

ABSTRACT
P53 is a critical tumor suppressor gene, activating p53 and its downstream targets to induce apoptosis is a promising way for cancer therapy. However, more than 50% of cancer patients have p53 mutations, which may cause cancer therapy resistance, and the underline mechanism is poorly understood. Here, we found that cell viability decrease and apoptosis induced by p53-dependent traditional drugs in colon cancer cells were eliminated in p53 mutant cells. Mutant p53 did not up-regulate the expression of its direct downstream targets PUMA and p21, due to the inhibition of PUMA transcription. Furthermore, mutant p53 could not bind to the promoter of PUMA to activate its transcription like WT p53 did, while overexpressed WT p53 rescued PUMA-induced subsequent apoptosis. In conclusion, our findings demonstrate mutant p53 may cause chemo-resistance of tumor because of inactivating PUMA transcription, which prompts some new insights for clinical therapy of cancer patients with mutant p53.
Abbreviations: CRC: Colorectal cancer; CDKs: Cyclin-dependent kinases; PUMA: p53 up-regulated modulator of apoptosis; PDGF: the platelet-derived growth factor; WT p53: wild-type p53 protein; mutp53: mutant p53 proteins; BAX: Bcl-2-associated X protein; NOXA: Phorbol-12-myristate-13-acetate-induced protein 1.
KEYWORDS: Mutant p53, chemotherapy resistance, colon cancer, loss-of-function, PUMA
Introduction
CRC, a common gastrointestinal tumor, is the second leading cause of cancer-related death worldwide in 2018 with 11.6% of the total cancer deaths [1]. Global CRC risk is predicted to increase by 60% in 2030 with more than 2.2 million new cases and 1.1 million deaths [2]. The incidence of CRC is high in developed countries, showing the increasing trend annually.
The p53 protein is an important transcription factor in human body [3]. Functionally, activated p53 governs an exquisitely complex anti-proliferative transcriptional program triggering a bewildering array of biological responses [4]. Upon p53 activation, the expression of many genes is essential in the cell division cycle and which will be repressed. For instance, overexpression of p53 blocks the activity of the cyclin family proteins and its kinase [5,6]. Additionally, apoptosis is another consequence of p53 activation [7]. The two accepted initiations of p53-mediated apoptosis are the exogenous pathway and the endogenous pathway, by activating its downstream genes, such as BAX, PUMA, and NOXA, which will initiate endogenous apoptosis pathway rather than the exogenous pathway. After activation, PUMA, one of the p53 binding targets, binds to the pro-apoptotic proteins of Bcl-2 family which localize to the mitochondria and induce cytochrome c/Apaf-1-dependent endogenous apoptosis [8–10]. The chemotherapeutic drugs 5-fluorouracil and Doxorubicin induce p53-dependent PUMA expression in human cancer cells, including CRC [11,12]. Besides, some targeted drugs such as idelalisib [13,14], pazopanib [15,16], ipatasertib [17,18], which have been extensively studied in recent years, also induce PUMA-mediate apoptosis in a p53-dependent or independent fashion.
The p53 tumor suppressor gene is mutant in half of the human tumors, including breast, brain, lung, and colorectum [19–21]. The occurrence of p53 mutation is frequent in a variety of tumors, which implies that p53 mutant proteins promote malignant development of tumors. The majority of tumor-associated p53 mutations are missense mutations and predominantly clustered in the DNA-binding domain (amino acids 102–292), including several hot spots at codons R175, G245, R248, R249, R273 and R282 [22–25]. Missense mutations of p53 deprived their original cancer-suppressing function and, more interestingly, in some instances, conferred their new carcinogenic activity (gain-of-function, GOF) [24,26,27]. A murine model data of pancreatic cancer harboring a latent mutant p53-R175H have firmly established that mutant p53 promoted invasion and metastasis through activating cell-autonomous PDGF receptor β Signaling [28]. There is a clear evidence that the ability of p53-R273H mutant can inhibit mitochondria-dependent apoptosis and constitutively drive PI3K/AKT signaling in human breast cancer [29]. An increase in transcriptional levels of NOXA and a decrease in MYC mRNA were demonstrated in CRC cell lines (HROC183, HT29, and SW480) harboring mutant p53 with pronounced sensitivity to RITA [30].
