

Abstract
Recently the selective C–H bond cleavage under mild conditions with weak oxidants was reported for fluorenyl-benzoates. This mechanism is based on multi-site concerted proton-coupled electron transfer (PCET) involving intermolecular electron transfer to an outer-sphere oxidant coupled to intramolecular proton transfer to a well-positioned proton acceptor. The electron transfer driving force depends predominantly on the oxidant, and the proton transfer driving force depends mainly on the basicity of the carboxylate, which is influenced by the substituent on the benzoate fragment. Experiments showed that the rate constants are much more sensitive to the carboxylate basicity than to the redox potential of the oxidant. Herein a vibronically nonadiabatic PCET theory is used to explain how changing the driving force for the electron and proton transfer components of the reaction through varying the oxidant and the substituent, respectively, impacts the PCET rate constant. In addition to increasing the driving force for proton transfer, enhancing the basicity of the carboxylate also decreases the equilibrium proton donor-acceptor distance, thereby facilitating the sampling of shorter proton donor-acceptor distances. This additional effect arising from the strong dependence of proton transfer on the proton donor-acceptor distance provides an explanation for the greater sensitivity of the rate constant to the carboxylate basicity than to the redox potential of the oxidant. These fundamental insights have broad implications for developing new strategies to activate C–H bonds, specifically by designing systems with shorter equilibrium proton donor-acceptor distances.
Graphical Abstract
Introduction
The selective activation and cleavage of ubiquitous but relatively inert C–H bonds have been challenging problems in synthetic chemistry. Traditional strategies for C–H bond activation include oxidative addition, sigma bond metathesis, 1,2 addition, electrophilic activation, and metal-radical activation, all of which require metal catalysts.1–5 Recently, selective C–H bond activation was reported for 2-(9H-fluoren-9-yl)benzoate and its substituted derivatives,6, 7 requiring only weak oxidants. This C–H bond cleavage mechanism is based on multi-site concerted proton-coupled electron transfer (PCET),8–15 involving intermolecular electron transfer to an outer-sphere oxidant coupled to intramolecular proton transfer to a well-positioned proton acceptor. Due to steric effects, the fluorenyl C–H bond and the carboxylate group in these fluorenyl-benzoates are positioned in a manner that facilitates proton transfer from the carbon to the oxygen. The C–H bond cleavage reactions were carried out for a range of ferrocenium (FeCp2+) and aminium (NAr3•+) oxidants6 and for different substituent groups on the fluorenyl-benzoates7 in acetonitrile at room temperature.
In our previous work,16 we presented a study of the unsubstituted 2-(9H-fluoren-9-yl)benzoate, corresponding to X = H in Figure 1, to explain the unusual relationship between the PCET rate constant and the oxidant redox potential, which directly influences the reaction free energy. We investigated this multi-site concerted PCET reaction with a vibronically nonadiabatic PCET theory9, 17–20 that incorporates proton transfer into Marcus theory21–22 for electron transfer. For a series of oxidants, a plot of the logarithm of the PCET rate constant, log kPCET, versus the logarithm of the equilibrium constant, log Keq, exhibited a shallow slope, denoted the Brønsted α, significantly smaller than 0.5. This shallow slope was explained by the high exoergicity of the dominant transitions between the reactant and product electron-proton vibronic states.
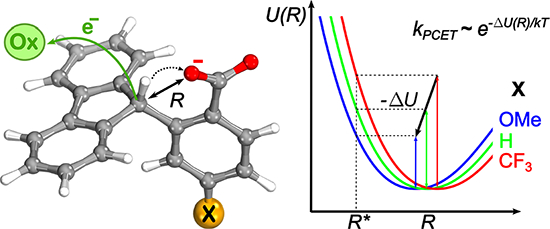
Figure 1.

Schematic depiction of the oxidative C–H bond cleavage of 2-(9H-fluoren-9-yl)-4-X-benzoate (X = CF3, H, OMe, NH2) via multi-site concerted PCET, leading to the formation of a radical product. This work focuses on this concerted PCET reaction, but experimentally this radical is found to rapidly undergo oxidative deprotonation and cyclization to form a lactone.
