

Abstract
The objective of this study was to investigate the processability of AquaSolve™ hydroxypropyl methylcellulose acetate succinate L grade (HPMCAS LG) via hot-melt extrusion and to examine the effect of pressurized carbon dioxide (P-CO2) on the physicomechanical properties of efavirenz (EFA)-loaded extrudates. To optimize the process parameters and formulations, various physical mixtures of EFA (30%, 40%, and 50%, w/w) and HPMCAS LG (70%, 60%, and 50%, w/w), respectively, were extruded using a co-rotating twin-screw extruder with a standard screw configuration, with P-CO2 injected into zone 8 of the extruder. Thermal characterization of the extrudates was performed using differential scanning calorimetry and thermogravimetric analysis. Scanning electron microscopy was employed to study the morphology and porosity of the formulations. Notably, the macroscopic morphology changed to a foam-like structure by P-CO2 injection resulting in an increased specific surface area, porosity, and dissolution rate. Thus, HPMCAS LG extrusion, coupled with P-CO2 injection, yielded faster dissolving extrudates. Stability studies indicated that HPMCAS LG was able to physically and chemically stabilize the amorphous state of high-dose EFA. Furthermore, the milling efficiency of the extrudates produced with P-CO2 injection improved because of their increased porosity.
Keywords: Hot-melt extrusion, Pressurized carbon dioxide, HPMCAS LG, Milling efficiency, Porosity
Graphical Abstract
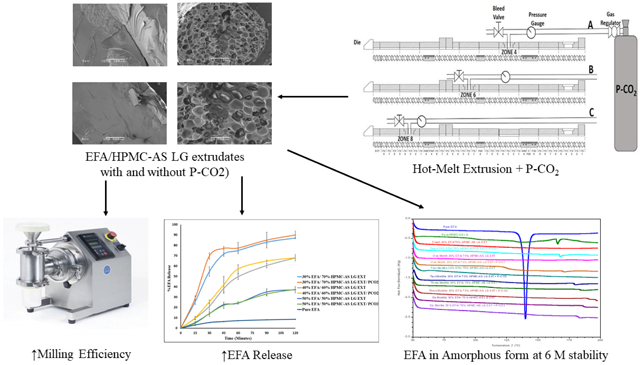
1. Introduction
Over a period of more than two decades, hot-melt extrusion (HME) has evolved into one of the most promising pharmaceutical processing technologies [1]. HME has several advantages over conventional pharmaceutical processing technologies, such as a short processing time and the capacity for continuous processing, which does not require the use of water or potentially toxic organic solvents [2,3]. HME is generally used for the solubility enhancement of active pharmaceutical ingredients (APIs) [4] and is suitable for manufacturing various dosage forms, such as pellets, tablets, and transmucosal products, or for various applications, such as controlled release formulations, targeted drug delivery, and taste-masking formulations [5,6].
Recently, several HME products have been commercially approved and marketed, resulting in a further increase of interest in this technology in the pharmaceutical industry. However, many pharmaceutical polymeric systems experience thermal and/or viscoelastic challenges during HME, which cannot be ignored. One thermal challenge is material degradation during processing, and a viscoelastic challenge is the feasibility of extrusion. Moreover, APIs exhibit their own individual limitations, such as high melting temperatures (Tms) and/or low degradation temperatures [7]. Physicochemical properties of APIs, including glass transition and melting point temperatures, degradation temperatures, and miscibility or solubility in a polymer carrier, have to be carefully considered since they have significant effects on HME processes as well as on the final product [8–11].
Polymeric carriers are usually the largest component in amorphous dispersion formulations. Thus, physicochemical properties of the polymer carrier, such as the molecular weight, viscosity, and glass transition temperature (Tg), can significantly influence HME processing conditions and the performance of the final drug products [12,13]. The degradation temperature and viscoelastic properties of polymers are major factors that must be considered for the HME process [14]. Therefore, determination of thermal and viscoelastic properties of various drug/polymer mixtures helps identify the optimal HME processing parameters (extrusion temperature, screw speed, feeding rate, motor load, and screw design) and provide insights into the properties of the extrudate [15].
More than 50% of common pharmaceutical polymers used for HME cannot be processed without a processing aid because these polymers have viscosities exceeding the defined maximum viscosity limit within the HME processing window. Often, the added API can plasticize polymers, which reduces viscosity of the complex and leads to smooth extrusion. If API cannot adequately plasticize polymeric carriers, alternative formulation or processing strategies are necessary to utilize these carriers [7].
One such strategy is the addition of plasticizers [16]. Typically, plasticizers increase the free volume between polymer chains, causing decreases in the Tg and melt viscosity [17]. Normally, plasticizers are utilized at a concentration of 5–30% (w/w) by being physically mixed with a pre-extrusion blend [18–20]. Plasticizer addition, therefore, results in significantly larger dosage forms [16]. Plasticizers also remain mobile and can lead to further physical and chemical changes, as well as to instability of the final drug product. Therefore, it would be preferable to find an alternative to traditional plasticizers for pharmaceutical extrusion. Recently, supercritical carbon dioxide (sc-CO2) and subcritical (pressurized) carbon dioxide (P-CO2) have been investigated as temporary plasticizers [21,22]. However, it has been observed that sc-CO2 and P-CO2 act as foaming agents [23], which increase the surface area and porosity of polymers, resulting in enhanced dissolution [24].
Hydroxypropyl methylcellulose acetate succinate (HPMCAS) has been shown to be a very effective crystallization inhibitor in amorphous solid dispersions (ASDs) [25,26]. HPMCAS polymers have a Tg of 120 °C but regularly require extrusion temperatures in excess of 170 °C, even with plasticization used to reduce the high motor torque, due to high melt viscosity [27]. HPMCAS L grade (LG) has the highest concentration of succinoyl groups compared with that in other grades (MG and HG), which results in a higher hydrophilicity. Succinoyl groups also play an important role in stabilizing ASDs by inducing high-affinity binding to the hydrophobic drug surface, leading to crystal growth inhibition of an API [28]. However, it has been reported that the succinoyl group shows sensitivity to a high temperature (above 160 °C) during the extrusion process by producing a free acid, which corresponds to a decrease in the succinoyl content. Considering the above factors, HPMCAS LG was selected as a polymer to investigate the effects of physical and mechanical properties of physical mixtures of a model API and HPMCAS LG on drug solubility and stability. The suitability of the combination and a correlation between drug/polymer properties and the HME process were assessed in this study. In addition, the effect of carbon dioxide as a foaming agent and its impact on the physicomechanical properties and performance of the extrudates were investigated.
2. Materials and methods
2.1. Materials
AquaSolve™ HPMCAS LG and efavirenz (EFA) were donated by Ashland Specialty Ingredients (Wilmington, DE). Carbon dioxide (CO2) was supplied in gas cylinders (pure clean) by Airgas (Tupelo, MS). All other chemicals and reagents used in the present study were of analytical grade and obtained from Fisher Scientific (Fair Lawn, NJ).
2.2. Methods
2.2.1. Thermogravimetric analysis
Thermogravimetric analysis (TGA) was performed for EFA and HPMCAS LG to determine the optimal processing temperatures for extrusion and stability studies using a Pyris 1 thermogravimetric analyzer with the Pyris manager software (PerkinElmer Life and Analytical Sciences, Shelton, CT). Approximately 5–7 mg of samples (API and polymer) was weighed in an aluminum pan and heated from 25 °C to 200 °C at 10 °C/min under a nitrogen atmosphere.