Using experiments we found some traditional chemotherapy drugs are the potential obstacle in effective chemotherapy of cancer, such as 5FU (5-fluorouracil), Dox (Doxorubicin) and Oxa (Oxaliplatin) [31]. However, it is worth noting mutant p53 as a promising cancer treatment target its molecular mechanism is not well understood. In this study, we identified mutant p53 could not interact with PUMA and p21, as an important p53-upregulated modulator of apoptosis. Mechanistically, mutations in p53, but not wild-type p53, inactivates PUMA transcription. In conclusion, we describe our work that apoptosis mediated by PUMA is no longer expression in CRC with mutant p53 in response to chemotherapy agents, which will help us better understand the carcinogenicity of p53 mutation.
Materials and methods
Cell culture and treatment
The human CRC cell lines expressing wild-type or mutant p53 (HCT-116, SW620, SW480), plus Pancreatic cancer cell (PANC-1) and Breast cancer cell (SKBR-3) were obtained from American type culture collection (ATCC). SW620 and SW480 were cultured in L-15 (KeyGEN BioTECH) modified media and HCT116 were routinely cultured in McCoy’s 5A (gibicol) modified media and other cell lines were cultured in DMEM (gibicol), supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/mL), and streptomycin (100 mg/mL) in 5% CO2 at 37°C in humidified incubator. All agents of 5-FU and Oxaliplation diluted with DMSO and Doxombicin diluted with water were added in the medium directly before detection. In the aspect of transfection experiments, the polyjet (Signgen Laboratories) transfection reagent was used following according to the supplier’s instructions. The medium was replaced with fresh culture medium after 5 h. Cells were then examined at 24–48 h after transfection.
Antibodies and reagents
Primary antibodies against p53, PUMA, Cleaved-Caspase3, Cleaved-Caspase1, β-actin were purchased from Cell Signaling Technology. Lipofectamine™ Reagent was purchased from Invitrogen. HRP-conjugated anti-rabbit and/or anti-mouse secondary antibodies an ECL-plus kit were from Advansta in America. All drugs including 5-FU, Oxaliplatin and Doxorubicin, and other chemicals were purchased from Sigma. CCK-8 kit was from 7 sea biotech (Shanghai, China).
P53 mutation and sequencing analysis
The latest CRC patient data that p53 protein has a high mutation frequency at sites 175 and 273 were from cBioPortal database.
Three CRC cell lines including HCT116, SW620 and SW480 were sent to the BBI Life Sciences Corporation for sequencing. Total RNA from the cells was purified using the Tri-Reagent (TIANGEN BIOTECH, Beijing, China). Then, 3μg of RNA was reversely transcribed into DNA that would be used to the conventional PCR performed by C1000 Thermal Cycler CFX96 Real-time PCR Detection System (Bio-Rad). Primer sequences used in our experiments were listed as follows: Proximal primers: 5′-ACAAGATGTTTTGCCAACTG-3′; Distal primers: 5′- ATTCTCCATCCAGTGGTTTC −3′.
Real-time reverse transcriptase (RT) PCR
Total RNA was extracted with Tri-Reagent (TIANGEN BIOTECH, Beijing, China) according to the manufacturer’s protocol. The amount and purity of the RNA were determined by spectrophotometry, and 3μg of RNA from the CRCs after 5-FU treatment were used in each RT reaction. The performance of Real-time qPCR was conducted by our previous report that we described on C1000 Thermal Cycler CFX96 Real-time PCR Detection System (Bio-Rad). Primer sequences used in our experiments were listed as follows:
PUMA Proximal primers: 5′-CGACCTCAACGCACAGTACGA-3′; PUMA Distal primers: 5′-AGGCACCTAATTGGGCTCCAT-3′. Human actin primers: Proximal primers: 5′-TGGCACCCAGCACAATGAA-3′; Distal primers: 5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′.