Herein we use this PCET theory to understand how the PCET rate constant changes with electron-donating (−OMe and −NH2) or electron-withdrawing (−CF3) substituents at the position para to the carboxylate group (Figure 1) in fluorenyl-benzoates. Recent experiments7 have shown that the rate constant depends more strongly on the pKa of the internal base than on the nature of the outer-sphere oxidant. These results were interpreted in the context of imbalanced transition states, implying asynchronous reaction paths, computed with density functional theory (DFT). However, these DFT calculations did not provide PCET rate constants and therefore could not reproduce the different dependence of the PCET rate constant on the external oxidant compared to the fluorenyl-benzoate substituent, corresponding to different Brønsted α values. Our calculations provide the PCET rate constants and produce these Brønsted α values for comparison to the experimental data. These calculations also provide a physical explanation for the significantly greater sensitivity of the PCET rate constant to the carboxylate basicity than to the redox potential of the oxidant.
Theory and Computational Methods
This section provides a brief summary of the vibronically nonadiabatic PCET theory9, 17–20 and the computational methods used to compute the input quantities to the rate constant expression. In this PCET theory, the electrons and the transferring proton are treated quantum mechanically, and the proton vibrational states are calculated for the reactant and product electronic states, resulting in two sets of electron-proton vibronic states. The PCET reaction is described in terms of nonadiabatic transitions between these reactant and product electron-proton vibronic states, which can be represented by two sets of stacked parabolic free energy curves along a collective solvent coordinate. Reorganization of the solute and solvent can lead to a degeneracy of two vibronic states, enabling simultaneous electron and proton transfer from their respective donors to their respective acceptors.
For a given proton donor-acceptor distance R, the PCET rate constant is expressed as17–18
| (1) |
where the summation runs over all pairs of reactant and product electron-proton vibronic states, denoted by indices μ and v, respectively, Pμ is the Boltzmann population of the reactant vibronic state μ, Sμv(R) is the overlap between the reactant and product proton vibrational wavefunctions μ and v, λ is the total reorganization energy, and V el is the electronic coupling. Moreover, is the free energy barrier for the transition from reactant vibronic state μ to product vibronic state v, and it is related to the reaction free energy for vibronic states (μ,v) through:
| (2) |
The reaction free energy can be expressed as , where , and ϵμ and ϵv are the energies of the reactant vibronic state μ and the product vibronic state v relative to their respective ground vibronic states (i.e., ).
The total rate constant kPCET is evaluated by calculating the rate constant k(R) for a range of proton donor-acceptor distances R and integrating over all R, weighting each value according to the probability distribution function P(R):
| (3) |
The probability distribution P(R) can be expressed as
| (4) |
where U(R) is the potential energy of the reactant relative to the potential energy U(R0) at its equilibrium proton donor-acceptor distance R0, CN is the normalization constant, which is assumed be approximately the same for all related systems studied, and . All rate constants and log Keq, which is computed as ΔG0/(2.303RT), were calculated at the temperature T = 298.15 K.
To evaluate the PCET rate constant k(R), we performed the following procedure for determining the required input quantities for each of the studied fluorenyl-benzoates (o-Flr)(p-X)C6H3COO−. For this procedure, density functional theory (DFT) calculations were performed with the B3LYP23–24 functional and the 6–31++G** basis set,25–27 as well as the conductor-like polarizable continuum model28–29 for solvent, using Gaussian 09.30 All input quantities were computed in acetonitrile, which was the solvent used in the experiments. First, the equilibrium structures for the reactant (anionic) and product (neutral) states were optimized for each fluorenyl-benzoate. Then constrained reactant and product geometry optimizations with a fixed value of the proton donor-acceptor C–O distance R were performed for a series of R values. The electronic energies U(R) obtained for the reactant state from these constrained optimizations were used to compute the probability distribution function P(R), as in Eq. (4). Furthermore, at each value of R, an average geometry in between the optimized reactant and product geometries, corresponding approximately to the crossing point between the reactant and product states along an inner-sphere solute coordinate, was obtained. For each of these average structures, a one-dimensional proton potential energy curve for the reactant and product states (i.e., for the anionic and neutral states, respectively) was computed. Finally, the one-dimensional Schrödinger equation for a proton moving in the reactant and product potentials was solved numerically to provide reactant and product proton vibrational energy levels ϵμ and ϵv and their corresponding wavefunctions. The overlap integrals Sμv(R) were computed numerically from these reactant and product proton vibrational wavefunctions.