2.2.2. Hot-melt extrusion
A co-rotating twin-screw intermeshing extruder (16-mm Prism Euro Lab; Thermo Fisher Scientific, Waltham, MA) was used to perform HME. The extruder is divided into 10-barrel zones adjacent to the gravimetric feeder. The standard manufacturer’s screw configuration was used for this study, which consists of four conveying zones and three mixing zones. P-CO2 was injected into the extruder using a high-pressure regulator connected to a flexible stainless-steel hose with armor casing. The other end of the hose was connected to a four-way connection and fitted with a pressure gauge, bleed valve, and check valve (ball type for a unidirectional flow of gas), with the latter connected to an injection port at zone 8 (conveying zone) of the extruder (Fig. 1). The P-CO2 flow rate was regulated using the regulator knob. Preliminary extrusion experiments were conducted to determine the processing temperature for the pure polymer over a temperature range of 140 to 190 °C at 75 and 100 rpm (Table 1). Based on these experiments, extrusion in the presence of P-CO2 was studied at 190 °C. Formulations were extruded at a drug load of 20%, 30%, 40%, and 50%, with and without P-CO2.
Figure 1.
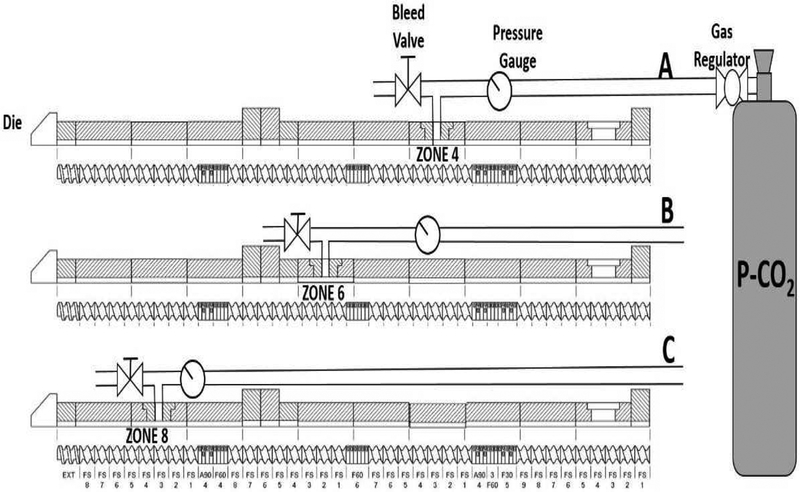
Schematic representation for the set-up of the combined HME/P-CO2 techniques; Injection of P-CO2 at (A) Zone 4; (B) Zone 6; (C) Zone 8
Table 1.
The preliminary processing trials of HPMC-AS LG polymer by hot-melt extrusion
| Formulations | Processing Temperature (°C) |
Screw Speed (rpm) |
Barrel Torque (%) |
|---|---|---|---|
| 100% HPMC-AS LG | 140 | 100 | Maximum |
| 100% HPMC-AS LG | 140 | 75 | Maximum |
| 100% HPMC-AS LG | 140 | 100 | Maximum |
| 100% HPMC-AS LG | 140 | 75 | Maximum |
| 100% HPMC-AS LG | 150 | 100 | Maximum |
| 100% HPMC-AS LG | 150 | 75 | Maximum |
| 100% HPMC-AS LG | 150 | 100 | Maximum |
| 100% HPMC-AS LG | 150 | 75 | Maximum |
| 100% HPMC-AS LG | 160 | 100 | 98 |
| 100% HPMC-AS LG | 160 | 75 | 88 |
| 100% HPMC-AS LG | 170 | 100 | 88-90 |
| 100% HPMC-AS LG | 170 | 75 | 82 |
| 100% HPMC-AS LG | 190 | 100 | 84-92 |
| 100% HPMC-AS LG | 190 | 75 | 80-90 |
| 100% HPMC-AS LG | 190 | 100 | 84-92 |
| 100% HPMC-AS LG | 190 | 75 | 80-90 |
2.2.3. Preparation of physical mixtures
After optimizing the parameters of the HME process, various physical mixtures were prepared with the following composition: EFA (30, 40, and 50%, w/w) and HPMCAS LG (70, 60, and 50%, w/w), respectively. The mixtures were blended using a V-shell blender ((MaxiBlend™, GlobePharma, North Brunswick, NJ) at 25 rpm for 15 minutes. All the formulations listed in (Table 2) were successfully extruded at the selected processing conditions.
Table 2.
Conditions and outcomes for optimized formulations during HME processes
| Formulations | Processing Temperature (°C) |
Screw Speed (rpm) |
Barrel Torque (%) |
P-CO2 (PSI) |
P-CO2 Injection Zone |
|---|---|---|---|---|---|
| 30% EFA/ 70% HPMC-AS LG EXT | 140 | 100 | 46 | 300-400 | Zone 8 |
| 30% EFA/ 70% HPMC-AS LG EXT | 140 | 100 | 46 | - | - |
| 40% EFA/ 60% HPMC-AS LG EXT | 140 | 100 | 33 | 300-400 | Zone 8 |
| 40% EFA/ 60% HPMC-AS LG EXT | 140 | 100 | 33 | - | - |
| 50% EFA/ 50% HPMC-AS LG EXT | 140 | 100 | 27 | 300-400 | Zone 8 |
| 50% EFA/ 50% HPMC-AS LG EXT | 140 | 100 | 27 | - | - |
| 30% EFA/ 70% HPMC-AS LG EXT* | 150 | 75 | 25-30 | 250-300 | Zone 8 |
| 30% EFA/ 70% HPMC-AS LG EXT* | 150 | 75 | 25-30 | - | - |
| 40% EFA/ 60% HPMC-AS LG EXT* | 150 | 75 | 20-25 | 250-300 | Zone 8 |
| 40% EFA/ 60% HPMC-AS LG EXT* | 150 | 75 | 20-25 | - | - |
| 50% EFA/ 50% HPMC-AS LG EXT* | 150 | 75 | 13-20 | 250-300 | Zone 8 |
| 50% EFA/ 50% HPMC-AS LG EXT* | 150 | 75 | 13-20 | - | - |
These formulations were selected for further studies.
2.2.4. Milling efficiency analysis
Prior to the analysis, HME extrudates were milled using a laboratory-scale FitzMill (model L1A; Fitzpatrick, Perth Amboy, NJ), and the particle size distribution and milling efficiency were studied using a vibrating sieve shaker (Performer III SS-3; Gilson). The fraction below 600 μm was used for further investigation. To study the milling efficiency of formulations before and after P-CO2 injection, 25 g of each melt-extruded sample was milled for 3 min at 3,000 rpm, and the particle size distribution was estimated using a vibrating sieve method. A set of sieves with known mesh sizes (75, 125, 250, 420, 600, and 840 μm) and known tare weights were stacked on one another, and a known amount of the powder was placed in the top sieve. The whole stack was placed on a vibrating plate at an amplitude of 1.5 mm for 10 minutes, after which each sieve was weighed to obtain the average distribution of particle sizes.
2.2.5. Drug content analysis
Assay methods detailed in the United States Pharmacopeia (USP) were used. The EFA content in extrudates was determined by dissolving extrudates in a phosphate buffer solution (pH 6.8) and performing ultraviolet (UV) spectrophotometric analysis against a blank buffer solution. The analysis was performed using a UV spectrophotometer (Genesys 6; Thermo Fisher Scientific, Madison, WI) at a wavelength of 247 nm, at which absorbance of the polymer is negligible. The measured drug content was compared to the calculated value. The experimental value was an average of triplicate measurements.