Cell viability and apoptosis assays
CRC cells were cultured in 96-well microplate at a density of 5 × 103 cells/well for 24 h. Cell viability was assessed with CCK-8 at indicated time post-treatment according to the manufacturer’s instructions. CCK-8 solution was added directly to the cells, which not required premix. To estimate the viability of the cells, the absorbance of 450 nm (OD450) was measured with a 96-well plate reader (DG5032, Hua Dong, Nanjing, China).
For analysis of apoptosis by Hoechst 33,258 (Invitrogen), colorectal cells were cultured on the coverslip of a chamber, rinsed with PBS, and then added in 500 mL of McCoy’s 5A or L-15 containing 5μg Hoechst 33,258, incubated at 37°C with 5% CO2 for 15 min. Apoptosis was detected through microscopic visualization of condensed chromatin and micro-nucleation.
For colony formation assays, equal number of cells after different treatments were plated into 6-well plates. Colonies were visualized by crystal violet staining 14 days after plat.
Western blotting
Protein samples were extracted with RIPA buffer (10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS. 140 mM NaCl). Equivalent protein samples (30 μg protein extract was loaded on each lane) were subjected to SDS-PAGE on 10% gel. The proteins were then transferred onto PVDF membranes (Millipore) and were blocked with 5% nonfat milk for 1 h at room temperature. The membranes, probed with the indicated primary antibodies, were incubated at 4°C overnight. Primary antibody was detected by binding horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse secondary antibody with an ECL plus kit. Detection was performed by using the Odyssey infrared imaging system (LI-COR, Lincoln, NE).
To detect the multimerization of BAX, the purified mitochondrial fraction was cross-linked with DSP (dithiobis (succinimidyl propionate)) (1 mM), followed by Western blotting analysis. Western blot detection of the protein expression levels of puma, p21, p53, Caspase-3, C-Caspase3, and C-caspase1 in cancer cells. Total cell protein was extracted by adopting cell lysis method, and the concentration of protein was measured by BCA method.
Flow cytometry
Human CRC cell line with HCT-116 (p53 wild-type), SW620 (p53 mutation) was suspended in 1 × 105 cells/mL, and 5 μL Annexin V and 5 μL propidium iodide staining solution were added to 100 μL of the cell suspension. Then, 400 μL Binding Buffer was added to cell suspension again after the cells were incubated at room temperature for 10 min in the dark, stained cells were assayed and quantified using a FACSort Flow Cytometer (Beckman Coulter, Brea, CA, USA). Cell debris was excluded from the analysis by an appropriate forward light scatter threshold setting. Compensation was used wherever necessary.
Chromatin immunoprecipitation
ChIP assay was performed by using the Chromatin Immunoprecipitation Assay kit (Millipore, Massachusetts, USA) in accordance with manufacturer’s instructions with minor modifications. All the solutions used come from this ChIP Assay kit unless otherwise stated.
Briefly, after 5-FU treatment, HCT-116 or SW620 were fixed with 1% formaldehyde and lysed in warm SDS lysis buffer. The genomic DNA was obtained and sheared to 200–1,000 bp by sonication on ice. Samples were precleared with Protein A-Agarose/Salmon Sperm DNA (50% Slurry) for 1 h at 4°C with agitation. Then, anti-p53 antibody was added and incubated overnight on a shaker at 4°C. Normal rabbit IgG (Invitrogen) was used as a negative control. The protein agarose/salmon sperm DNA (50% slurry) bead was then added to precipitate the antibody/protein/DNA complexes. After washed with serial wash buffers, DNA-protein immunocomplexes were eluted from the beads by elution buffer (1% SDS, 0.1 M NaHCO3) for 30 min. Finally, the protein-DNA cross-links were reversed to release DNA by incubation with 0.2 M NaCl at 65°C for 4 h.
The total DNA was finally recovered from the samples by phenol/chloroform extraction and ethanol precipitation. Semi-quantitative PCR was then performed as described above. The precipitates were analyzed by PCR using primers 5′-GACTGTCCCGGTGTCTGG-3′ and 5′-GGGCCCGCTCCAAAGC-3′ to amplify a PUMA promoter fragment containing putative p53 sites.
