

Abstract
Single-module nonribosomal peptide synthetases (NRPSs) and NRPS-like enzymes activate and transform carboxylic acids in both primary and secondary metabolism; and are of great interests due to their biocatalytic potentials. The single-module NRPS IvoA is essential for fungal pigment biosynthesis. Here, we show that IvoA catalyzes ATP-dependent unidirectional stereoinversion of l-tryptophan to d-tryptophan with complete conversion. While the stereoinversion is catalyzed by the epimerization (E) domain, the terminal condensation (C) domain stereoselectively hydrolyzes d-tryptophanyl-S-phosphopantetheine thioester and thus represents a noncanonical C domain function. Using IvoA, we demonstrate a biocatalytic stereoinversion/deracemization route to access a variety of substituted d-tryptophan analogs in high enantiomeric excess.
Graphical Abstract
Nonribosomal peptide synthetases (NRPSs) are modular enzymes employing an assembly-line logic to synthesize a myriad of peptide-based secondary metabolites with diverse structures and biological activities.1 Single-module NRPS and NRPS-like enzymes adopt similar thiotemplated enzymology with a single set of adenylation (A) and thiolation (T) domain. These enzymes have important functions in transforming carboxylic acid substrates in primary and secondary metabolism;2 and have increased interests as biocatalysts due to their functional diversity (Figure 1).3 Following selection and thermodynamic activation of the carboxylic acid by the A domain, the substrate is preserved as phosphopantetheine (Ppant) thioester on the T domain. Depending on the type of downstream releasing domains, the thioester intermediates are subjected to a broad range of modifications (Figure 1a), including but not limited to: esterification/amidation by a condensation (C) domain in A-T-C;4 Dieckman/aldol condensation or cyclization by a thioesterase (TE) domain in A-T-TE;5,6 2- or 4-electron reduction by reductase (R) domain in either A-T-R or A-T-R-R;2b,7 2-electron reduction followed by PLP-dependent aldol condensation in A-T-R-P.8 In natural product biosynthetic pathways, these enzymes generate natural product scaffolds with structural diversity that complements the chemical space of canonical nonribosomal peptides, such as dihydroxybenzoquinone, furanone, butyrolactone, and dihydropyrazine (Figure 1A).
Figure 1.

Diverse functions of single-module NRPS and NRPS-like enzymes. A) Characterized examples. B) IvoA studied in this work.
Recently, a single-module NRPS, encoded by the gene ivoA from Aspergillus nidulans with an unusual domain architecture annotated as A-T-C-C* was proposed to acetylate l-tryptophan.9 The enzymatic product N-acetyl-l-tryptophan was suggested to be further oxidized by a P450 enzyme IvoC and a phenol oxidase IvoB en route to the conidiophore pigment (Figure 1B). Although genetic studies provided compelling evidence implicating ivoA in the biosynthesis of N-acetyl-hydroxytryptophan,10,11 the proposed acetyltransferase activity of IvoA is an unlikely fit for a single-module NRPS. Furthermore, the mechanistic proposal for IvoA is at odds with accepted logic of NRPS enzymology for the following reasons:1) It is metabolically wasteful to activate the carboxy group of a substrate at the expense of one equivalent of ATP in order to accomplish N-acetylation by acetyl-CoA; 2) It is against the NRPS directionality rule for a downstream C domain to carry out a condensation reaction (acetylation here) with an upstream T domain as the acceptor.1,12
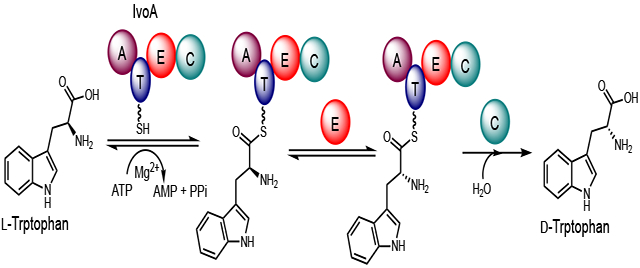
To elucidate the enzymatic function of IvoA, we first reanalyzed its domain architecture. Since epimerization (E) domains show sequence and structure homology to C domains, and are often inserted between T and C domains in the NRPS assembly lines,12 we hypothesized that the true domain organization of IvoA is A-T-E-C. Embedding a functional E domain could rationalize the necessity of involving NRPS machinery: activation of the α-carboxy group can lower the pKa of the Cα proton, thereby facilitating stereoinversion. To test this hypothesis, we overexpressed IvoA by using S. cerevisiae JHY686 strain as a heterologous host.13 Consistent with the previous report, we were able to detect N-acetyltryptophan formation. However, the purified product from yeast cell culture was found to be exclusively d-enantiomeric (ee > 99%) as confirmed by chiral-HPLC analysis (Figure 2A). The inverted stereochemistry of tryptophan supports our hypothesis that an E domain is present within IvoA.
