangularis possessing a tetraploid genome has been implied based on the genome size expansion and the increase in chromosome numbers. However, more studies should be carried out to confirm the ploidy type. The information obtained in this study forms a foundation upon which additional phylogenetic and evolutionary studies can be carried out.
Supplemental Information
Additional Mitotic metaphase chromosomes compliments from Cissus quandrangularis
The data obtained from the pairwise comparison of short-read sequences are used to construct graphs with vertices representing the sequence reads and overlapping reads connected with edges. The edge weight is used to express the similarity scores for reads. Where the sequence genome coverage is low (<0.5 ×), presence of single copy sequences is low and therefore low overlapping resulting in detached nodes with no connections to other parts of the graph. Presence of repetitive sequences is represented by mutually connected nodes as a result of pooling of overlapping reads. Examination of graph topology facilitates separation and identification of clusters of reads frequently connected representing individual repetitive element families.
Acknowledgments
We would like express our gratitude to Prof. Jaroslav Dolezel of the Institute of Experimental Botany. The Centre of Plant Structural and Functional Genomics Czech Republic provided the standard reference material seeds for cytometry. We appreciate the help from Bin Liu (Institute of Botany, Chinese Academy of Sciences) in counting the chromosomes.
Funding Statement
This work was supported by the Sino-Africa Joint Research Center and the Chinese Academy of Sciences (SAJC201613). Computational resources were provided by the ELIXIR-CZ project (LM2015047), part of the international ELIXIR infrastructure. There was no additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
Lu Ma is employed by Shenzhen Tobeacon Technology Co. Ltd.
Author Contributions
Duncan Kiragu Gichuki and Lu Ma conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Zhenfei Zhu performed the experiments, analyzed the data, prepared figures and/or tables, authored or reviewed drafts of the paper, approved the final draft.
Chang Du performed the experiments, analyzed the data, prepared figures and/or tables, approved the final draft.
Qingyun Li analyzed the data, prepared figures and/or tables, approved the final draft.
Guangwan Hu, Zhixiang Zhong, Honglin Li, Qingfeng Wang and Haiping Xin conceived and designed the experiments, contributed reagents/materials/analysis tools, authored or reviewed drafts of the paper, approved the final draft.
DNA Deposition
The following information was supplied regarding the deposition of DNA sequences:
Raw Illumina reads of Cissus quandrangularis are available at Figshare: Gichuki, Duncan Kiragu; Ma, Lu; Zhu, Zhenfei; Du, Chang; Hu, Guangwan; Zhong, Zhixiang; et al. (2019): Characters of repetitive elements in Cissus quadrangularis R1. figshare. Dataset. 10.6084/m9.figshare.7712219.v1.
Ma, Lu; Gichuki, Duncan Kiragu; Zhu, Zhenfei; Du, Chang; Hu, Guangwan; Zhong, Zhixiang; et al. (2019): Characters of repetitive elements in Cissus quadrangularis R2. figshare. Dataset. 10.6084/m9.figshare.7712222.v1.
Gichuki, Duncan Kiragu; Ma, Lu; Zhu, Zhenfei; Du, Chang; Hu, Guangwan; Zhong, Zhixiang; et al. (2019): RepeatExplorer output of Cissus quandrangularis. figshare. Dataset. 10.6084/m9.figshare.8115695.v1.
Grape reads are available at SRA: SRR5627797.
Cissus discolor reads are available at SRA: SRX1323033.
Cissus microcarpa reads are available at SRA: SRX1322892.
Cissus trifoliata reads are available at SRA: SRX1322890.
Cissus tuberosa reads are available at SRA: SRX1322889.
Data Availability
The following information was supplied regarding data availability:
Cissus quadrangularis raw sequencing data are available at SRA: SRR8573652.