Results
P53 protein has high-frequency mutation at position 175 and 273
CRC is one of the first tumor types to be recognized as a genetic disease, in which the accumulation of genetic alterations leads to highly proliferative and invasive cancers [32]. With the advent of genome-wide experiments and bioinformatics analyses, the latest data that genomic alterations had identified in p53 in metastatic CRC patients revealed in cBioPortal database [33]. Those data showed that CRC patients had a high missense mutation, mainly situated within the DNA-binding domain (amino acids 102–292) [34], especially in amino acids 175 and 273 (Figure 1(a)). Next, we sequenced the cell genome, which manifested that the mutation occurred at the 881 bp site and caused G mutated to A (in this case, an arginine mutated to a tryptophan at position 273 in the p53 protein) in SW620 and SW480 cell lines (Figure 1(b)).
Figure 1.
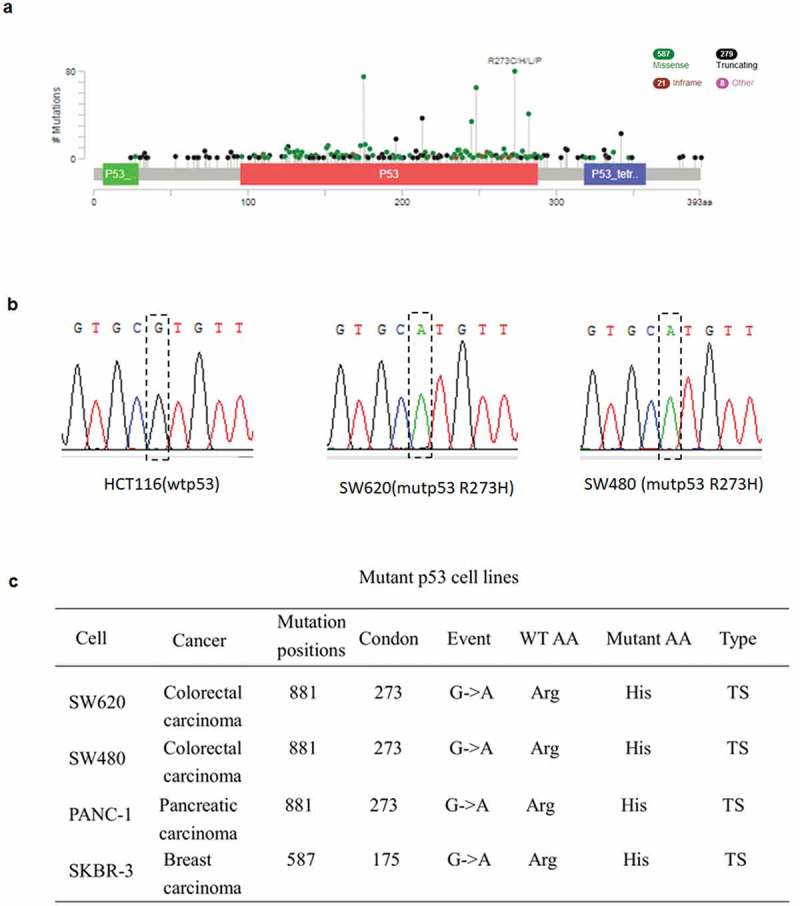
Frequency and the position of R273H and R175H mutation. a The relative frequency of R273H and R175H mutation in CRC. Data were obtained from the cBioPortal database (2018). b Gene sequencing manifested that the mutation occurred at the 881 site and causing G mutated to A in SW620 cells as well as in SW480 cells. c The mutant cell lines used in this study and their mutation status.
5FU dramatically suppressed proliferation of CRC cells with wtp53 but not mutp53
As a classical anticancer drug, 5FU is the most widely used anti-pyrimidine drug in the clinical therapy of human cancers. To establish a proper dose of 5-FU in our system, HCT116 cells were treated with various doses of 5-FU. Cell viability was analyzed by using Cell Counting Kit-8 at 24, 48 and 72 h, respectively. Consistent with our previous study [12], treated cells show a decrease in viability with increasing 5-FU dose and post-treatment time (Figure 2(a)), which suggests that the effect of 5-FU on cell viability is dose- and time dependent.