Figure 2.

Characterization of IvoA activity. A) Stereochemistry determination for isolated N-acetyl-d-tryptophan. B) Mass spectrometry shows the mass shift of tryptophan when the assay was performed in D2O. C) 1H-NMR spectra indicate incorporation of deuterium at the α position: 1) the change of splitting pattern of the diastereotopic β proton signal due to smaller coupling constant (3JH-D)); 2) the disappearance of α proton signal. D) Chiral HPLC resolution of tryptophan enantiomers from IvoA reaction demonstrated complete stereoinversion of l-tryptophan to d-tryptophan.
To interrogate the function of IvoA, particularly the cryptic acetyltransferase activity, we purified IvoA from S. cerevisiae and assayed its activity in vitro (Figure S1). We first examined the substrate specificity of IvoA adenylation domain (Figure S2). As expected, l-tryptophan is the preferred substrate, while d-tryptophan is activated with 64% lower efficiency (Figure S2). We were unable to detect formation of acetylated tryptophan starting from either l/d-tryptophan in the presence of ATP, provided with either acetyl-CoA or acetyl-phosphate as the acetyl-donor. Increasing enzyme concentration to > 50 μM, plus prolonged overnight incubation only led to trace amount of N-acetyl-d-tryptophan (kobsapp < 0.1 h−1, Figure S3).
When we performed the assay ([E] = 2 μM) in D2O and analyzed the reaction mixture by LC-MS, we readily observed a gradual +1 Da mass shift of tryptophan (Figure 2B). This deuterium “wash-in” result is phosphopantetheinylation-dependent as inactivating the T domain (IvoA-T0, S785A mutation) completely abolished the incorporation. Monitoring the reaction by 1H-NMR confirmed that the hydrogen-deuterium exchange took place at the Cα–H (Figure 2C), indicating that epimerization of tryptophanyl-S-Ppant occurred. The complete hydrogen-deuterium exchange is also consistent with the “two-base” mechanism proposed for NRPS E domain.14
We next followed the reaction by using chiral-HPLC and complete conversion of l-tryptophan (1 mM) to d-tryptophan was observed in 3 hours (Figure 2D). Chiral-resolution allowed us to determine the apparent steady-state kinetics by quantifying d-tryptophan formation (Table 1, Figure S4). The apparent kcat (38 min−1) is similar to that of other characterized single-module NRPSs and NRPS-like enzymes,4,7 while the Km (~50 μM) is close to the intracellular level of l-tryptophan (12 μM).15 Both E and C domains are catalytically important for IvoA, as inactivating either domain by mutating the catalytic histidine residues (H963A and H1428A) substantially compromised the apparent turnover number kcat (420-fold by E0 while 4700-fold by C0). In contrast, the apparent Michaelis constants were not changed, suggesting that substrate binding (at the A domain) was not affected.
Table 1.