2.2.6. Dissolution studies
In vitro dissolution testing was conducted on milled and #40-sieved extrudate samples to investigate the influence of P-CO2 injection to the polymer matrix on drug release from HPMCAS LG extrudates. The results were compared with those obtained for pure crystalline EFA. A USP dissolution apparatus II (Hanson SR8; Hanson Research, Chatsworth, CA) was used with a paddle rotation speed of 50 rpm, and samples were incubated in pH 6.8 phosphate buffer at 37 ± 0.5 °C for 2 h. At predetermined time intervals, 2-mL aliquots were collected and replaced with an equal volume of a fresh dissolution medium. The aliquots withdrawn were filtered through a 10-μm filter, and EFA was measured by UV spectrophotometry (Genesys 6; Thermo Fisher Scientific, Madison, WI) at 247 nm. All dissolution tests were carried out in triplicate, and the mean ± standard deviation was determined.
2.2.7. Differential scanning calorimetry
The solid-state nature of pure EFA, pure HPMCAS LG, their physical mixtures, and the corresponding extrudates was assessed by differential scanning calorimetry (DSC) to determine the impact of P-CO2 on thermal properties during formulation processing. The analysis was preformed using a differential scanning calorimeter (DSC 25 Discovery series; TA Instruments, New Castle, DE) coupled with a refrigerated cooling system. Samples of 4–8 mg were weighed using a Mettler Toledo scale and then placed in non-perforated aluminum pans, which were crimped before testing, with an empty crimped aluminum pan used as a reference cell. Calorimetry scans were carried out from 50 to 200 °C at a scanning rate of 10 °C/min. Volatiles were removed from the purging head with a nitrogen flow at 20 mL/min. Calibration of the instrument was performed using indium as the standard. After each scan completed, the melting points were analyzed, and the Trois® manager software was used for data analysis.
2.2.8. Powder X-ray diffraction measurements
The degree of crystallinity of pure EFA, pure HPMCAS LG, EFA in physical mixtures, and the respective extrudates was investigated using powder X-ray diffraction (XRD) with a Bruker D8 Focus X-ray diffractometer, operated at a voltage of 40 kV and a current of 40 mA. The powder was packed into a sample holder. Data for each sample were collected in a 2θ angle range of 4–40° over 10 min in a continuous detector scan mode. A scanning step of 0.02° and a scanning step duration of 0.3 s were used.
2.2.9. Scanning electron microscopy (SEM)
Morphological characteristics and the degree of porosity of extrudates with different drug loads (30, 40, and 50%), with and without P-CO2 treatment, were studied using a scanning electron microscope (JEOL JSM-5600) at a 25-, 50-, and 100-fold magnification and an accelerating voltage of 5 kV. Each specimen was fixed using a conductive, double-sided carbon adhesive tape and coated with gold using a Hummer® 6.2 sputtering system (Anatech, Ltd., Springfield, VA) in a high-vacuum evaporator prior to the test to avoid electrostatic charging.
2.2.10. Density and porosity estimation
True densities of milled extrudates for all formulations, as well as that of the pure polymer, were measured, with and without P-CO2 injection, using an AccuPyc 1330 gas pycnometer S/N-4011 (Micromeritics Instrument Corp., Norcross, GA). Prior to each run, calibration was performed. The sample was filled in a 10-cm3 sample cup, and the weight of the sample was recorded. The true density was measured at an equilibration rate of 0.0050 psig/min, and the number of purges was set to 10. Bulk and tapped densities were calculated by measuring the volume of 5 g of a milled extrudate in a 10-mL graduated cylinder. The porosity was calculated by using the following equation [29]:
2.2.11. Physical and chemical stability tests
Stability studies were conducted for 24 weeks by monitoring the crystalline content (using DSC), chemical stability (drug content estimation), in vitro dissolution, and physical appearance of samples. Extrudate samples were stored in 20-mL clear glass scintillation vials, which were sealed with screw caps, at 25 °C and 60% relative humidity. The samples were analyzed on the first day and after 1, 2, 3, and 6 months. Drug release profiles were obtained for samples removed from the stability chamber after 12 and 24 weeks.
3. Results and discussion
3.1. Hot-melt extrusion
Physicochemical properties of an API and polymeric carrier need to be considered carefully since they have a significant impact on the HME process, as well as on the final output [8]. Therefore, the process parameters of HME should be adjusted based on the physicochemical properties of each formulation component. Theoretically, the extrusion temperature should be set at 10–20 °C above the Tg of the polymer to facilitate the extrudability of materials during the extrusion process. In addition, there are many other properties that can be critical for HME, such as the polymer melt viscosity, molecular weight, and miscibility. Furthermore, the plasticization effect of the drug on the polymer may significantly affect the extrudability of materials [15]. Besides, many research studies have shown that P-CO2 plasticizes some pharmaceutical polymers [30,31].
In this study, the extrusion temperature and drug/polymer ratio were found to have a major influence on the HME process. Extrusion of the pure polymer was impossible when the extrusion temperature was set below 160 °C because the maximum motor load was exceeded. At 160, 165, and 170 °C, the motor load approached the maximum. This could be attributed to a high melt viscosity of the polymer. To reduce the motor load, the screw speed of the extruder was reduced to 75 rpm at all studied temperatures (Table 3); however, the impact was very limited. Moreover, high torque values were observed, with P-CO2 injection showing no plasticizing effect on HPMCAS LG. Therefore, it was not reasonable to inject P-CO2 at such a high motor load. In fact, the extruder exhibited an increase in the motor load even with an increasing extruder temperature, which could be explained by the inability of the P-CO2 stream to penetrate the polymer. Table 3 shows the results and conditions used for the investigation of the extruder parameters, i.e., the polymer/drug ratios in the HME processes with and without P-CO2 injection. Injecting P-CO2 at zones 4 and 6 provided relatively long periods of contact between the polymer and the injected P-CO2 before the extruder die was reached. Additionally, at zones 4 and 6 (Fig. 1), the possibility of a back-pressure issue in the extruder was high, which spread the materials out of the feeding zone. Upon injection of P-CO2 at zone 8 (Fig. 1), the formulation components and P-CO2 were in contact only at zones 9 and 10. The injection of P-CO2 at zone 8 was desirable to successfully produce foamy, porous extrudates (Fig. 1).
Table 3.