References
- Adams et al. (2016).Adams NE, Collinson M, Smith S, Bamford M, Forest F, Malakasi P, Marone F, Sykes D. X-rays and virtual taphonomy resolve the first Cissus (Vitaceae) macrofossils from Africa as early-diverging members of the genus. American Journal of Botany. 2016;103(9):1657–1677. doi: 10.3732/ajb.1600177. [DOI] [PubMed] [Google Scholar]
- Aird et al. (2011).Aird D, Ross MG, Chen W-S, Danielsson M, Fennell T, Russ C, Jaffe DB, Nusbaum C, Gnirke A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biology. 2011;12(2):R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul et al. (1990).Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Barghini et al. (2015).Barghini E, Mascagni F, Natali L, Giordani T, Cavallini A. Analysis of the repetitive component and retrotransposon population in the genome of a marine angiosperm, Posidonia oceanica (L.) Delile. Marine Genomics. 2015;24(3):397–404. doi: 10.1016/j.margen.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Bennetzen et al. (2004).Bennetzen JL, Coleman C, Liu R, Ma J, Wusirika R. Consistent over-estimation of gene number in complex plant genomes. Current Opinion in Plant Biology. 2004;7(6):732–736. doi: 10.1016/j.pbi.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Chandler & Brendel (2002).Chandler VL, Brendel V. The maize genome sequencing project. Plant Physiology. 2002;130(4):1594–1597. doi: 10.1104/pp.015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi et al. (2008).Choi H-W, Kim J-S, Lee S-H, Bang J-W. Physical mapping by FISH and GISH of rDNA loci and discrimination of genomes A and B in Scilla scilloides complex distributed in Korea. Journal of Plant Biology. 2008;51(6):408–412. doi: 10.1007/BF03036061. [DOI] [Google Scholar]
- Chu et al. (2018).Chu Z, Wen J, Yang Y-P, Nie Z-L, Meng Y. Genome size variation and evolution in the grape family Vitaceae: genome size variation in Vitaceae. Journal of Systematics and Evolution. 2018;56:273–282. [Google Scholar]
- Dodsworth et al. (2015).Dodsworth S, Chase MW, Kelly LJ, Leitch IJ, Macas J, Novak P, Piednoël M, Weiss-Schneeweiss H, Leitch AR. Genomic repeat abundances contain phylogenetic signal. Systematic Biology. 2015;64(1):112–126. doi: 10.1093/sysbio/syu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodsworth et al. (2016).Dodsworth S, Chase MW, Särkinen T, Knapp S, Leitch AR. Using genomic repeats for phylogenomics: a case study in wild tomatoes (Solanum section Lycopersicon: Solanaceae) Biological Journal of the Linnean Society. 2016;117(1):96–105. doi: 10.1111/bij.12612. [DOI] [Google Scholar]
- Dolezel et al. (2003).Dolezel J, Bartos J, Voglmayr H, Greilhuber J. Nuclear DNA content and genome size of trout and human. Cytometry. Part A: The Journal of the International Society for Analytical Cytology. 2003;51A(2):127–128. doi: 10.1002/cyto.a.10013. [DOI] [PubMed] [Google Scholar]
- Dolezel, Greilhuber & Suda (2007).Dolezel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nature Protocols. 2007;2(9):2233–2244. doi: 10.1038/nprot.2007.310. [DOI] [PubMed] [Google Scholar]
- Du et al. (2010).Du J, Tian Z, Hans CS, Laten HM, Cannon SB, Jackson SA, Shoemaker RC, Ma J. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. The Plant Journal: For Cell and Molecular Biology. 2010;63(4):584–598. doi: 10.1111/j.1365-313X.2010.04263.x. [DOI] [PubMed] [Google Scholar]