Figure 2.
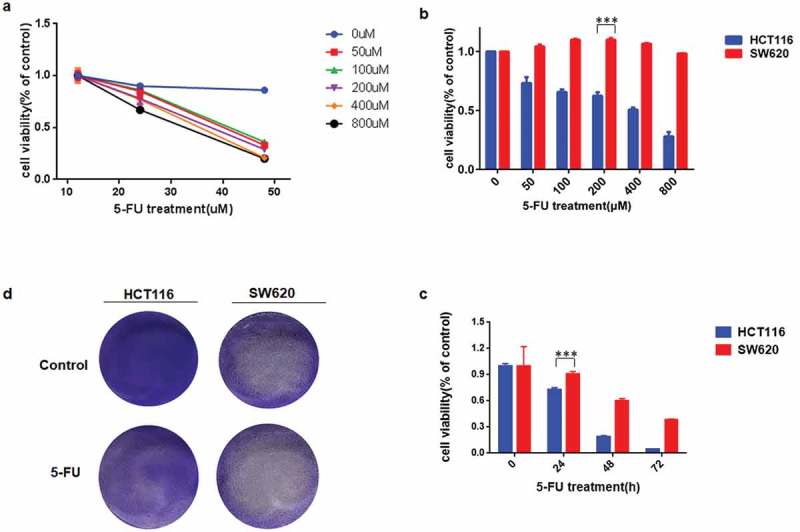
CRC cells with mutant p53 were more resistant to 5FU stimulus than the wild-type p53.a-c, (a) CCK-8 was used to analyze HCT116 cell viability after 5FU treatment. Same test method was designed to analyzed cell viability in HCT116 and SW620 after 50-800μM (b) or 24–72 h (c) 5FU treatments, respectively. d Colony formation was detected using crystal violet staining in HCT116 and SW620 after 2 weeks of 5FU treatment.
To further investigate the influence of 5FU on cell viability with wild-type or mutant p53, HCT116 and SW620 cells were treated with varied doses of 5FU for 24 h (Figure 2(b)) or 200μM 5FU at indicated time points (Figure 2(c)). As a result, cell viability declined significantly after 5-FU treatment in HCT116, but had nearly no change in SW620 cells. Next, colony formation assay was conducted to estimate the effect of 5-FU on cell proliferation for a relatively long period. As shown in Figure 2(d), proliferation was suppressed by 5-FU in HCT116, but not in SW620 cells. These results suggested that mutant p53 makes SW620 cells arise resistance to 5-FU treatments.
P53-mutated cells escape apoptosis and reduce sensitivity to 5-FU
For many malignancies, especially CRC, 5FU is a mainstream chemotherapy drug in long term, which effectively kills tumor cells by causing DNA damage and inducing apoptosis. So whether mutant p53 caused apoptosis as WT p53 did was next investigated. Flow cytometry analysis shows that 5-FU stimulation led to obvious apoptosis in HCT116 cells, but in the contrast, there was no apoptosis happened in SW620 cells (Figure 3(a,b)). To evaluate apoptosis directly, morphological examination was performed with Hoechst 33258 staining. As shown in Figure 3(c), chromatin condensation was observed in HCT116 cells. While nuclei were significantly enlarged at the same magnification after 5FU treatment in SW620 cells (Figure 3(c)).
Figure 3.