Extrusion conditions and parameters of various polymer/ drug ratio compositions with and without P-CO2 injection
| Formulations | Processing Temperature |
Screw Speed |
Barrel Torque |
P-CO2 Injection |
P-CO2 (PSI) |
Back Pressure |
Feeding Zone |
HME Process |
||
|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | (rpm) | (%) | Zone |
Blockage | ||||||
| 4 | 6 | 8 | ||||||||
| 100% HPMC-AS LG | 140 | 100 | Maximum * | - | - | - | - | - | - | Failed |
| 100% HPMC-AS LG | 140 | 75 | Maximum | - | - | - | - | - | - | Failed |
| 100% HPMC-AS LG | 140 | 100 | Maximum | √ | √ | - | - | High | - | Failed |
| 100% HPMC-AS LG | 140 | 75 | Maximum | √ | √ | - | - | High | - | Failed |
| 100% HPMC-AS LG | 150 | 100 | Maximum | - | - | - | - | - | - | Failed |
| 100% HPMC-AS LG | 150 | 75 | Maximum | - | - | - | - | - | - | Failed |
| 100% HPMC-AS LG | 150 | 100 | Maximum | √ | √ | - | - | High | - | Failed |
| 100% HPMC-AS LG | 150 | 75 | Maximum | √ | √ | - | - | High | - | Failed |
| 100% HPMC-AS LG | 160 | 100 | 98H.TQ | √ | √ | - | - | High | - | Successful** |
| 100% HPMC-AS LG | 160 | 75 | 88H.TQ | √ | √ | - | - | High | - | Successful** |
| 100% HPMC-AS LG | 170 | 100 | 88-90H.TQ | √ | √ | - | - | High | - | Successful** |
| 100% HPMC-AS LG | 170 | 75 | 82H.TQ | √ | √ | - | - | High | - | Successful** |
| 100% HPMC-AS LG | 190H.T | 100 | 84-92H.TQ | √ | √ | - | - | High | - | Successful** |
| 100% HPMC-AS LG | 190H.T | 75 | 80-90H.TQ | √ | √ | - | - | High | - | Successful** |
| 100% HPMC-AS LG | 190H.T | 100 | 84-92H.TQ | - | - | √ | 300-400 | Low | No | Successful** |
| 100% HPMC-AS LG | 190H.T | 75 | 80-90H.TQ | - | - | √ | 300-400 | Low | No | Successful** |
| 80% HPMC-AS LG/% 20 EFA | 140 | 100 | 54 | - | - | - | - | - | - | Successful |
| 70% HPMC-AS LG/% 30 EFA | 140 | 100 | 46 | - | - | - | - | - | - | Successful |
| 60% HPMC-AS LG/% 40 EFA | 140 | 100 | 33 | - | - | - | - | - | - | Successful |
| 50% HPMC-AS LG/% 50 EFA | 140 | 100 | 27 | - | - | - | - | - | - | Successful |
| 80% HPMC-AS LG/% 20 EFA | 150 | 75 | 44 | - | - | - | - | - | - | Successful |
| 70% HPMC-AS LG/% 30 EFA | 150 | 75 | 25-30 | - | - | - | - | - | - | Successful |
| 60% HPMC-AS LG/% 40 EFA | 150 | 75 | 20-25 | - | - | - | - | - | - | Successful |
| 50% HPMC-AS LG/% 50 EFA | 150 | 75 | 13-20 | - | - | - | - | - | - | Successful |
| 80% HPMC-AS LG/% 20 EFA | 140 | 100 | 54 | - | - | √ | 300-400 | Low | No | Successful |
| 70% HPMC-AS LG/% 30 EFA | 140 | 100 | 46 | - | - | √ | 300-400 | Low | No | Successful |
| 60% HPMC-AS LG/% 40 EFA | 140 | 100 | 33 | - | - | √ | 300-400 | Low | No | Successful |
| 50% HPMC-AS LG/% 50 EFA | 140 | 100 | 27 | - | - | √ | 300-400 | Low | No | Successful |
| 80% HPMC-AS LG/% 20 EFA | 150 | 75 | 44 | - | - | √ | 250-350 | Low | No | Successful |
| 70% HPMC-AS LG/% 30 EFA | 150 | 75 | 25-30 | - | - | √ | 250-350 | Low | No | Successful |
| 60% HPMC-AS LG/% 40 EFA | 150 | 75 | 20-25 | - | - | √ | 250-350 | Low | No | Successful |
| 50% HPMC-AS LG/% 50 EFA | 150 | 75 | 13-20 | - | - | √ | 250-350 | Low | No | Successful |
The motor load reached the maximum level in which it was impossible to move on.
Very limited success to obtain the final product, but with very high motor load and high temperature.
H.T: Very high temperature.
H.TQ: Very high motor load (High Torque).
3.2. TGA analysis and drug content
The TGA data demonstrated that all formulations used in this study were stable at the employed processing temperature (data not shown). UV analysis showed acceptable drug content uniformity in the extrudates, indicating no loss of EFA as a function of the temperature and pressure during HME, with and without P-CO2 injection. This confirmed that EFA was uniformly distributed in the formulations, with a high yield. The experimentally obtained drug contents corresponded to the theoretical values, ranging from 96% to 102%.
3.3. Differential scanning calorimetry
As shown in Fig. 2,, the DSC thermograms of pure EFA showed a characteristic endothermic peak at approximately 140 °C. The sharp EFA peak disappeared in all processed formulations but was retained in the respective physical mixtures, which showed smaller endothermic peaks (lower Tm), with a lower enthalpy of fusion. The absence of the peak indicated that all processed formulations had an amorphous nature after HME coupled with P-CO2 processing.
Figure 2.

DSC thermograms of pure EFA, pure HPMC-AS LG, physical mixtures (PM) and extrudates (EXTs) with and without P-CO2
3.4. Powder X-ray diffraction measurements
The diffraction patterns of pure EFA, pure HPMCAS LG, all processed formulations, and their respective physical mixtures are shown in Fig. 3. Analysis of the XRD pattern of the pure EFA sample showed characteristic peaks, indicating the crystalline nature of EFA. On the other hand, the diffraction pattern of pure HPMCAS LG did not show any peaks (hollow band), which indicated that its polymeric structure was amorphous. The XRD patterns of the physical mixtures of EFA and HPMCAS LG showed less intense peaks, owing to the partial presence of EFA in the crystalline form. Meanwhile, the EFA/HPMCAS LG extrudates did not show these characteristic peaks, indicating the transformation of crystalline EFA into an amorphous form, which was consistent with the DSC data. Finally, comparison of the diffractograms of the processed formulations, obtained with and without P-CO2 treatment, confirmed that the treatment caused no modifications.
Figure 3.
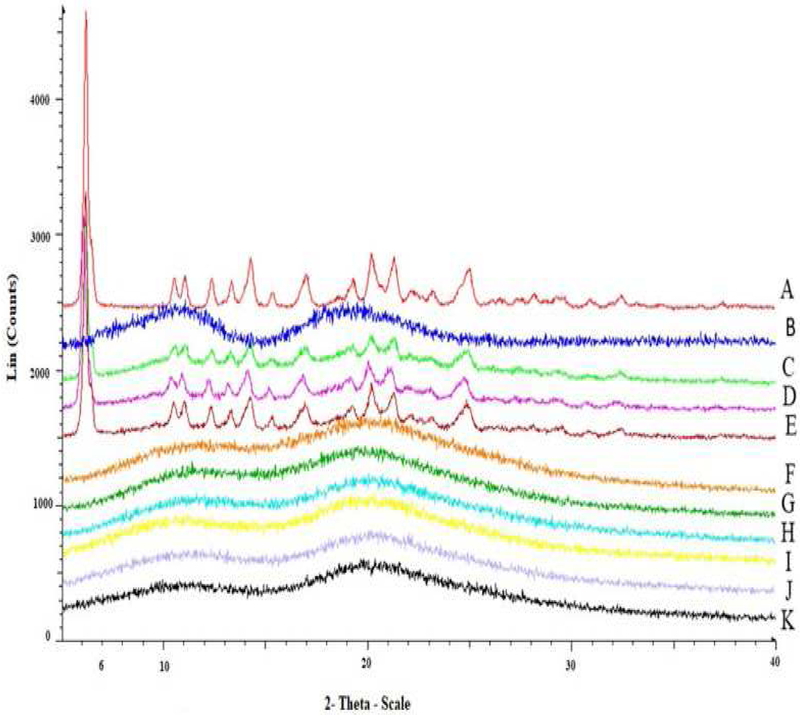
(A): PXRD diffractograms of pure crystalline EFA, (B): PXRD diffractograms of pure HPMC-AS LG; (C, D, and E): PXRD diffractograms of %30, %40, and %50 EFA/ HPMC-AS LG PM; (F, G, and H): PXRD diffractograms of %30, %40, and %50 EFA/ HPMC-AS LG without P-CO2; (I, J, and K): PXRD diffractograms of %30, %40, and %50 EFA/ HPMC-AS LG with P-CO2
3.5. Dissolution studies
HPMCAS LG enhanced the EFA dissolution rate in proportion to the HPMCAS LG content in the formulations. Fig. 4 shows the dissolution of EFA from various extrudates in the dissolution medium (pH 6.8). The order of improvement in the drug dissolution for the formulations processed by this coupled technology (HME/P-CO2) was 30% EFA/70% HPMCAS LG > 40% EFA/60% HPMCAS LG > 50% EFA/50% HPMCAS LG. These results can be attributed to the dissolution medium, which was more suitable for the HPMCAS LG polymer, and to the presence of a high percentage of the polymer.