The reorganization energy λ was calculated as the sum of the inner-sphere reorganization energy31–32 λi, which is associated with the solute and oxidant, and the outer-sphere or solvent reorganization energy λs. The computed inner-sphere reorganization energies ranged from 27.4 kcal/mol for (o-Flr)(p-NH2)C6H3COO− to 29.6 kcal/mol for (o-Flr)(p-CF3)C6H3COO−. The large inner-sphere reorganization energy computed for the fluorenyl-benzoates can be explained by a significant rotation of the sizeable fluorenyl fragment due to the change in hybridization of the donor carbon from sp3 in the reactant to sp2 in the product. The inner-sphere reorganization energies for the oxidants were relatively small: 0.2 kcal/mol for FeCp*2+ and 2.6 kcal/mol for N(ArOMe)3•+. The solvent reorganization energy was estimated based on the Marcus two-sphere model22 and was found to be 21.2–21.4 and 18.6–18.8 kcal/mol for FeCp*2+ and N(ArOMe)3•+, respectively. The total reorganization energies ranged from 48.9 kcal/mol for FeCp*2+ and 48.6 kcal/mol for N(ArOMe)3•+ in the case of (o-Flr)(p-NH2)C6H3COO− to 51.0 kcal/mol for FeCp*2+ and 50.8 kcal/mol for N(ArOMe)3•+ in the case of (o-Flr)(p-CF3)C6H3COO−. Because the total reorganization energy is ~50 kcal/mol (Table S12) for all cases, we assumed it to be the same (i.e., 50 kcal/mol) for all oxidants and all substituted fluorenyl-benzoates to retain simplicity. Moreover, as the two-sphere model can overestimate the solvent reorganization energy, we computed the PCET rate constants for two different values of the total reorganization energy λ: 50 kcal/mol, which is the computed value, and 30 kcal/mol, which represents the approximate inner-sphere reorganization energy for all fluorenyl-benzoate molecules studied. The general conclusions are the same for both values of the total reorganization energy.
As shown in our previous work, the effect of changing the oxidant can be simulated by varying the reaction free energy ΔG0 for a given fluorenyl-benzoate.16 The range of ΔG0 corresponding to the experimental conditions of these reactions was established using experimentally determined redox potentials of the oxidants and the reaction free energy of the oxidation of (o-Flr)(p-H)C6H3COO− by FeCp*2+. The latter was estimated from experimentally measured effective bond dissociation free energies and used as a reference for reactions with other oxidants. Similarly, to determine ΔG0 for the oxidation of para-substituted fluorenyl-benzoates, we computed the reaction free energy for each (o-Flr)(p-X)C6H3COO− species and added a correction corresponding to the difference between the experimentally determined and calculated reaction free energies for (o-Flr)(p-H)C6H3COO−. For the analysis below, these free energies are computed relative to the species with the electron-withdrawing substituent CF3:
| (5) |
Table S1 provides the ΔΔG0 values for the series of para-substituted fluorenyl-benzoates (o-Flr)(p-X)C6H3COO−.
The experimentally measured second-order rate constant is the product of the equilibrium constant for the formation of a reactive complex between the fluorenyl-benzoate and the oxidant and the first-order PCET rate constant kPCET within this complex. The equilibrium constant is expected to be dominated by electrostatic interactions between the positively charged oxidant and the negatively charged fluorenyl-benzoate. Therefore, this equilibrium constant is not expected to be significantly influenced by different substituents on the fluorenyl-benzoate or by different oxidants, as long as the charge on each remains the same. Thus, our analysis assumes that the equilibrium constant for the formation of the reactive complex remains constant and does not impact the relative rate constants studied herein. Although this approximation appears to be reasonable for these systems, it is important to note that the variations in the experimentally observed rate constants may not be due solely to the concerted PCET step but could potentially be influenced by the bimolecular association of the reactants in solution.