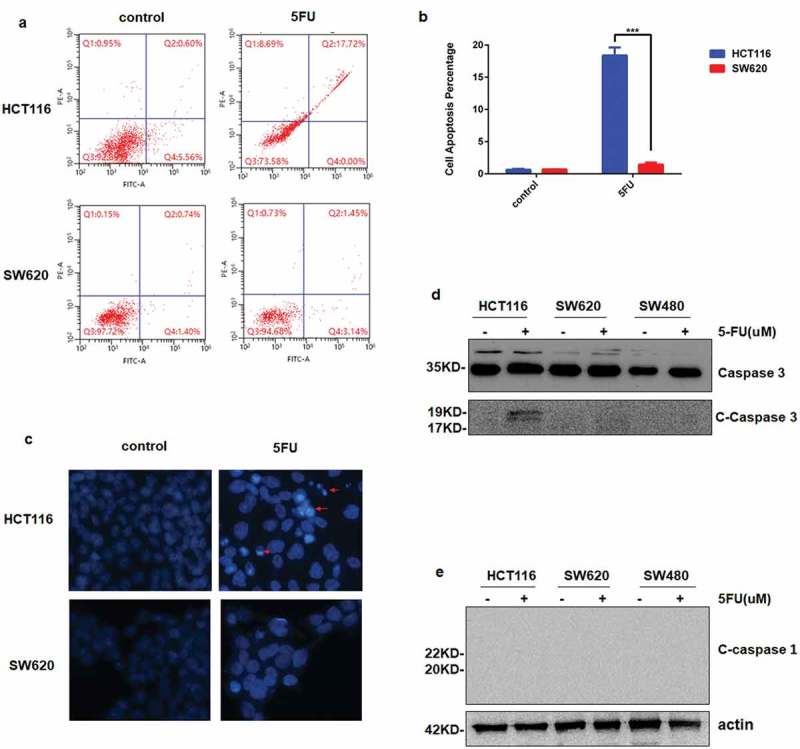
Apoptosis is not induced in CRC cells with mutp53 after 5FU stimuli. a and b, a Fluorescence-activated cell sorting (FACS) assays were used to analyze cell apoptosis after HCT116 WT and SW620 cells were treated with 200 µM 5FU for 24 h. b The percentage of apoptotic cells was calculated from FACS analysis. Data represent the mean±SD of three independent experiments. ***P < 0.001 vs WT. c Hoechst 33,258 morphological examination of apoptosis in HCT116 WT and SW620. Cells were treated with 200 µM 5FU and incubated for 24 h, then stained with Hoechst 33,258. d and e The expression of caspase-3, cleave-caspase 3 (d) and cleave caspase 1 (e) are detected by Western blotting in tree CRC cell lines after 5FU for 24 h.
Furthermore, the expression of cleaved-caspase 3 was up-regulated in HCT116 and completely undetectable in p53 mutant colorectal cells (SW620 and SW480) (Figure 3(d)). Caspase-1, as a marker for pyroptosis, activates inflammatory cytokines, and induces programmed death as opposed to apoptosis [35], was not cleaved in both wild-type and p53 mutant cells (Figure 3(e)). These results indicated that mutant p53 helps cells to resist apoptosis induced by 5-FU, and pyroptosis did not happen in this process.
Mutant p53 could not up-regulate PUMA or p21
As a direct downstream target of WT p53 and an important regulator for apoptosis, PUMA activation was analyzed in this present study. Figure 4(a,c) shows the expression of p53 increased markedly in the presence of 5-FU in HCT116 cells, which was dose- and time dependent. As downstream targets of p53, PUMA and p21 show similar trends. However, the basal level of mutant p53 was much higher in SW620 cells and had nearly no change after 5-FU stimulation (Figure 4(b,d)). More interestingly, PUMA and p21 were not detectable at all (Figure 4(b,d)). Almost identical results were observed in SW480 cells, which is another p53 mutant cell line (Figure 4(e)). That means mutant p53 is over-activated under normal circumstance, but it could not up-regulate the direct downstream targets of WT p53.
Figure 4.