Apparent steady-state kinetic constants of l-tryptophan stereoinversion by IvoA and mutants.
| Enzyme | kcatapp (min−1) | Kmapp (μM) | (kcat/Km)app (M−1 s−1) |
|---|---|---|---|
| WTa | 38 ± 3 | 50 ± 10 | 1.3 × 104 |
| T0(S785A) | inactive | inactive | inactive |
| E0(H963A) | 0.09 ± 0.03 | 50 ± 10 | 30 |
| C0(H1428A) | 0.008 ± 0.002 | 40 ± 10 | 3.5 |
Substrate inhibition was observed with Ki app of 4 ± 1 (mM).
Taken together, these data indicate that IvoA lacks acetyltransferase activity in vitro, but instead is a bona fide ATP-dependent enzyme catalyzing enantioselective stereoinversion of l-tryptophan to d-tryptophan. The observed acetylation of d-tryptophan in vivo must be carried out by an endogenous acetyltransferase. Because yeast histone acetyltransferase Hpa3 is known to act as a d-amino acid N-acetyltransferase for detoxification of d-amino acids,16 we overexpressed IvoA in the hpa3-deleted yeast strain constructed by replacing hpa3 with leu2 (SI Methods, Figure S5). Consequently, the culture medium was devoid of N-acetyltryptophan, whereas free d-tryptophan (ee = 98%) were still accumulated inside the cells (Figure S6-S7). Therefore, we conclude that IvoA does not acetylate tryptophan and the origin of the negligible acetyltransferase activity of IvoA observed in vitro may derive from trace amount of contaminated yeast Hpa3.
Distinct from common PLP-dependent or PLP-independent amino acid racemases (Scheme 1), which often catalyze bidirectional stereoinversion and also inevitably lead to racemization (equilibrium constant approaches unity),17 IvoA catalyzes unidirectional stereoinversion, completely converting l-tryptophan to its enantiomer d-tryptophan. The complete conversion is driven by coupled ATP hydrolysis, which is thermodynamically favored (Scheme 1),18 and is enabled by the thiotemplate enzymology of IvoA (Figure 3). We reason that the activated tryptophan is delivered to the E domain as tryptophanyl-S-Ppant thioester, which undergoes epimerization to give a mixture of d/l-tryptophanyl-S-Ppant diastereoisomers in equilibrium. We propose that dynamic kinetic resolution may be accomplished by the C domain in a releasing step, which stereoselectively hydrolyzes the d-tryptophanyl-S-Ppant thioester to achieve irreversible conversion.
Scheme 1.

Stereoinversion of amino acid enantiomer into its mirror-image counterpart by common racemases and IvoA.
Figure 3.

Working model of IvoA.
As mentioned earlier, even though IvoA A domain prefers l-tryptophan, d-tryptophan can still be adenylated and thioesterified (Figure S2). In addition, the loaded d-tryptophanyl-S-Ppant underwent epimerization by IvoA E domain as evidenced by similar, yet slower deuterium “wash-in” behavior under multiple-turnover condition (Figure S8). The slower turnover measured by deuterium incorporation reflects the lower adenylation efficiency of d-tryptophan. Nonetheless, the occurrence of hydrogen-deuterium exchange at d-tryptophanyl-S-Ppant Cα position not only suggests that epimerization is faster than the C-domain catalyzed d-specific tryptophanyl thioester hydrolysis, but also indicates that the d/l-tryptophanyl-S-Ppant equilibrium can be approached from either direction (Figure 3). However, IvoA cannot convert d-tryptophan to l-tryptophan, which suggests that the l-tryptophanyl-S-Ppant is not hydrolyzed by the C domain. A d-specific hydrolytic releasing C domain is therefore the key for unidirectional complete stereoinversion.