Figure 4.

In-Vitro release profiles of extrudates with and without P-CO2 (A USP dissolution apparatus II, 50 rpm, pH 6.8 phosphate buffer at 37 ± 0.5 °C for 2 h)
Although the 50% EFA extrudate was highly porous compared with the 30% EFA extrudate (Fig. 5), the dissolution was the highest for the 30% EFA extrudate, which confirmed a high solubility of the HPMCAS LG polymer in the dissolution medium. Furthermore, it was observed that the HPMCAS substitution level (L grade) played an important role in enhancing the dissolution rate of EFA, which was released at a faster rate than pure EFA did. The higher succinoyl content in the L-grade polymer results in an increased hydrophilicity and erodibility, thereby improving the release of EFA from extrudates with a higher HPMCAS LG content. The dissolution profiles demonstrated a relatively higher release of EFA from the CO2-treated materials. These extrudates were all amorphous, indicating that the release could be controlled as a function of the carbon dioxide treatment [24]. The HPMCAS LG matrix controlled the dissolution rate of the extrudates. The HPMCAS LG polymer was eroded, and the EFA molecules were simultaneously dissolved, owing to their presence in an amorphous state. Thus, there was no need to overcome the crystalline lattice energy.
Figure 5.

SEM images of HPMC-AS LG Extrudates with and without P-CO2; A, B, and C: 30 % Extrudate (30% EFA / 70% HPMC-AS LG without P-CO2) D, E, and F: 30 % Extrudate (30% EFA / 70% HPMC-AS LG with P-CO2) G, H, and I: 40 % Extrudate (40% EFA / 60% HPMC-AS LG without P-CO2) J, K, and L: 40 % Extrudate (40% EFA / 60% HPMC-AS LG with P-CO2) M, N, and O: 50 % Extrudate (50% EFA / 50% HPMC-AS LG without P-CO2) P, Q, and R: 50 % Extrudate (50% EFA / 50% HPMC-AS LG with P-CO2)
The dissolution rate depends on the surface area of the material exposed to dissolution media [32]. Although the foamed extrudates had an increased surface area, the unmilled foams showed a significantly slower dissolution than did regular, milled extrudates.
Moreover, although the porous matrix exhibited a high surface area, the P-CO2-processed 50% EFA/50% HPMCAS LG formulation, with a lower density, higher porosity (Table 4), and a larger pore size (Fig. 5P–R), had almost the same dissolution profile as the 50% EFA/50%HPMCAS LG formulation without P-CO2 treatment. This result was attributed to visually observed particles that were floating and accumulating on the surface of the media, making these particles inaccessible to the media.
Table 4.
Bulk, tapped, true density and porosity of pure EFA, pure HPMC-AS LG, Physical Mixture (PM) and Extrudates (EXT) with and without P-CO2 (g/ml)
| Formulations | Bulk Density |
Tapped Density |
True Density |
%Porosity |
|---|---|---|---|---|
| EFA | 0.225 | 0.338 | 1.450 | 84.482 |
| HPMC-AS LG | 0.462 | 0.470 | 1.289 | 64.158 |
| 30% EFA / HPMC-AS LG PM | 0.369 | 0.447 | 1.416 | 73.940 |
| 40% EFA / HPMC-AS LG PM | 0.351 | 0.421 | 1.474 | 76.187 |
| 50% EFA / HPMC-AS LG PM | 0.317 | 0.404 | 1.532 | 79.308 |
| 30% EFA / HPMC-AS LG EXT | 0.598 | 0.629 | 1.347 | 55.605 |
| 40% EFA / HPMC-AS LG EXT | 0.612 | 0.668 | 1.362 | 55.066 |
| 50% EFA / HPMC-AS LG EXT | 0.620 | 0.652 | 1.381 | 55.104 |
| 30% EFA / HPMC-AS LG EXT+P-CO2 | 0.494 | 0.493 | 1.350 | 63.407 |
| 40% EFA / HPMC-AS LG EXT+P-CO2 | 0.461 | 0.519 | 1.357 | 66.028 |
| 50% EFA / HPMC-AS LG EXT+P-CO2 | 0.350 | 0.466 | 1.382 | 74.674 |
In contrast, the P-CO2-processed 30% and 40% EFA/HPMCAS LG formulations sank into the dissolution media, allowing the dissolution process to proceed faster through the fully accessible surfaces [33]. Since P-CO2-treated extrudates are more hygroscopic [24], water penetrated faster into the matrix compared with its penetration into extrudates not treated with P-CO2.
3.6. Scanning electron microscopy
The surface of all processed extrudates subjected to P-CO2 treatment, which are shown in the SEM images in Fig. 5, was quite different from that of extrudates prepared without P-CO2. The latter extrudates were smoother than were the foamed and porous extrudates treated with P-CO2. The effective surface area was larger in the case of the foamed samples, where EFA particles were exposed to the dissolution media and exhibited an increased release [33]. The internal and external porous structures of the solid dispersions shown in Fig. 5 can alter the physicomechanical properties of such formulations.
On the other hand, as shown in Fig. 5, the porous matrix exhibited a mesh-like framework, indicating that P-CO2 uniformly penetrated during the process through the HPMCAS LG matrix. Thus, these images suggested that a homogeneous or heterogeneous porosity behavior could occur, depending on the drug load (30% to 50%). Homogeneous pores were formed during processing of the 40% and 50% EFA dispersions with P-CO2 treatment. In contrast, in the case of the 30% drug load, heterogeneous pores were formed during processing of the dispersion with P-CO2 treatment. The highly porous nature of the extrudates may be attributed to the plasticizing effect of EFA, which led to less viscous matrices and facilitated the homogeneous penetration of P-CO2 throughout the matrix during processing [33]. This could be visually confirmed in images of extrudates processed without P-CO2, which showed smoother surfaces.
3.7. Density and porosity
As shown in Table 4, all density values decreased with P-CO2 injection because of the formation of foamy and porous matrices. Furthermore, the porosity values were higher in samples prepared with P-CO2 treatment. This resulted in a larger surface area, which led to improved dissolution profiles of high-HPMCAS LG content extrudates [22]. In addition, P-CO2 treatment exhibited a high free surface energy, which aided in stabilizing the amorphous form of the drug [37–40].
3.8. Milling efficiency
The influence of P-CO2 on the milling efficiency of extrudates was evaluated. The extrudates without P-CO2 treatment were glassy, which made them very difficult to mill to obtain a suitable particle size distribution. Table 5 displays the results of this study, which confirmed that the amounts of particles with selected sizes (<600, 250, and 125 μm), obtained after milling P-CO2-treated formulations, were higher than those of the corresponding formulations obtained without P-CO2 treatment. Moreover, the torque of the milling instrument during processing of P-CO2-treated formulations was less than that needed for the corresponding formulations obtained without P-CO2 treatment. The enhancement in the milling efficiency was due to the morphological changes of extrudates into foam-like structures, which was the result of P-CO2 injection.
Table 5.