Results and Discussion
The use of oxidants with different redox potentials, Eox, changes the electron transfer (ET) component of the free energy of the PCET reaction. To investigate the impact of changing the oxidant for a given fluorenyl-benzoate, we calculated the PCET rate constant as a function of the driving force. Figure 2 depicts the correlation between log kPCET and ΔG0/(2.303RT)for four different para-substituents computed with two different values of the total reorganization energy (30 and 50 kcal/mol). The computed Brønsted α, defined as the slope of log kPCET versus log Keq, or equivalently, the scaled free energy ΔG0/(2.303RT), is higher than the experimental range of 0.19–0.22. However, the Brønsted α values obtained from the PCET theory are significantly lower than the value of ~0.5 typically observed for hydrogen atom transfer reactions involving C–H bonds33 and PCET reactions involving polar O–H or N–H bonds34–36 and thus qualitatively reproduce the experimental observations. The Brønsted α is slightly lower for the electron-donating substituents (i.e., NH2 and OMe) than for H and the electron-withdrawing substituent (i.e., CF3), although these differences are smaller than the precision of the calculations and the experiments. As the results do not depend significantly on the reorganization energy within this physically reasonable range, we use λ = 30 kcal/mol for the remainder of the analysis.
Figure 2.

Computed correlation between the logarithm of the PCET rate constant and the scaled reaction free energy for λ=30 kcal/mol (left) and λ=50 kcal/mol (right). The rate constants were computed for the reaction free energy ranging from −1.4 to −27 kcal/mol, the estimated range of the driving forces for the oxidants used in the experiments.6–7
For a given oxidant, such as FeCp*2+, changing the functional group X at the position para- to the carboxylate group significantly affects the proton transfer (PT) component of the free energy of the PCET reaction. The dependence of the PCET rate constant kPCET on the electronic properties of the substituent can be explained in terms of a general PCET reaction involving an external oxidant Ox+, an acid A–H, and a base B−:
For the oxidative PCET of fluorenyl-benzoates, A—H is associated with the C—H bond of the fluorenyl fragment, and B—H is associated with the O—H bond of the benzoate fragment (Figure 1). The reaction free energy ΔG0 is a function of Eox, the redox potential of Ox+, as well as EEPT, the proton-coupled redox potential of the fluorenyl fragment A—H, corresponding to the removal of an electron and the transfer of a proton to B−. Thus, the driving force, −ΔG0, corresponding to the oxidation of these fluorenyl-benzoates can be expressed as
| (6) |
where F is the Faraday constant.
To simplify the analysis, we express the total rate constant given by the integral in Eq. (3) in terms of the dominant proton donor-acceptor distance R*, which is defined as the distance corresponding to the maximum of the integrand. Specifically, the total rate constant is expressed as the product of the integrand k(R)P(R) at R* and an effective width δR:
| (7) |
Combining Eqs. (3), (4), and (7), the natural logarithm of the total PCET rate constant can be expressed as
| (8) |
Moreover, U(R) can be treated as a function of EEPT because both quantities depend on the Hammett constant σ that characterizes the electronic properties of the substituent and thus influences the basicity of the benzoate. Assuming that δR is also a function of Eox and EEPT (or, equivalently, the Hammett constant of the substituent), the total PCET rate constant kPCET can be expressed as a function of Eox and EEPT.