To directly demonstrate the stereoselectivity of IvoA C domain, we purified the standalone IvoA-C and assayed its activity in vitro. Addition of IvoA-C in equimolar to either IvoA(C0) mutant or IvoA-ΔC truncation mutant successfully rescued the impaired stereoinversion activity, which proved that the standalone IvoA-C is active (Figure S9). We then synthesized both d- and l-tryptophanyl-S-N-acetylcysteamine as surrogate substrates mimicking the IvoA T domain bound tryptophanyl-S-Ppant intermediates. However, the enzyme did not catalyze hydrolysis significantly above the background nonenzymatic rate (Figure S10). Using d-tryptophanyl-S-pantetheine (d-Trp-pant) also did not improve enzymatic hydrolysis. We reason that the protein:protein interaction between T and C domain is important for substrate recognition, which has been shown in other studies of C domains.19 Hence, we chose to enzymatically load d/l-tryptophan to IvoA-ΔC(E0) by taking advantage of the promiscuous A domain. It is imperative to inactivate the E domain in this truncation mutant in order to minimize the epimerization. The formation of corresponding d/l-tryptophanyl-S-Ppant of IvoA-ΔC was confirmed by intact protein mass spectrometry (Figure S11). Free excess d/l-tryptophan substrates were quickly removed from IvoA-ΔC(E0) by using desalting spin columns and the loaded d/l-tryptophanyl-S-IvoA-ΔC(E0) were immediately subjected to IvoA-C catalyzed hydrolysis. The liberated free tryptophan was then quantified by LC-MS. As shown in Figure 4, IvoA-C stereoselectively hydrolyzed d-tryptophanyl-S-IvoA-ΔC(E0) over l-tryptophanyl-S-IvoA-ΔC(E0). NRPS C domains that have thioesterase activity are rare, and to date only one example from crocacin PKS-NRPS hybrid assembly-line was known, but did not show stereoselectivity.20 Therefore, the IvoA-C characterized here represents a novel C domain and we classify it as a DCH2O subtype according to the universally acknowledged nomenclature.12
Figure 4.

Characterization of IvoA-C activity in vitro by LC-MS.
The verified stereoinversion activity of IvoA prompted us to explore its biocatalytic potential. d-tryptophan and its substituted analogues are important building blocks for many peptide pharmaceuticals, such as FDA approved lanreotide, pasireotide, octreotide, macimorelin, triptorelin, etc. Recently, there is growing interest in developing biocatalytic processes for syntheses of substituted d-tryptophans by stereoinversion and deracemization from the l-enantiomers and rac-tryptophans, respectively.21 However, to overcome the entropically unfavorable deracemization process (ΔG o = 0.4 kcal/mol),22 the current methods are based on multi-step cascade reactions to establish non-equilibrium conditions for enrichment of d-enantiomers.21 In contrast, IvoA offers a concise one-step, direct nonredox stereoinversion/deracemization process, and allows us to access a library of d-tryptophan analogues in high enantiomeric excess (ee >99%) at millimolar level. Different substitution groups, either electron-withdrawing or electron-donating, at most positions (4, 5, 6 and 7) on the indole ring can be tolerated (Table 2). No conversion of 2-Me-dl-tryptophan is due to inefficient activation by A domain (Figure S12), which suggests that substitution at 2-position may interfere with substrate recognition. The poor substrates are generally those with larger substituents (e.g. 5-NO2, 5-CN, 6-Br, 7-Br), which reflects the size limit by IvoA A domain. In light of recent success in A domain engineering,23 it is conceivable that the substrate scope can be expanded in the future by enlarging the substrate binding pocket of A domain.
Table 2.