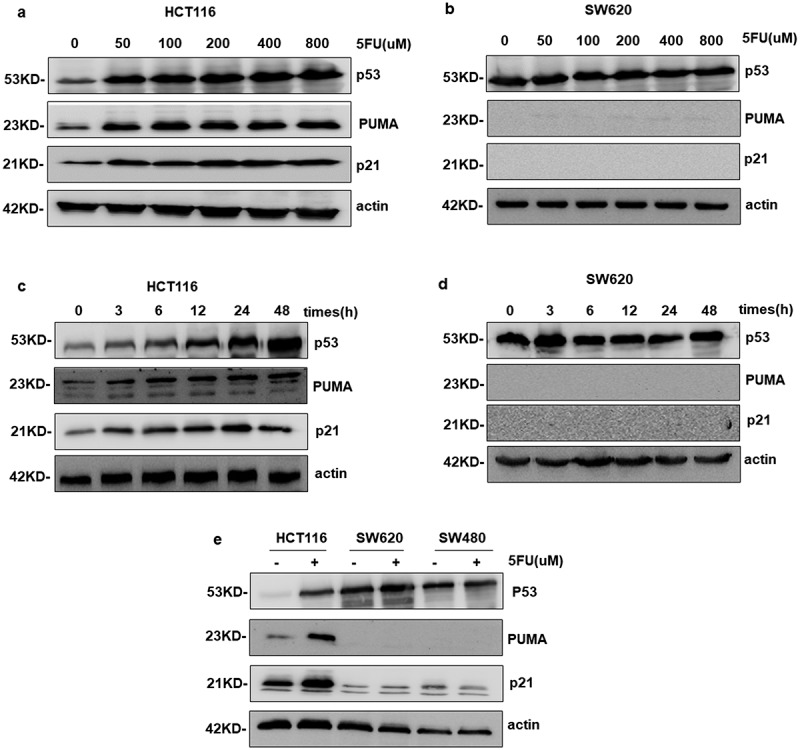
SW620 cells mutated p53 no longer express PUMA and p21. a-e Western blotting analysis the expression levels of p53, PUMA and p21. a and c PUMA and p21 were upregulated with 50–800 μM 5FU treatment for indicated times in HCT116. b and d The gene of PUMA and p21 detected in SW620 was not expressed in the presence of 5FU or not.
PUMA was not activated by p53-dependent stress in p53 mutant cells
The agents of 5FU (5-fluorouracil), Dox (Doxorubicin) and Oxa (Oxaliplatin) are the common choices for tumor chemotherapy, which induce p53-dependent apoptosis. So next, we would like to know except for 5-FU, whether other drugs could activate p53 downstream targets in p53 mutant colon cancer cells, even in other types of cancer cells. As depicted in Figure 5(a), PUMA and p21 were not induced by 5-FU in p53 mutant pancreatic and breast cancer cells (PANC-1 and SKBR-3). Dox and Oxa made a significant induction of p53, PUMA and p21 in HCT116 cells (Figure 5(b)). However, this phenomenon completely disappeared in SW620, SW480, PANC-1 and SKBR-3 cells (Figure 5(b–d)). These results demonstrated drugs that were dependent on p53 activation could not up-regulate its downstream targets in p53 mutant cells, which suggested that cancer patients with mutant p53 may appear more resistance to these kinds of drugs.
Figure 5.
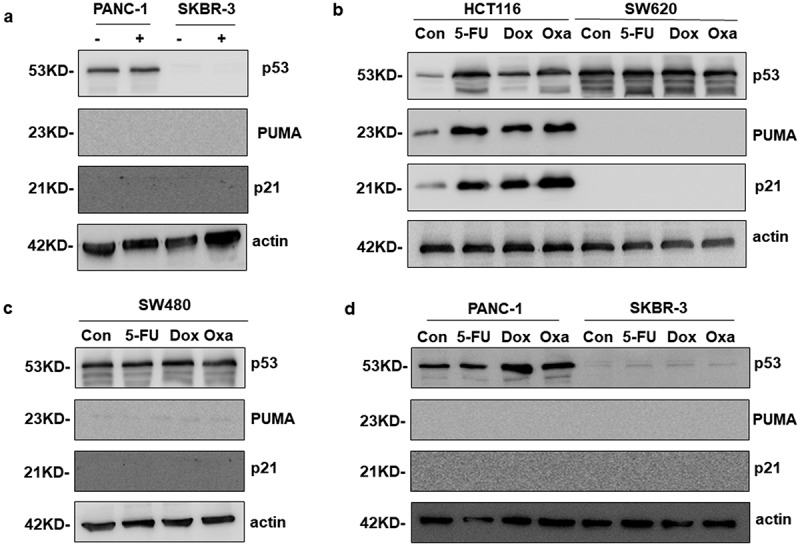
PUMA was not activated by chemotherapy drugs in various cancer cells with mutp53. a Western blotting analysis of PUMA expression after the treatment of 200 μM 5FU in PANC-1 and SKBR-3 cells. b-d The expression of PUMA was tested in multiple cancer cell lines treated with three chemotherapeutics alone.