To compute the Brønsted α associated with a series of oxidants for a given para-substituent on the fluorenyl-benzoate, we calculate the partial derivative of log kPCET with respect to log Keq at fixed EEPT. This partial derivative, which is denoted αox, can be expressed as
| (9) |
because the normalization factor CN and the potential energy U(R) only depend on the properties of the fluorenyl-benzoate and not on the oxidant. Note that R* is also assumed to be approximately independent of the oxidant, and this assumption is shown to be reasonable in Table S9. To compute the Brønsted α associated with a series of para-substituents for a given oxidant, we calculate the partial derivative of log kPCET with respect to log Keq at fixed Eox. This partial derivative, which is denoted ασ, can be expressed as
| (10) |
Eqs. (9) and (10) describe the free energy relationships between the logarithm of the PCET rate constant and the scaled driving force for changes in oxidant strength and changes in the para-substituent, respectively.
The Brønsted α for changing the oxidant, αox, is estimated by the first term in Eq. (9), which was shown in previous work16 to be:
| (11) |
where ωμv is the percentage contribution from the reactant/product vibronic state pair (μ,v) to the overall rate constant. Note that the slope given by Eq. (11) depends on ΔG0 and therefore on Eox and EEPT, leading to a range of Brønsted α values for different oxidants and para-substituents. With total reorganization energy λ = 30 kcal mol−1, Eq. (11) produces an estimated Brønsted α of 0.27–0.38 (Table S17) for the oxidant FeCp2+, where the range reflects the differences among the substituents. These values are in reasonable agreement with the slopes of the linear fits of the Brønsted plots (0.31–0.37) obtained using the full PCET rate constant expression given in Eq. (3) (Figure 2). This agreement indicates that the second term in Eq. (9) is small, i.e., the effective width δR varies only slightly with the oxidant potential Eox.
Similarly, the Brønsted α for changing the substituent, ασ, is estimated as the sum of the first two terms of Eq. (10):
| (12) |
The first term in Eq. (12) is the same as the only term in Eq. (11) with a different quantity fixed. Because this term depends on ΔG0, which in turn depends on both Eox and EEPT, in practice it is computed with both quantities fixed. Thus, ασ can be estimated as the sum of αox, which is estimated by Eq. (11), and an additional second term, . Table 1 provides the Eox equilibrium reactant proton donor-acceptor distance, R0, the dominant proton donor-acceptor distance, R*, the reactant potential energy at R* relative to that at R0, U(R*), and the relative reaction free energy, ΔΔG0, as defined in Eq. (5), for each substituent. Using a total reorganization energy of λ = 30 kcal mol−1, Eq. (12) produces an estimated Brønsted α of 0.46–0.53 for the oxidant FeCp2+, where the range reflects the differences among the four substituents. The Brønsted plot obtained using the full PCET rate constant expression given in Eq. (3) for the four para-substituents with the oxidant FeCp2+ produces a linear fit with a slightly larger slope that is in good agreement with the experimentally measured value of 0.58±0.10 (Figure 3). This deviation provides an indication of the magnitude of the last two terms of Eq. (10), which are small but not negligible.
Table 1.
Parameters and Estimated Brønsted α for Changing the Substituent with FeCp2+as the Oxidant and λ=30 kcal/mol
| Substituent X | R0, Å | R*, Åa | U(R*),b kcal/mol | ΔΔG0,c kcal/mol | d | c | Brønsted α (fixed Eox)e |
|---|---|---|---|---|---|---|---|
| NH2 | 2.96 | 2.69 | 1.81 | −7.38 | 0.27 | 0.22 | 0.49 |
| OMe | 2.97 | 2.68 | 2.08 | −5.69 | 0.30 | 0.23 | 0.53 |
| H | 3.03 | 2.65 | 2.96 | −4.03 | 0.35 | 0.11 | 0.46 |
| CF3 | 3.07 | 2.64 | 3.39 | 0.00 | 0.37 | N/A | N/A |
The dominant proton-donor acceptor distance R* was determined as the position of the maximum of k(R)P(R) fitted to Gaussian distribution (see SI for more details).
U(R*) was determined by cubic spline interpolation of U(R), and U(R*) is defined relative to U(R0) (i.e., U(R0) = 0). Physically, U(R*) is the energy required to access the dominant proton donor-acceptor distance from the equilibrium distance in the reactant state.