Biocatalytic stereoinversion or deracemization of substituted tryptophans.a
 | |||
|---|---|---|---|
| Entry | Substrate | ee (%) | |
| Stereochemistry | R | ||
| 1 | L | H | >99 |
| 2 | L | 5-OMe | >99 |
| 3 | rac | 5-CN | 63 |
| 4 | rac | 5-NO2 | 44 |
| 5 | rac | 4-F | >99 |
| 6 | rac | 5-F | >99 |
| 7 | rac | 6-F | >99 |
| 8 | rac | 5-Cl | >99 |
| 9 | rac | 6-Cl | 98 |
| 10 | rac | 5-Br | >99 |
| 11 | rac | 6-Br | 75 |
| 12 | rac | 7-Br | 87 |
| 13 | rac | 2-Me | 1.3 |
| 14 | rac | 4-Me | >99 |
| 15 | rac | 5-Me | >99 |
| 16 | rac | 6-Me | 97 |
| 17 | rac | 7-Me | >99 |
Expt. Cond.: 1.5 mM substrates, 5 μM IvoA, 5 mM ATP, 10 mM MgCl2, 100 mM K2HPO4 buffer, pH 7.5.
In summary, our biochemical study uncovered the unusual activity of IvoA, and our findings expand the function diversity of single-module NRPSs. The reassigned function of IvoA also provides insight to fungal pigment biosynthesis. By inverting the chirality of tryptophan, IvoA perhaps can modulate amino acid flux to pigment biosynthesis in vivo. Considering the proposed role of IvoB and IvoC, one can speculate that the d-configuration generated by IvoA may be retained in the final uncharacterized conidiophore pigment.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH 1R35GM118056 to YT. YH is a Life Sciences Research Foundation fellow sponsored by the Mark Foundation for Cancer Research. MJ is the recipient of a BBSRC Future Leader Fellowship (BB/R01212/1). The Bruker MaXis II instrument used in this study was funded by the BBSRC (BB/M017982/1). We thank Dr. Dewei Gao for helpful discussion regarding organic synthesis.
Footnotes
Supporting Information.
This material is available free of charge via the Internet at http://pubs.acs.org. Experimental procedures, chromatograms, and spectroscopic data.
No competing financial interests have been declared.
REFERENCES
- (1).(a) Fischbach MA and Walsh CT Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev 2006, 106, 3468–3496. [DOI] [PubMed] [Google Scholar]; (b) Sieber SA and Mrahiel MA Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem. Rev 2005, 105, 715–738. [DOI] [PubMed] [Google Scholar]
- (2).(a) Ehmann DE; Gehring AM; Walsh CT Lysine biosynthesis in Saccharomyces cerevisiae: mechanism of a-aminoadipate reductase (Lys2) involves posttranslational phosphopantetheinylation by Lys2. Biochemistry 1999, 38, 6176–6177. [DOI] [PubMed] [Google Scholar]; (b) Hai Y; Huang AM; Tang Y Structure-guided function discovery of an NRPS-like glycine betaine reductase for choline biosynthesis in fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 21402–21407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Richardt A; Kemme T; Wagner S; Schwarzer D; Marahiel MA; Hovemann BT Ebonyl, a novel nonribosomal peptide synthetase for β-alanine conjugation with biogenic amines in Drosophila. J. Biol. Chem 2003, 278, 41160–41166. [DOI] [PubMed] [Google Scholar]; (d) Wang M; Beissner M; Zhao H Aryl-aldehyde formation in fungal polyketides: Discovery and characterization of a distinct biosynthetic mechanism. Chem. Biol 2014, 21, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Winkler M Carboxylic acid reductase enzymes (CARs). Curr. Opin. Chem. Biol 2018, 43, 23–29. [DOI] [PubMed] [Google Scholar]; (b) Ramsden JI, Heath RS, Derrington SR, Montgomery SL, Mangas-Sanchez J, Mulholland KR, Turner NJ Biocatalytic N-alkylation of amines using either primary alcohols or carboxylic acids via reductive aminase cascade. J. Am. Chem. Soc 2019, 141, 1201–1206. [DOI] [PubMed] [Google Scholar]