Results of the Milling Efficiency
| Formulations | Torque (Amps) |
Particles < 600 um (%) |
Particles < 250 um (%) |
Particles < 125 um (%) |
|---|---|---|---|---|
| 30% EFA / HPMC-AS LG EXT | 1.08 | 78.44 | 9.57 | 1.03 |
| 40% EFA / HPMC-AS LG EXT | 0.85 | 84.99 | 15.33 | 2.74 |
| 50% EFA / HPMC-AS LG EXT | 0.79 | 77.90 | 3.37 | 0.98 |
| 30% EFA / HPMC-AS LG EXT P-CO2 | 0.79 | 92.14 | 14.88 | 1.97 |
| 40% EFA / HPMC-AS LG EXT P-CO2 | 0.74 | 97.70 | 24.41 | 4.75 |
| 50% EFA / HPMC-AS LG EXT P-CO2 | 0.72 | 98.08 | 13.64 | 1.79 |
3.9. Physical and chemical stability
For solid dispersion systems to be applicable commercially, stability issues have to be resolved [41]. During processing or storage, the amorphous state of a formulation may potentially transform to a crystalline state [42]. The impact of moisture on stability of amorphous formulations during storage is also an important concern because it may increase the polymer mobility and promote drug crystallization.
The DSC thermograms of EFA (30%, 40%, and 50%) in the extrudates confirmed that none of the formulations, with and without P-CO2 treatment, displayed any sign of recrystallization after storage for 6 months (Fig. 6A-C). The physical appearance of the formulations was examined during storage, and photographs were taken and compared. The photographs showed no signs of changes in the color, morphology, and physical state of the formulations, both with and without P-CO2 injection, as shown in (Fig. 7A-C). Further, the drug release results revealed no significant changes in the release profiles after a long-term storage, for 6 months (Fig. 8A-C). The similarity factor values (f2) for the release profiles between fresh and 6-month-old samples were more than 50 for the EFA formulations (30%, 40%, and 50%). The drug content after storage of all formulations was in an acceptable range (97.0% and 106.0%), according to USP, as shown in Fig. 9. The drug content of EFA in the extrudates indicated that EFA was chemically stable, with no sign of degradation or weight loss after storage for 6 months.
Figure 6.
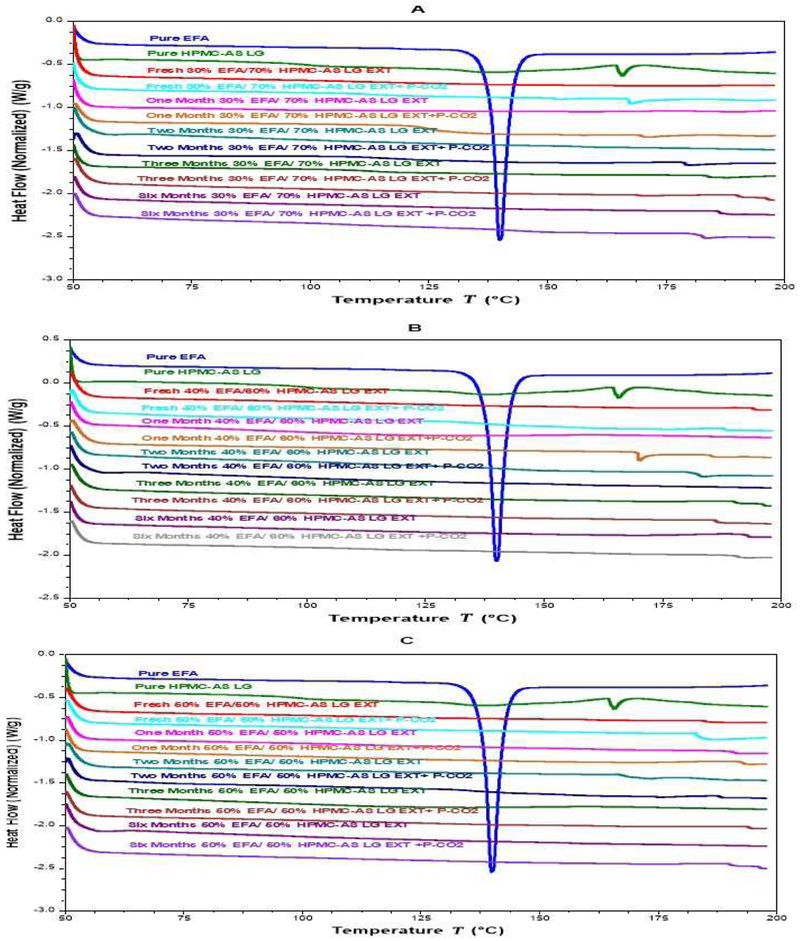
DSC thermograms of extrudates with and without P-CO2 Stored at 25°C/60% RH. (A): formulations 30 % Extrudate (30% EFA / 70% HPMC-AS LG); (B): formulations 40 % Extrudate (40% EFA / 60% HPMC-AS LG); and (C): formulations 50 % Extrudate (50% EFA / 50% HPMC-AS LG)
Figure 7.

Physical appearance of extrudates with and without P-CO2 Stored at 25°C/60% RH. (A): formulation 30 % Extrudate (30% EFA / 70% HPMC-AS LG), (B): formulations 40 % Extrudate (40% EFA / 60% HPMC-AS LG), and (C): formulations 50 % Extrudate (50% EFA / 50% HPMC-AS LG)
Figure 8.
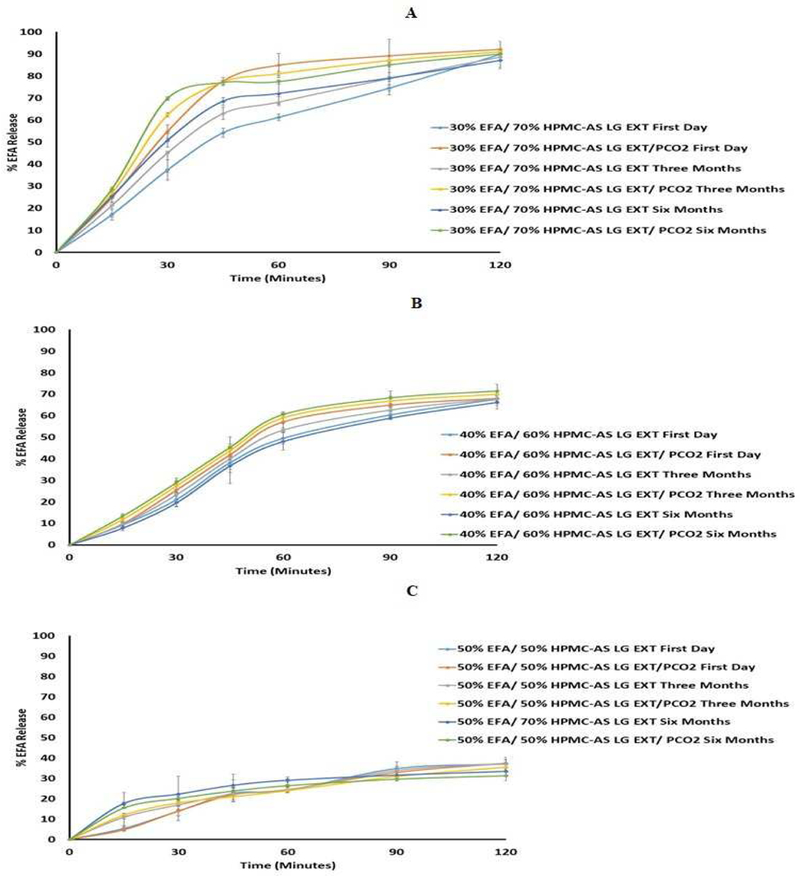
In-Vitro release profile of extrudates with and without P-CO2 (A USP dissolution apparatus II, 50 rpm, pH 6.8 phosphate buffer at 37 ± 0.5 °C for 2 h) stored at 25°C/60% RH. (A): formulation 30 % Extrudate (30% EFA / 70% HPMC-AS LG), (B): formulation 40 % Extrudate (40% EFA / 60% HPMC-AS LG), and (C): formulation 50 % Extrudate (50% EFA / 50% HPMC-AS LG)
Figure 9.