- Feng et al. (2017).Feng R, Wang X, Tao M, Du G, Wang Q. Genome size and identification of abundant repetitive sequences in Vallisneria spinulosa. PeerJ. 2017;5:e3982. doi: 10.7717/peerj.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuillet et al. (2011).Feuillet C, Leach JE, Rogers J, Schnable PS, Eversole K. Crop genome sequencing: lessons and rationales. Trends in Plant Science. 2011;16(2):77–88. doi: 10.1016/j.tplants.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Galbraith et al. (1983).Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science. 1983;220(4601):1049–1051. doi: 10.1126/science.220.4601.1049. [DOI] [PubMed] [Google Scholar]
- Ganguly, Ganguly & Banerjee (2018).Ganguly A, Ganguly D, Banerjee SK. Topical phytotherapeutic treatment: management of normalization of elevated levels of biochemical parameter during osteoarthritic disorders: a prospective study. Journal of Orthopaedic Rheumatology. 2018;5(1):14. [Google Scholar]
- Gebre et al. (2016).Gebre YG, Bertolini E, Pe ME, Zuccolo A. Identification and characterization of abundant repetitive sequences in Eragrostis tef cv. Enatite genome. BMC Plant Biology. 2016;16:39. doi: 10.1186/s12870-016-0725-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs & Semir (1988).Gibbs P, Semir J. A taxonomic revision of the genus Ceiba Mill (Bombacaceae) Anales del Jardín Botánico de Madrid. 1988;60(2):259–300. [Google Scholar]
- Haga & Noda (1976).Haga T, Noda S. Cytogenetics of the Scilla scilloides complex. Genetica. 1976;46(2):161–176. doi: 10.1007/BF00121032. [DOI] [Google Scholar]
- Hawkins et al. (2006).Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Research. 2006;16(10):1252–1261. doi: 10.1101/gr.5282906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang et al. (2013).Huang S, Ding J, Deng D, Tang W, Sun H, Liu D, Liu D, Zhang L, Niu X, Zhang X, Meng M, Yu J, Liu J, Han Y, Shi W, Zhang D, Cao S, Wei Z, Cui Y, Xia Y, Zeng H, Bao K, Lin L, Min Y, Zhang H, Miao M, Tang X, Zhu Y, Sui Y, Li G, Sun H, Yue J, Sun J, Liu F, Zhou L, Lei L, Zheng X, Liu M, Huang L, Song J, Xu C, Li J, Ye K, Zhong S, Lu BR, He G, Xiao F, Wang HL, Zheng H, Fei Z, Liu Y. The draft genome of the kiwifruit Actinidia chinensis. Nature Communications. 2013;4(1):2640. doi: 10.1038/ncomms3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husband, Baldwin & Suda (2013).Husband BC, Baldwin SJ, Suda J. In: The incidence of polyploidy in natural plant populations: major patterns and evolutionary processes BT—plant genome diversity volume 2: physical structure, behaviour, and evolution of plant genomes. Greilhuber J, Dolezel J, Wendel JF, editors. Springer Vienna; Vienna: 2013. pp. 255–276. [Google Scholar]
- Indran & Raj (2015).Indran S, Raj RE. Characterization of new natural cellulosic fiber from Cissus quadrangularis stem. Carbohydrate Polymers. 2015;117:392–399. doi: 10.1016/j.carbpol.2014.09.072. [DOI] [PubMed] [Google Scholar]
- Jackes (1988).Jackes BR. Revision of the Australian Vitaceae, 3. Cissus L. Austrobaileya. 1988;2(5):481–505. [Google Scholar]
- Jaillon et al. (2007).Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyère C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pè ME, Valle G, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J, Quétier F, Wincker P. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449(7161):463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- Karkamkar, Patil & Misra (2010).Karkamkar S, Patil SG, Misra SC. Cyto—morphological studies and their significance in the evolution of family Vitaceae. The Nucleus. 2010;53(1–2):37–43. doi: 10.1007/s13237-010-0009-6. [DOI] [Google Scholar]
- Kejnovsky et al. (2006).Kejnovsky E, Kubat Z, Hobza R, Lengerova M, Sato S, Tabata S, Fukui K, Matsunaga S, Vyskot B. Accumulation of chloroplast DNA sequences on the Y chromosome of Silene latifolia. Genetica. 2006;128(1–3):167–175. doi: 10.1007/s10709-005-5701-0. [DOI] [PubMed] [Google Scholar]