- (4).(a) Gao X; Chooi Y-H; Ames BD; Wang P; Walsh CT; Tang Y Fungal indole alkaloid biosynthesis: genetic and biochemical investigation of the tryptoquialanine pathway in Penicillium aethiopicum. J. Am. Chem. Soc 2011, 133, 2729–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shinohara Y; Takahashi S; Osada H; Koyama Y Identification of a novel sesquiterpene biosynthetic machinery involved in astellolide biosynthesis. Sci. Rep 2016, 6, 32865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Balibar CJ; Howard-Jones AR; Walsh CT Terrequinone A biosynthesis through L-tryptophan oxidation, dimerization and bisprenylation. Nat. Chem. Biol 2007, 3, 584–592. [DOI] [PubMed] [Google Scholar]; (b) Schneider P; Weber M; Rosenberger K; Hoffmeister D A one-pot chemoenzymatic synthesis for the universal precursor of antidiabetes and antiviral bisindolylquinones. Chem. Biol 2007, 14, 635–644. [DOI] [PubMed] [Google Scholar]
- (6).Hühner E; Öqvist K; Li S-M Design of a-keto carboxylic acid dimers by domain recombination of nonribosomal peptide synthetase (NRPS)-like enzymes. Org. Lett 2019, 21, 498–502. [DOI] [PubMed] [Google Scholar]
- (7).Yu X; Liu F; Zou Y; Tang M-C; Hang L; Houk KN; Tang Y Biosynthesis of strained piperazine alkaloids: uncovering the concise pathway of herquline A. J. Am. Chem. Soc 2016, 138, 13529–13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Baccile JA; Spraker JE; Le HH; Brandenburger E; Gomez C; Bok JW; Macheleidt J; Brakhage AA; Hoffmeister D; Keller NP; Schroeder FC Plant-like biosynthesis of isoquinoline alkaloids in Aspergillus fumigatus. Nat. Chem. Biol 2016, 12, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sung CT; Chang S-L; Entwistle R; Ahn G; Lin T-S; Petrova V; Yeh H-H; Praseuth MB; Chiang YM; Oakley BR; Wang CCC Overexpression of a three-gene conidial pigment biosynthetic pathway in Aspergillus nidulans reveals the first NRPS known to acetylate tryptophan. Fungal Genet. Biol 2017, 12, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Clutterbuck AJ A mutational analysis of conidial development in Aspergillus nidulans. Genetics, 1969, 63, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McCorkindale NJ; Hayes D; Johnston GA; Clutterbuck AJ N-Acetyl-6-hydroxytryptophan a natural substrate of a monophenol oxidase from Aspergillus nidulans. Phytochemistry, 1983, 22, 1026–1028. [Google Scholar]
- (12).Bloudoff K and Schmeing TM Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: discovery, dissection and diversity. Biochem. Biophys. Acta, 2017, 1865, 1587–1604. [DOI] [PubMed] [Google Scholar]
- (13).Harvey CJB; Tang M; Schlecht U; Horecka J; Fischer CR; Lin HC; Naughton B; Cherry J; Miranda M; Li YF; Chu AM; Hennessy JR; Vandova GA; Inglis D; Aiyar RS; Steinmetz LM; Davis RW; Medema MH; Sattely E; Khosla C; St Onge RP, Tang Y; Hillenmeyer ME Hex: A heterologous expression platform for the discovery of fungal natural products. Sci. Adv 2018, 11, eaar5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Stachelhaus T; Walsh CT Mutational analysis of the epimerization domain in the initiation module PheATE of gramicidin S synthetase. Biochemistry 2000, 39, 5775–5787. [DOI] [PubMed] [Google Scholar]