ΔΔG0 and ΔU(R*) are defined to be computed relative to their values for X = CF3, i.e., ΔΔG0 =ΔG0(X) -ΔG0(CF3) and ΔU(R*) =ΔU(R*, X) - ΔU(R*, CF3).
This term is equivalent to the Brønsted α for changing the oxidant, evaluated using Eq. (11) with ΔG0 for the fluorenyl-benzoate with substituent X, the FeCp2+ oxidant, and λ = 30 kcal/mol. The individual contributions are given in Table S11.
The Brønsted α for changing the substituent, evaluated using Eq. (12) with ΔG0 for the fluorenyl-benzoate with substituent X, the FeCp2+ oxidant, and λ = 30 kcal/mol. Each value in this column is the sum of the values in the previous two columns, where the second term in Eq. (12) is approximated by ΔU(R*)/ΔΔG0. Note that the accuracy of the various components in this table depends on factors such as the DFT functional and the solvent model, and the accuracy of the Brønsted α also depends on approximations underlying the vibronically nonadiabatic PCET theory. Thus, the qualitative trends are more meaningful than the quantitative values.
Figure 3.

Computed and experimental correlation between the logarithm of the PCET rate constant and the scaled free energy for four different para-substituents with the oxidant FeCp2+ for λ = 30 kcal/mol. Note that the calculations used the experimentally determined driving force for the H substituent as a reference and the experimentally determined rate constant for the CF3 substituent as a reference to clearly illustrate the slopes.
The Brønsted α for changing the para-substituent can be understood in terms of the electron-withdrawing or electron-donating properties of the substituents. The electron-donating groups (NH2 and OMe) decrease the equilibrium reactant proton donor-acceptor distance R0 because of additional electron density at the carboxylate oxygen that enhances its basicity and strengthens the hydrogen-bonding interaction with the C—H group. Conversely, the electron-withdrawing group CF3 increases this equilibrium distance R0. The substituents exert the opposite effect to a lesser extent on the dominant distance R*, which corresponds to the maximum of k(R)P(R). The dominant distance is determined by a balance among many factors that are not easily disentangled, preventing a straightforward interpretation of this trend. The most important trend is that the difference between R* and R0 increases as the substituent becomes less electron-donating or more electron-withdrawing. As a result, more energy is required to access the dominant proton donor-acceptor distance, leading to a larger U(R*), for the more electron-withdrawing substituent (see Figure S1). Furthermore, the reaction free energy for proton transfer is lower (i.e., proton transfer is more thermodynamically favorable) for the electron-donating substituents, leading to a larger ΔΔG0 for the more electron-withdrawing (and less electron-donating) substituents. On the basis of these trends across the series of substituents, ΔU(R*)/ΔΔG0, which approximates in Eq. (12), is positive, as indicated by the calculated data in Table 1. As a result of this additional positive term, the Brønsted α is larger when changing the substituent (Figure 3) compared to its value when changing the oxidant (Figure 2).
A simpler explanation of this phenomenon is that the Brønsted α is approximated by the derivative of an effective free energy barrier with respect to the driving force, and the effective free energy barrier is the sum of two terms. The first term depends on both the oxidant and the benzoate substituent, which are associated with the ET and PT driving forces, respectively. The second term is related to the energy required to access shorter proton donor-acceptor distances that allow proton transfer and depends on only the benzoate substituent, which impacts the proton transfer interface. The sum of the two terms leads to a larger Brønsted α associated with varying the substituent compared to varying the oxidant because the second term only contributes to the former.
Conclusion
This work provides an explanation for the greater sensitivity of the PCET rate constant of these fluorenyl-benzoate systems to the carboxylate basicity than to the redox potential of the oxidant. Enhancing the basicity of the carboxylate not only lowers the reaction free energy for proton transfer, making it more thermodynamically favorable, but also decreases the equilibrium proton donor-acceptor distance, making the shorter proton donor-acceptor distances required for proton transfer more energetically accessible. This combined impact of changing the basicity of the carboxylate is greater than the impact of changing the redox potential of the oxidant, which influences the reaction free energy for electron transfer but not the proton donor-acceptor distance. An important distinction between electron transfer and proton transfer or PCET reactions is the much stronger dependence of proton transfer and PCET on the proton donor-acceptor distance. This analysis highlights the complexity of PCET reactions and the variety of strategies that can be used to tune PCET rate constants. In particular, altering the proton donor-acceptor distance provides an additional route for tuning the rates of PCET reactions compared to pure electron transfer reactions. These fundamental insights have broad implications for developing new strategies to activate C–H bonds, specifically by designing systems with shorter equilibrium proton donor-acceptor distances.