Drug contents of EFA extrudates with and without P-CO2 Stored at 25°C/60% RH
In a recent study, it has been reported that a higher content of succinoyl substitution in HPMCAS polymers resulted in a strong affinity toward hydrophobic drug surfaces, leading to the inhibition of the crystal growth of an API [43]. HPMCAS LG has the highest succinoyl content and the lowest acetyl content compared with those in the other HPMCAS grades (MG and HG).
HPMCAS LG enhanced the physical stability of highly loaded amorphous EFA (30%, 40%, and 50%) in all formulations by increasing the Tg of the miscible mixture, which resulted in a reduction in molecular mobility during storage. Moreover, the miscibility between HPMCAS LG and EFA had a very significant impact on the stabilization of extrudates in the case of a high EFA load [44]. Further, the high viscosity of HPMCAS LG had a very desirable influence on the stability of EFA at a high load [45].
4. Conclusion
Highly loaded EFA extrudates were successfully processed in the presence of P-CO2. However, there was no significant plasticizing effect of P-CO2 on the HPMCAS LG polymer with EFA. The use of HPMCAS LG resulted in good release profiles for all EFA-loaded extrudates, with and without P-CO2 injection. However, the EFA-loaded extrudates obtained with P-CO2 injection exhibited a relatively higher drug release than did those obtained without P-CO2. Morphological changes in extrudates, with the formation of a foam-like structure after P-CO2 injection, resulted in an increased porosity and surface area. Thus, the milling efficiency of the extrudates improved. Stability studies demonstrated that EFA retained its amorphous form, indicating stability of the drug within extrudates. In summary, it was demonstrated that HPMCAS LG is a promising carrier for the production of physically and chemically stable ASD systems via the HME technology.
Acknowledgements
This work was supported by Ashland Speciality Ingredients. This project was also partially supported by Grant Number P20GM104932 from the National Institute of General Medical Sciences and the Biopharmaceutics-Clinical and Translational Core E of the COBRE, a component of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Crowley MM et al. , “Pharmaceutical applications of hot-melt extrusion: Part I,” Drug Dev. Ind. Pharm., vol. 33, no. 9, pp. 909–926, 2007. [DOI] [PubMed] [Google Scholar]
- [2].Pimparade MB et al. , “Development of taste masked caffeine citrate formulations utilizing hot melt extrusion technology and in vitro-in vivo evaluations,” Int. J. Pharm., vol. 487, no. 1–2, pp. 167–176, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Morott JT et al. , “The effects of screw configuration and polymeric carriers on hot-melt extruded taste-masked formulations incorporated into orally disintegrating tablets,” J. Pharm. Sci., vol. 104, no. 1, pp. 124–134, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Maniruzzaman M, Rana MM, Boateng JS, Mitchell JC, and Douroumis D, “Dissolution enhancement of poorly water-soluble APIs processed by hot-melt extrusion using hydrophilic polymers,” Drug Dev. Ind. Pharm., vol. 39, no. 2, pp. 218–227, 2013. [DOI] [PubMed] [Google Scholar]
- [5].Repka MA, Majumdar S, Battu SK, Srirangam R, and Upadhye SB, “Applications of hot-melt extrusion for drug delivery,” Expert Opin. Drug Deliv., vol. 5, no. 12, pp. 1357–1376, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Thomas S and Weimin Y, Advances in polymer processing. Woodhead Publishing Ltd, 2009. [Google Scholar]
- [7].LaFountaine JS, McGinity JW, and Williams RO, “Challenges and Strategies in Thermal Processing of Amorphous Solid Dispersions: A Review,” AAPS PharmSciTech, vol. 17, no. 1, pp. 43–55, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Repka MA et al. , “Melt extrusion: process to product,” Expert Opin. Drug Deliv., vol. 9, no. 1, pp. 105–125, Jan. 2012. [DOI] [PubMed] [Google Scholar]
- [9].Qian F, Huang J, and Hussain MA, “Drug–Polymer Solubility and Miscibility: Stability Consideration and Practical Challenges in Amorphous Solid Dispersion Development,” J. Pharm. Sci., vol. 99, no. 7, pp. 2941–2947, 2010. [DOI] [PubMed] [Google Scholar]
- [10].Marsac PJ, Shamblin SL, and Taylor LS, “Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility,” Pharm. Res., vol. 23, no. 10, pp. 2417–2426, 2006. [DOI] [PubMed] [Google Scholar]
- [11].Kyeremateng SO, Pudlas M, and Woehrle GH, “A fast and reliable empirical approach for estimating solubility of crystalline drugs in polymers for hot melt extrusion formulations,” J. Pharm. Sci., vol. 103, no. 9, pp. 2847–2858, 2014. [DOI] [PubMed] [Google Scholar]
- [12].Shah S, Maddineni S, Lu J, and Repka MA, “Melt extrusion with poorly soluble drugs,” Int. J. Pharm., vol. 453, no. 1, pp. 233–252, 2013. [DOI] [PubMed] [Google Scholar]
- [13].Gupta SS, Solanki N, and Serajuddin ATM, “Investigation of Thermal and Viscoelastic Properties of Polymers Relevant to Hot Melt Extrusion, IV: Affinisol™ HPMC HME Polymers,” AAPS PharmSciTech, vol. 17, no. 1, pp. 148–157, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kolter K, Karl M, and Gryczke A, Introduction to Solid Dispersions. 2012. [Google Scholar]
- [15].Chokshi RJ, Sandhu HK, Iyer RM, Shah NH, Malick AW, and Zia H, “Characterization of physico-mechanical properties of indomethacin and polymers to assess their suitability for hot-melt extrusion processs as a means to manufacture solid dispersion/solution,” J. Pharm. Sci., vol. 94, no. 11, pp. 2463–2474, 2005. [DOI] [PubMed] [Google Scholar]
- [16].Verreck G et al. , “The effect of pressurized carbon dioxide as a temporary plasticizer and foaming agent on the hot stage extrusion process and extrudate properties of solid dispersions of itraconazole with PVP-VA 64,” Eur. J. Pharm. Sci., vol. 26, no. 3–4, pp. 349–358, 2005. [DOI] [PubMed] [Google Scholar]
- [17].Verreck G, “The Influence of Plasticizers in Hot-Melt Extrusion,” in Hot-Melt Extrusion: Pharmaceutical Applications, Chichester, UK: John Wiley & Sons, Ltd, 2012, pp. 93–112. [Google Scholar]
- [18].Wu C and McGinity JW, “Influence of methylparaben as a solid-state plasticizer on the physicochemical properties of Eudragit® RS PO hot-melt extrudates,” Eur. J. Pharm. Biopharm., vol. 56, no. 1, pp. 95–100, 2003. [DOI] [PubMed] [Google Scholar]