- Kelly et al. (2015).Kelly LJ, Renny-Byfield S, Pellicer J, Macas J, Novak P, Neumann P, Lysak MA, Day PD, Fay MF, Nichols RA, Leitch AR, Leitch IJ. Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. The New Phytologist. 2015;208(2):596–607. doi: 10.1111/nph.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell (2002).Kidwell MG. Transposable elements and the evolution of genome size in eukaryotes. Genetica. 2002;115(1):49–63. doi: 10.1023/A:1016072014259. [DOI] [PubMed] [Google Scholar]
- Kirov et al. (2014).Kirov I, Divashuk M, Van Laere K, Soloviev A, Khrustaleva L. An easy SteamDrop method for high-quality plant chromosome preparation. Molecular Cytogenetics. 2014;7(1):21. doi: 10.1186/1755-8166-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latiff (2001).Latiff A. Diversity of the Vitaceae in the Malay Archipelago. Malayan Nature Journal. 2001;55:467–478. [Google Scholar]
- Li et al. (2017).Li S-F, Su T, Cheng G-Q, Wang B-X, Li X, Deng C-L, Gao W-J. Chromosome evolution in connection with repetitive sequences and epigenetics in plants. Gene. 2017;8(10):E290. doi: 10.3390/genes8100290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al. (2013).Liu X-Q, Ickert-Bond SM, Chen L-Q, Wen J. Molecular phylogeny of Cissus L. of Vitaceae (the grape family) and evolution of its pantropical intercontinental disjunctions. Molecular Phylogenetics and Evolution. 2013;66(1):43–53. doi: 10.1016/j.ympev.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Lombardi (2015).Lombardi J. New combinations for the South American Cissus striata clade (Vitaceae) Phytotaxa. 2015;227(3):295–298. doi: 10.11646/phytotaxa.227.3.10. [DOI] [Google Scholar]
- Loureiro et al. (2006).Loureiro J, Rodriguez E, Dolezel J, Santos C. Flow cytometric and microscopic analysis of the effect of tannic acid on plant nuclei and estimation of DNA content. Annals of Botany. 2006;98(3):515–527. doi: 10.1093/aob/mcl140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro et al. (2007a).Loureiro J, Rodriguez E, Doležel J, Santos C. Two new nuclear isolation buffers for plant DNA flow cytometry: a test with 37 species. Annals of Botany. 2007a;100(4):875–888. doi: 10.1093/aob/mcm152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro et al. (2007b).Loureiro J, Rodriguez E, Gomes A, Santos C. Genome size estimations on Ulmus minor Mill., Ulmus glabra Huds. and Celtis australis L. using flow cytometry. Plant Biology. 2007b;9(4):541–544. doi: 10.1055/s-2007-965165. [DOI] [PubMed] [Google Scholar]
- Ma et al. (2010).Ma L, Vu GTH, Schubert V, Watanabe K, Stein N, Houben A, Schubert I. Synteny between Brachypodium distachyon and Hordeum vulgare as revealed by FISH. Chromosome Research: An International Journal on the Molecular, Supramolecular and Evolutionary Aspects of Chromosome Biology. 2010;18(7):841–850. doi: 10.1007/s10577-010-9166-3. [DOI] [PubMed] [Google Scholar]
- Macas et al. (2011).Macas J, Kejnovský E, Neumann P, Novák P, Koblížková A, Vyskot B. Next generation sequencing-based analysis of repetitive DNA in the model dioecious plant Silene latifolia. PLOS ONE. 2011;6(11):e27335. doi: 10.1371/journal.pone.0027335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas, Neumann & Navrátilová (2007).Macas J, Neumann P, Navrátilová A. Repetitive DNA in the pea (Pisum sativum L.) genome: comprehensive characterization using 454 sequencing and comparison to soybean and Medicago truncatula. BMC genomics. 