Supplementary Material
Acknowledgment
We thank Jim Mayer, Julia Darcy, Zach Goldsmith, and Alexander Soudackov for helpful discussions. This work was supported by the National Institutes of Health Grant GM056207.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Computational details and supplementary tables and figures.
References
- 1.Labinger JA; Bercaw JE, Understanding and Exploiting C-H Bond Activation. Nature 2002, 417, 507–14. [DOI] [PubMed] [Google Scholar]
- 2.Giri R; Shi BF; Engle KM; Maugel N; Yu JQ, Transition Metal-Catalyzed C-H Activation Reactions: Diastereoselectivity and Enantioselectivity. Chem. Soc. Rev 2009, 38, 3242–72. [DOI] [PubMed] [Google Scholar]
- 3.Hartwig JF, Evolution of C-H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc 2016, 138, 2–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newton CG; Wang SG; Oliveira CC; Cramer N, Catalytic Enantioselective Transformations Involving C-H Bond Cleavage by Transition-Metal Complexes. Chem. Rev 2017, 117, 8908–8976. [DOI] [PubMed] [Google Scholar]
- 5.Gandeepan P; Muller T; Zell D; Cera G; Warratz S; Ackermann L, 3d Transition Metals for C-H Activation. Chem. Rev 2019, 119, 2192–2452. [DOI] [PubMed] [Google Scholar]
- 6.Markle TF; Darcy JW; Mayer JM, A New Strategy to Efficiently Cleave and Form C-H Bonds Using Proton-Coupled Electron Transfer. Sci. Adv 2018, 4, eaat5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darcy JW; Kolmar SS; Mayer JM, Transition State Asymmetry in C-H Bond Cleavage by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2019, 141, 10777–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stubbe J; Nocera DG; Yee CS; Chang MC, Radical Initiation in the Class I Ribonucleotide Reductase: Long-Range Proton-Coupled Electron Transfer? Chem. Rev 2003, 103, 2167–201. [DOI] [PubMed] [Google Scholar]
- 9.Hammes-Schiffer S; Soudackov AV, Proton-Coupled Electron Transfer in Solution, Proteins, and Electrochemistry. J. Phys. Chem. B 2008, 112, 14108–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dempsey JL; Winkler JR; Gray HB, Proton-Coupled Electron Flow in Protein Redox Machines. Chem. Rev 2010, 110, 7024–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ, Proton-Coupled Electron Transfer. Chem. Rev 2012, 112, 4016–93. [DOI] [PubMed] [Google Scholar]
- 12.Bourrez M; Steinmetz R; Ott S; Gloaguen F; Hammarstrom L, Concerted Proton-Coupled Electron Transfer from a Metal-Hydride Complex. Nat. Chem 2014, 7, 140–5. [DOI] [PubMed] [Google Scholar]
- 13.Hammes-Schiffer S, Proton-Coupled Electron Transfer: Moving Together and Charging Forward. J. Am. Chem. Soc 2015, 137, 8860–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller DC; Tarantino KT; Knowles RR, Proton-Coupled Electron Transfer in Organic Synthesis: Fundamentals, Applications, and Opportunities. Top Curr Chem (Cham) 2016, 374, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu G; Knowles RR, Rate-Driving Force Relationships in the Multisite Proton-Coupled Electron Transfer Activation of Ketones. J. Am. Chem. Soc 2019, 141, 2721–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sayfutyarova ER; Goldsmith ZK; Hammes-Schiffer S, Theoretical Study of C-H Bond Cleavage Via Concerted Proton-Coupled Electron Transfer in Fluorenyl-Benzoates. J. Am. Chem. Soc 2018, 140, 15641–15645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soudackov A; Hammes-Schiffer S, Derivation of Rate Expressions for Nonadiabatic Proton-Coupled Electron Transfer Reactions in Solution. J. Chem. Phys 2000, 113, 2385–2396. [Google Scholar]