- [19].Repka MA, Gerding TG, Repka SL, and McGinity JW, “Influence of plasticizers and drugs on the physical-mechanical properties of hydroxypropylcellulose films prepared by hot melt extrusion,” Drug Dev. Ind. Pharm., vol. 25, no. 5, pp. 625–633, 1999. [DOI] [PubMed] [Google Scholar]
- [20].Mehuys E, Vervaet C, and Remon JP, “Hot-melt extruded ethylcellulose cylinders containing a HPMC-Gelucire® core for sustained drug delivery,” J. Control. Release, vol. 94, no. 2–3, pp. 273–280, 2004. [DOI] [PubMed] [Google Scholar]
- [21].Sauceau M, Fages J, Common A, Nikitine C, and Rodier E, “New challenges in polymer foaming: A review of extrusion processes assisted by supercritical carbon dioxide,” Prog. Polym. Sci., vol. 36, no. 6, pp. 749–766, 2011. [Google Scholar]
- [22].Ashour EA et al. , “Influence of pressurized carbon dioxide on ketoprofen-incorporated hot-melt extruded low molecular weight hydroxypropylcellulose,” Drug Dev. Ind. Pharm., vol. 42, no. 1, pp. 123–130, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Verreck G et al. , “The effect of pressurized carbon dioxide as a plasticizer and foaming agent on the hot melt extrusion process and extrudate properties of pharmaceutical polymers,” J. Supercrit. Fluids, vol. 38, no. 3, pp. 383–391, 2006. [DOI] [PubMed] [Google Scholar]
- [24].Verreck G et al. , “The effect of supercritical CO2as a reversible plasticizer and foaming agent on the hot stage extrusion of itraconazole with EC 20 cps,” J. Supercrit. Fluids, vol. 40, no. 1, pp. 153–162, 2007. [Google Scholar]
- [25].Curatolo W, Nightingale JA, and Herbig SM, “Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu,” Pharm. Res., vol. 26, no. 6, pp. 1419–1431, 2009. [DOI] [PubMed] [Google Scholar]
- [26].Ueda K, Higashi K, Yamamoto K, and Moribe K, “Inhibitory Effect of Hydroxypropyl Methylcellulose Acetate Succinate on Drug Recrystallization from a Supersaturated Solution Assessed Using Nuclear Magnetic Resonance Measurements,” Mol. Pharm., vol. 10, no. 10, pp. 3801–3811, 2013. [DOI] [PubMed] [Google Scholar]
- [27].Warashina S, Maruyama N, Quadir A, Obara S, editors, “Evaluation of a new development grade of hypromellose acetate succinate (HPMCAS) for solid dispersion by hot melt extrusion,” AAPS Annu. Meet., 2014. [Google Scholar]
- [28].Alshahrani SM, Lu W, Park J-B, Morott JT, Alsulays BB, Majumdar S, Langley N, Kolter K, Gryczke A, and Repka MA, "Stability-enhanced hot-melt extruded amorphous solid dispersions via combinations of Soluplus® and HPMCAS-HF," AAPS PharmSciTech. 2015. August;16(4):824–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Joshi DC, Das SK, and Mukherjee RK, “Physical Properties of Pumpkin Seeds,” J. Agric. Eng. Res., vol. 54, no. 3, pp. 219–229, Mar. 1993. [Google Scholar]
- [30].Verreck G et al. , “Hot stage extrusion of p-amino salicylic acid with EC using CO2as a temporary plasticizer,” Int. J. Pharm., vol. 327, no. 1–2, pp. 45–50, 2006. [DOI] [PubMed] [Google Scholar]
- [31].Lyons JG et al. , “Preparation of monolithic matrices for oral drug delivery using a supercritical fluid assisted hot melt extrusion process,” Int. J. Pharm., vol. 329, no. 1–2, pp. 62–71, 2007. [DOI] [PubMed] [Google Scholar]
- [32].Verreck G, Six K, Van den Mooter G, Baert L, Peeters J, and Brewster ME, “Characterization of solid dispersions of itraconazole and hydroxypropylmethylcellulose prepared by melt extrusion—part I,” Int. J. Pharm., vol. 251, no. 1–2, pp. 165–174, 2003. [DOI] [PubMed] [Google Scholar]
- [33].Nagy ZK et al. , “Use of supercritical CO2-aided and conventional melt extrusion for enhancing the dissolution rate of an active pharmaceutical ingredient,” Polym. Adv. Technol., vol. 23, no. 5, pp. 909–918, 2012. [Google Scholar]
- [34].Almutairy BK et al. , “Development of Floating Drug Delivery System with Superior Buoyancy in Gastric Fluid using Hot-Melt Extrusion Coupled with Pressurized CO2,” Pharmazie, vol. 71, pp. 1–6, 2015. [PubMed] [Google Scholar]
- [35].Lee M, Tzoganakis C, and Park CB, “Extrusion of PE/PS blends with supercritical carbon dioxide,” Polym. Eng. Sci., vol. 38, no. 7, pp. 1112–1120, 1998. [Google Scholar]
- [36].Park CB, Baldwin DF, and Suh NP, “Effect of the pressure drop rate on cell nucleation in continuous processing of microcellular polymers,” Polym. Eng. Sci., vol. 35, no. 5, pp. 432–440, 1995. [Google Scholar]
- [37].Laitinen R, Löbmann K, Strachan CJ, Grohganz H, and Rades T, “Emerging trends in the stabilization of amorphous drugs,” Int. J. Pharm., vol. 453, no. 1, pp. 65–79, 2013. [DOI] [PubMed] [Google Scholar]
- [38].Jackson CL and McKenna GB, “Vitrification and Crystallization of Organic Liquids Confined to Nanoscale Pores,” Chem. Mater., vol. 8, no. 8, pp. 2128–2137, 1996. [Google Scholar]
- [39].Prestidge CA, Barnes TJ, Lau C-H, Barnett C, Loni A, and Canham L, “Mesoporous silicon: a platform for the delivery of therapeutics,” Expert Opin. Drug Deliv., vol. 4, no. 2, pp. 101–110, 2007. [DOI] [PubMed] [Google Scholar]
- [40].Qian KK and Bogner RH, “Application of Mesoporous Silicon Dioxide and Silicate in Oral Amorphous Drug Delivery Systems,” J. Pharm. Sci., vol. 101, no. 2, pp. 444–463, 2012. [DOI] [PubMed] [Google Scholar]
- [41].Vasconcelos T, Sarmento B, and Costa P, “Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs,” Drug Discov. Today, vol. 12, no. 23–24, pp. 1068–1075, Dec. 2007. [DOI] [PubMed] [Google Scholar]
- [42].Johari GP, Kim S, and Shanker RM, “Dielectric studies of molecular motions in amorphous solid and ultraviscous acetaminophen,” J. Pharm. Sci., vol. 94, no. 10, pp. 2207–2223. [DOI] [PubMed] [Google Scholar]
- [43].Ueda K, Higashi K, Yamamoto K, and Moribe K, “Inhibitory Effect of Hydroxypropyl Methylcellulose Acetate Succinate on Drug Recrystallization from a Supersaturated Solution Assessed Using Nuclear Magnetic Resonance Measurements,”Mol. Pharm., vol.10, no. 10, pp. 3801–3811, 2013. [DOI] [PubMed] [Google Scholar]
- [44].Marsac PJ, Rumondor ACF, Nivens DE, Kestur US, Stanciu Lia., and Taylor LS, “Effect of temperature and moisture on the miscibility of amorphous dispersions of felodipine and poly(vinyl pyrrolidone),” J. Pharm. Sci., vol. 99, no. 1, pp. 169–185, 2010. [DOI] [PubMed] [Google Scholar]
- [45].Chokshi R and Zia H, “Hot-Melt Extrusion Technique: A Review,” Iran. J. Pharm. Res., vol. Volume 3, no. Number 1, pp. 3–16, 2010. [Google Scholar]