2007;8:427. doi: 10.1186/1471-2164-8-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macas et al. (2015).Macas J, Novák P, Pellicer J, Čížková J, Koblížková A, Neumann P, Fukova I, Dolezel J, Kelly LJ, Leitch IJ. In-depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLOS ONE. 2015;10(11):e0143424. doi: 10.1371/journal.pone.0143424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascagni et al. (2015).Mascagni F, Barghini E, Giordani T, Rieseberg LH, Cavallini A, Natali L. Repetitive DNA and plant domestication: Variation in copy number and proximity to genes of LTR-retrotransposons among wild and cultivated sunflower (Helianthus annuus) genotypes. Genome Biology and Evolution. 2015;7(12):3368–3382. doi: 10.1093/gbe/evv230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer et al. (2012).Mayer KFX, Waugh R, Brown JWS, Schulman A, Langridge P, Platzer M, Fincher GB, Muehlbauer GJ, Close TJ, Stein N. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491(7426):711–716. doi: 10.1038/nature11543. [DOI] [PubMed] [Google Scholar]
- Michael & VanBuren (2015).Michael TP, VanBuren R. Progress, challenges and the future of crop genomes. Current Opinion in Plant Biology. 2015;24:71–81. doi: 10.1016/j.pbi.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Mirzaghaderi (2010).Mirzaghaderi G. Full-length research paper, a simple metaphase chromosome preparation from meristematic root tip cells of wheat for karyotyping or in situ hybridization. African Journal of Biotechnology. 2010;9(3):314–318. [Google Scholar]
- Mishra, Srivastava & Nagori (2010).Mishra G, Srivastava S, Nagori BP. Pharmacological and therapeutic activity of Cissus quadrangularis an overview. International Journal of PharmTech Research. 2010;2(2):1298–1310. [Google Scholar]
- Murata & Motoyoshi (1995).Murata M, Motoyoshi F. Floral chromosomes of Arabidopsis thaliana for detecting low-copy DNA sequences by fluorescence in situ hybridization. Chromosoma. 1995;104(1):39–43. doi: 10.1007/BF00352224. [DOI] [PubMed] [Google Scholar]
- Nakamura et al. (2011).Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, Ishikawa S, Linak MC, Hirai A, Takahashi H, Altaf-Ul-Amin M, Ogasawara N, Kanaya S. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Research. 2011;39(3):e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak et al. (2014).Novak P, Hribova E, Neumann P, Koblizkova A, Dolezel J, Macas J. Genome-wide analysis of repeat diversity across the family Musaceae. PLOS ONE. 2014;9(6):e98918. doi: 10.1371/journal.pone.0098918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák, Neumann & Macas (2010).Novák P, Neumann P, Macas J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics. 2010;15(11):387. doi: 10.1186/1471-2105-11-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak et al. (2013).Novak P, Neumann P, Pech J, Steinhaisl J, Macas J. RepeatExplorer: a galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics. 2013;29(6):792–793. doi: 10.1093/bioinformatics/btt054. [DOI] [PubMed] [Google Scholar]
- Nystedt et al. (2013).Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin Y-C, Scofield DG, Vezzi F, Delhomme N, Giacomello S, Alexeyenko A, Vicedomini R, Sahlin K, Sherwood E, Elfstrand M, Gramzow L, Holmberg K, Hällman J, Keech O, Klasson L, Koriabine M, Kucukoglu M, Käller M, Luthman J, Lysholm F, Niittylä T, Olson A, Rilakovic N, Ritland C, Rosselló JA, Sena J, Svensson T, Talavera-López C, Theißen G, Tuominen H, Vanneste K, Wu ZQ, Zhang B, Zerbe P, Arvestad L, Bhalerao R, Bohlmann J, Bousquet J, Gil RGarcia, Hvidsten TR, De Jong P, MacKay J, Morgante M, Ritland K, Sundberg B, Thompson SL, Van de Peer Y, Andersson B, Nilsson O, Ingvarsson PK, Lundeberg J, Jansson S. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;479:579–584. doi: 10.1038/nature12211. [DOI] [PubMed] [Google Scholar]