- (15).Bennett BD; Kimball EH; Gao M; Osterhout R; Van Dien SJ; Rabinowitz JD Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol 2009, 5, 593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Yow GY; Uo T; Yoshimura T; Esaki N d-amino acid N-acetyltransferase of Saccharomyces cerevisiae: a close hoologue of histone acetyltransferase Hpa2p acting exclusively on free d-amino acids. Arch. Microbiol 2004, 182, 396–403. [DOI] [PubMed] [Google Scholar]; (b) Yow GY; Uo T; Yoshimura T; Esaki N Physiological role of d-amino acid N-acetyltransferase of Saccharomyces cerevisiae: detoxification of d-amino acids. Arch. Microbiol 2006, 185, 39–46. [DOI] [PubMed] [Google Scholar]; (c) Sampath V; Liu B; Tafrov S; Srinivasan M; Rieger R; Chen EI; Sternglanz R Biochemical characterization of Hpa2 and Hpa3, two small closely related acetyltransferases from Saccharomyces cerevisiae. J. Biol. Chem 2013, 288, 21506–21513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Tanner ME Understanding Nature’s strategies for enzyme-catalyzed racemization and epimerization. Acc. Chem. Res 2002, 35, 237–246. [DOI] [PubMed] [Google Scholar]; (b) Fischer C; Ahn Y-C; Vederas JC Catalytic mechanism and properties of pyridoxal 5’-phosphate independent racemases: how enzymes alter mismatched acidity and basicity. Nat. Prod. Rep 2019, in press, DOI: 10.1039/c9np00017h. [DOI] [PubMed] [Google Scholar]
- (18).Calculated under 0.25 M ionic strength at pH 7 according to Alberty, R. A. Calculating apparent equilibrium constants of enzyme-catalyzed reactions at pH 7. Biochem. Educ 2000, 28, 12–17. [PubMed] [Google Scholar]
- (19).(a) Hai Y and Tang Y Biosynthesis of long-chain N-acyl amide by a truncated polyketide synthase–nonribosomal peptide synthetase hybrid megasynthase in fungi. J. Am. Chem. Soc 2018, 140, 1271–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao X; Haynes SW; Ames BD; Wang P; Vien LP; Walsh CT; Tang Y Cyclization of fungal nonribosomal peptides by a terminal condensation-like domain. Nat. Chem. Biol 2012, 8, 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Müller S; Rachid S; Hoffmann T; Surup F; Volz C; Zaburannyi N; Müller R Biosynthesis of crocacin involves an unusual hydrolytic release domain showing similarity to condensation domains. Chem. Biol. Catal 2014, 21, 855–865. [DOI] [PubMed] [Google Scholar]
- (21).(a) Schnepel C; Kemker I; Sewald N One-pot synthesis of D-halotryptophans by dynamic stereoinversion using a specific l-amino acid oxidase. ACS Catal 2019, 9, 1149–1158. [Google Scholar]; (b) Parmeggiani F; Casamajo AR; Walton CJW; Galman JL; Turner J; Chica RA One-pot biocatalytic synthesis of substituted d-tryptophans from indoles enabled by an engineerred aminotransferase. ACS Catal 2019, 9, 3482–3486. [Google Scholar]; (c) Matsuyama A; Mitsuhashi K; Tokuyama S; Yamamoto H d-aminoacylase and gene encoding the same. U. S. Patent 6,887,697 B2, 2005. [Google Scholar]
- (22).Gruber CC; Lavandera I; Faber K; Kroutil W From a racemate to a single enantiomer: deracemization by stereoinversion. Adv. Synth. Catal 2006, 348, 1789–1805. [Google Scholar]
- (23).(a) Ishikawa F; Miyanaga A; Kitayama H; Nakamura S; Nakanishi I; Kudo F; Eguchi T; Tanabe G An engineered aryl acid adenylation domain with an enlarged substrate binding pocket. Angew. Chem. Int. Ed. Engl 2019, 58, 6906–6910. [DOI] [PubMed] [Google Scholar]; (b) Niquille DL; Hansen DA; Mori T; Fercher D; Kries H; Hilvert D Nonribosomal biosynthesis of backbone-modified peptides. Nat. Chem 2018, 10, 282–287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.