- 18.Soudackov A; Hatcher E; Hammes-Schiffer S, Quantum and Dynamical Effects of Proton Donor-Acceptor Vibrational Motion in Nonadiabatic Proton-Coupled Electron Transfer Reactions. J. Chem. Phys 2005, 122, 14505. [DOI] [PubMed] [Google Scholar]
- 19.Hammes-Schiffer S; Stuchebrukhov AA, Theory of Coupled Electron and Proton Transfer Reactions. Chem. Rev 2010, 110, 6939–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huynh MT; Mora SJ; Villalba M; Tejeda-Ferrari ME; Liddell PA; Cherry BR; Teillout A-L; Machan CW; Kubiak CP; Gust D; Moore TA; Hammes-Schiffer S; Moore AL, Concerted One-Electron Two-Proton Transfer Processes in Models Inspired by the Tyr-His Couple of Photosystem Ii. ACS Cent. Sci 2017, 3, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcus RA, Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem 1964, 15, 155–196. [Google Scholar]
- 22.Marcus RA, On the Theory of Oxidation‐Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys 1956, 24, 966–978. [Google Scholar]
- 23.Becke AD, Density-Functional Thermochemistry. Iii. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- 24.Lee C; Yang W; Parr RG, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 25.Hehre WJ; Ditchfield R; Pople JA, Self-Consistent Molecular Orbital Methods. Xii. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys 1972, 56, 2257–2261. [Google Scholar]
- 26.Clark T; Chandrasekhar J; Spitznagel GW; Schleyer PVR, Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. Iii. The 3–21+G Basis Set for First-Row Elements, Li–F. J. Comput. Chem 1983, 4, 294–301. [Google Scholar]
- 27.Hariharan PC; Pople JA, The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- 28.Miertuš S; Scrocco E; Tomasi J, Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of Ab Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys 1981, 55, 117–129. [Google Scholar]
- 29.Tomasi J; Mennucci B; Cammi R, Quantum Mechanical Continuum Solvation Models. Chem. Rev 2005, 105, 2999–3093. [DOI] [PubMed] [Google Scholar]
- 30.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich A; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Jr JAM; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Keith T; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ, Gaussian 09 Revision D.01. 2013. [Google Scholar]
- 31.Nelsen SF; Blackstock SC; Kim Y, Estimation of Inner Shell Marcus Terms for Amino Nitrogen Compounds by Molecular Orbital Calculations. J. Am. Chem. Soc 1987, 109, 677–682. [Google Scholar]
- 32.Auer B; Fernandez LE; Hammes-Schiffer S, Theoretical Analysis of Proton Relays in Electrochemical Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2011, 133, 8282–92. [DOI] [PubMed] [Google Scholar]
- 33.Mayer JM, Hydrogen Atom Abstraction by Metal–Oxo Complexes: Understanding the Analogy with Organic Radical Reactions. Acc. Chem. Res 1998, 31, 441–450. [Google Scholar]
- 34.Morris WD; Mayer JM, Separating Proton and Electron Transfer Effects in Three-Component Concerted Proton-Coupled Electron Transfer Reactions. J. Am. Chem. Soc 2017, 139, 10312–10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markle TF; Rhile IJ; Dipasquale AG; Mayer JM, Probing Concerted Proton-Electron Transfer in Phenol-Imidazoles. Proc. Natl. Acad. Sci. U S A 2008, 105, 8185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhile IJ; Markle TF; Nagao H; DiPasquale AG; Lam OP; Lockwood MA; Rotter K; Mayer JM, Concerted Proton-Electron Transfer in the Oxidation of Hydrogen-Bonded Phenols. J. Am. Chem. Soc 2006, 128, 6075–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.