- Ovodov (1998).Ovodov IS. [Polysaccharides of flower plants: structure and physiological activity] Bioorganicheskaia Khimiia. 1998;24(7):483–501. [PubMed] [Google Scholar]
- Paterson et al. (2009).Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob-ur Rahman P, Ware D, Westhoff P, Mayer KF, Messing J, Rokhsar DS. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(7229):551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- Pfosser et al. (1995).Pfosser M, Heberle-Bors E, Amon A, Lelley T. Evaluation of the sensitivity of flow cytometry in detecting aneuploidy in wheat using disomic and ditelosomic wheat-rye addition lines. Cytometry. 1995;21(4):387–393. doi: 10.1002/cyto.990210412. [DOI] [PubMed] [Google Scholar]
- Raghavan (1957).Raghavan RS. Chromosome numbers in Indian medicinal plants. Proceedings of the Indian Academy of Sciences—Section B. 1957;45(6):294–298. [Google Scholar]
- Roa & Guerra (2012).Roa F, Guerra M. Distribution of 45S rDNA sites in chromosomes of plants: structural and evolutionary implications. BMC Evolutionary Biology. 2012;12(1):225. doi: 10.1186/1471-2148-12-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roark et al. (2010).Roark LM, Hui AY, Donnelly L, Birchler JA, Newton KJ. Recent and frequent insertions of chloroplast DNA into maize nuclear chromosomes. Cytogenetic and Genome Research. 2010;129(1–3):17–23. doi: 10.1159/000312724. [DOI] [PubMed] [Google Scholar]
- Robert et al. (2001).Robert GW, Qing-feng W, Yong W, You-hao G. A taxonomic investigation of variation within Cissus quadrangularis L. (Vitaceae) in Kenya. Wuhan University Journal of Natural Sciences. 2001;6(3):715–724. doi: 10.1007/BF02830291. [DOI] [Google Scholar]
- Rodrigues, Lombardi & Lovato (2014).Rodrigues J, Lombardi J, Lovato M. Phylogeny of Cissus (Vitaceae) focusing on South American species. Taxon. 2014;63(2):33. [Google Scholar]
- SanMiguel et al. (1996).SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, Bennetzen JL. Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996;274(5288):765–768. doi: 10.1126/science.274.5288.765. [DOI] [PubMed] [Google Scholar]
- Sato et al. (2012).Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, Kaneko T, Nakamura Y, Shibata D, Aoki K, Egholm M, Knight J, Bogden R, Li C, Shuang Y, Xu X, Pan S, Cheng S, Liu X, Ren Y, Wang J, Albiero A, Dal Pero F, Todesco S, Van Eck J, Buels RM, Bombarely A, Gosselin JR, Huang M, Leto JA, Menda N, Strickler S, Mao L, Gao S, Tecle IY, York T, Zheng Y, Vrebalov JT, Lee J, Zhong S, Mueller LA, Stiekema WJ, Ribeca P, Alioto T, Yang W, Huang S, Du Y, Zhang Z, Gao J, Guo Y, Wang X, Li Y, He J, Li C, Cheng Z, Zuo J, Ren J, Zhao J, Yan L, Jiang H, Wang B, Li H, Li Z, Fu F, Chen B, Feng Q, Fan D, Wang Y, Ling H, Xue Y, Ware D, McCombie WR, Lippman ZB, Chia JM, Jiang K, Pasternak S, Gelley L, Kramer M, Anderson LK, Chang SB, Royer SM, Shearer LA, Stack SM, Rose JK, Xu Y, Eannetta N, Matas AJ, McQuinn R, Tanksley SD, Camara F, Guigó R, Rombauts S, Fawcett J, Van de Peer Y, Zamir D, Liang C, Spannagl M, Gundlach H, Bruggmann R, Mayer K, Jia Z, Zhang J, Ye Z, Bishop GJ, Butcher S, Lopez-Cobollo R, Buchan D, Filippis I, Abbott J, Dixit R, Singh M, Singh A, Pal JK, Pandit A, Singh PK, Mahato AK, Gaikwad VD, Sharma RR, Mohapatra T, Singh NK, Causse M, Rothan C, Schiex T, Noirot C, Bellec A, Klopp C, Delalande C, Berges H, Mariette J, Frasse P, Vautrin S, Zouine M, Latché A, Rousseau C, Regad F, Pech JC, Philippot M, Bouzayen M, Pericard P, Osorio S, Carmen AFernandezdel, Monforte A, Granell A, Fernandez-Muñoz R, Conte M, Lichtenstein G, Carrari F, De Bellis G, Fuligni F, Peano C, Grandillo S, Termolino P, Pietrella M, Fantini E, Falcone G, Fiore A, Giuliano G, Lopez L, Facella P, Perotta G, Daddiego L, Bryan G, Orozco M, Pastor X, Torrents D, Van Schriek MG, Feron RM, Van Oeveren J, De Heer P, DaPonte L, Jacobs-Oomen S, Cariaso M, Prins M, Van Eijk MJ, Janssen A, Van Haaren MJ, Jungeun Kim SH, Kwon SY, Kim S, Koo DH, Lee S, Hur CG, Clouser C, Rico A, Hallab A, Gebhardt C, Klee K, Jöcker A, Warfsmann J, Göbel U, Kawamura S, Yano K, Sherman JD, Fukuoka H, Negoro S, Bhutty S, Chowdhury P, Chattopadhyay D, Datema E, Klein Lankhorst RM. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel et al. (2009).Schnabel PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J, Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K, Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, Kudrna D, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D, Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L, Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S, Faga B, Levy MJ, McMahan L, Van Buren P, Vaughn MW, Ying K, Yeh CT, Emrich SJ, Jia Y, Kalyanaraman A, Hsia AP, Barbazuk WB, Baucom RS, Brutnell TP, Carpita NC, Chaparro C, Chia JM, Deragon JM, Estill JC, Fu Y, Jeddeloh JA, Han Y, Lee H, Li P, Lisch DR, Liu S, Liu Z, Nagel DH, McCann MC, SanMiguel P, Myers AM, Nettleton D, Nguyen J, Penning BW, Ponnala L, Schneider KL, Schwartz DC, Sharma A, Soderlund C, Springer NM, Sun Q, Wang H, Waterman M, Westerman R, Wolfgruber TK, Yang L, Yu Y, Zhang L, Zhou S, Zhu Q, Bennetzen JL, Dawe RK, Jiang J, Jiang N, Presting GG, Wessler SR, Aluru S, Martienssen RA, Clifton SW, McCombie WR, Wing RA, Wilson RK. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- Schwarzacher & Heslop-Harrison (2000).Schwarzacher T, Heslop-Harrison P. Practical in situ hybridization. BIOS Scientific Publishers; Oxford: 2000. p. 203. [Google Scholar]
- Sessegolo, Burlet & Haudry (2016).Sessegolo C, Burlet N, Haudry A. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biology Letters. 2016;12(8):20160407. doi: 10.1098/rsbl.2016.0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis et al. (2003).Soltis DE, Soltis PS, Bennett MD, Leitch IJ. Evolution of genome size in the angiosperms. American Journal of Botany. 2003;90(11):1596–1603. doi: 10.3732/ajb.90.11.1596. [DOI] [PubMed] [Google Scholar]
- Stohs & Ray (2012).Stohs SJ, Ray SD. A review and evaluation of the efficacy and safety of Cissus quadrangularis extracts. Phytotherapy Research. 2012;27(8):1107–1114. doi: 10.1002/ptr.4846. [DOI] [PubMed] [Google Scholar]
- Suda (2004).Suda J. Thesis. 2004. An employment of flow cytometry into plant biosystematics; p. 55. [Google Scholar]
- Ting, Sternberg & Deniro (1983).Ting IP, Sternberg LO, Deniro MJ. Variable photosynthetic metabolism in leaves and stems of Cissus quadrangularis L. Plant Physiology. 1983;71(3):677–679. doi: 10.1104/pp.71.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedha, Bn & Devi (2013).Vedha S, Bn H, Devi R. Review-pharmacological activities based on different extracts of Cissus quadrangularis. International Journal of Pharmacognosy and Phytochemical Research. 2013;5(2):128–133. [Google Scholar]
- Wang et al. (2014).Wang W, Haberer G, Gundlach H, Glasser C, Nussbaumer T, Luo MC, Lomsadze A, Borodovsky M, Kerstetter RA, Shanklin J, Byrant DW, Mockler TC, Appenroth KJ, Grimwood J, Jenkins J, Chow J, Choi C, Adam C, Cao XH, Fuchs J, Schubert I, Rokhsar D, Schmutz J, Michael TP, Mayer KF, Messing J. The Spirodela polyrhiza genome reveals insights into its neotenous reduction in fast growth and aquatic lifestyle. Nature Communications. 2014;5:3311. doi: 10.1038/ncomms4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen (2007).Wen J. Vitaceae. In: Kubitzki K, editor. The families and genera of vascular plants. 1st Edition Springer; Berlin Heidelberg: 2007. pp. 467–472. [Google Scholar]
- Wen et al. (2018).Wen J, Lu LM, Liu XQ, Zhang N, Ickert-Bond SM, Gerrath J, Manchester SR, Boggan J, Chen ZD. A new phylogenetic tribal classification of the grape family (Vitaceae) Journal of Systematics and Evolution. 2018;56:262–272. doi: 10.1111/jse.12427. [DOI] [Google Scholar]
- Wicker et al. (2007).Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH. A unified classification system for eukaryotic transposable elements. Nature Reviews Genetics. 2007;8(12):973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Wilson (1991).Wilson JB. A comparison of biogeographic models: migration, vicariance and panbiogeography. Global Ecology and Biogeography Letters. 1991;1(3):84–87. doi: 10.2307/2997494. [DOI] [Google Scholar]
- Xu et al. (2011).Xu X, Pan S, Cheng S, Zhang B, Mu D, Ni P, Zhang G, Yang S, Li R, Wang J, Orjeda G, Guzman F, Torres M, Lozano R, Ponce O, Martinez D, De la Cruz G, Chakrabarti SK, Patil VU, Skryabin KG, Kuznetsov BB, Ravin NV, Kolganova TV, Beletsky AV, Mardanov AV, Di Genova A, Bolser DM, Martin DM, Li G, Yang Y, Kuang H, Hu Q, Xiong X, Bishop GJ, Sagredo B, Meja N, Zagorski W, Gromadka R, Gawor J, Szczesny P, Huang S, Zhang Z, Liang C, He J, Li Y, He Y, Xu J, Zhang Y, Xie B, Du Y, Qu D, Bonierbale M, Ghislain M, Herrera Mdel R, Giuliano G, Pietrella M, Perrotta G, Facella P, O’Brien K, Feingold SE, Barreiro LE, Massa GA, Diambra L, Whitty BR, Vaillancourt B, Lin H, Massa AN, Geoffroy M, Lundback S, DellaPenna D, Buell CR, Sharma SK, Marshall DF, Waugh R, Bryan GJ, Destefanis M, Nagy I, Milbourne D, Thomson SJ, Fiers M, Jacobs JM, Nielsen KL, Snderkr M, Iovene M, Torres GA, Jiang J, Veilleux RE, Bachem CW, De Boer J, Borm T, Kloosterman B, Van Eck H, Datema E, Bt Hekkert, Goverse A, Van Ham RC, Visser RGF. Genome sequence and analysis of the tuber crop potato. Nature. 2011;475:189–195. doi: 10.1038/nature10158. [DOI] [PubMed] [Google Scholar]
- Zuccolo et al. (2007).Zuccolo A, Sebastian A, Talag J, Yu Y, Kim H, Collura K, Kudrna D, Wing RA. Transposable element distribution, abundance, and role in genome size variation in the genus Oryza. BMC Evolutionary Biology. 2007;7:152. doi: 10.1186/1471-2148-7-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Mitotic metaphase chromosomes compliments from Cissus quandrangularis
The data obtained from the pairwise comparison of short-read sequences are used to construct graphs with vertices representing the sequence reads and overlapping reads connected with edges. The edge weight is used to express the similarity scores for reads. Where the sequence genome coverage is low (<0.5 ×), presence of single copy sequences is low and therefore low overlapping resulting in detached nodes with no connections to other parts of the graph. Presence of repetitive sequences is represented by mutually connected nodes as a result of pooling of overlapping reads. Examination of graph topology facilitates separation and identification of clusters of reads frequently connected representing individual repetitive element families.
Data Availability Statement
The following information was supplied regarding data availability:
Cissus quadrangularis raw sequencing data are available at SRA: SRR8573652.