Mutant p53 cannot bind to PUMA promotor
Previous study showed direct induction of PUMA by endogenous p53 after 5FU treatment in CRC cells [36]. While our results above show that mutant p53 could not activate PUMA, without the underline mechanism. Our hypothesis is, after mutation, the configuration of p53 has been changed, so it may lose the ability of binding to PUMA’s promoter. To verify it, Real-time qPCR experiments and Chromatin Immunoprecipitation (ChIP) assay were performed. As shown in Figure 6(a), 5-FU stimuli enhanced the transcriptional level of PUMA in HCT116 cells, but not in p53 mutant cells (SW620). To further confirm it, WT p53 (YFP-p53) was overexpressed in SW620 cells, which increased PUMA expression obviously (Figure 6(b,c)). Finally, ChIP assay shows WT p53 had a direct interaction with PUMA’s promoter in HCT116 cells, which was expected (Figure 6(d)). While in SW620 cells, mutant p53 did not bind to PUMA’s promoter at all (Figure 6(d)).
Figure 6.
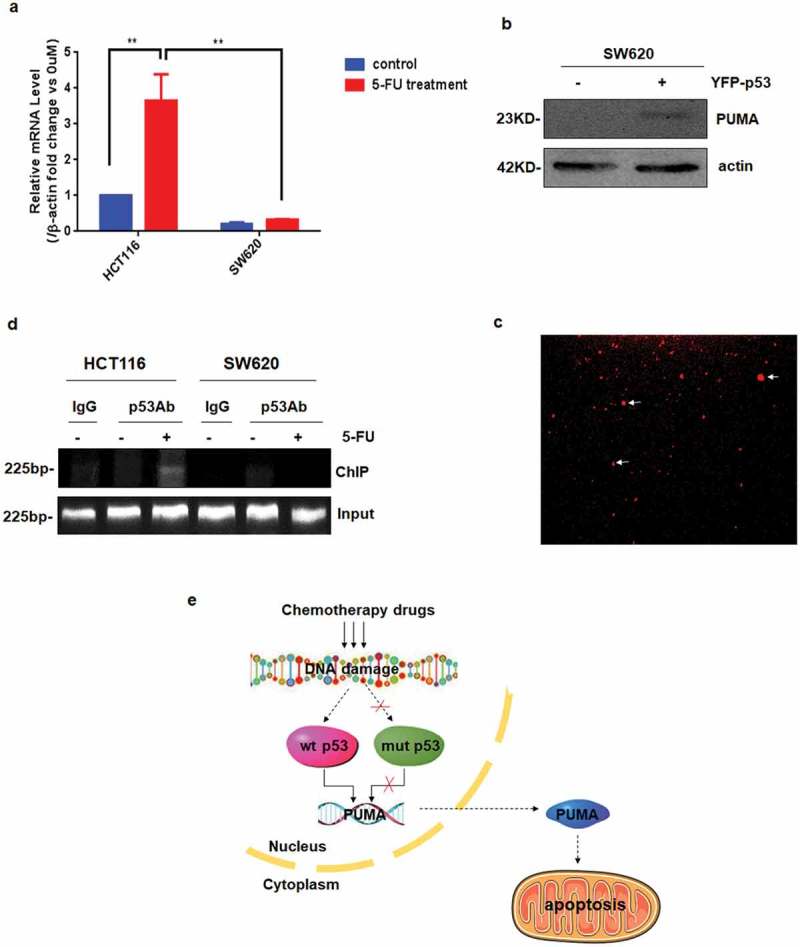
Mutant p53 cannot bind to PUMA promotor. a PUMA mRNA induction by 5FU was analyzed in HCT116 and SW620 by Real-time qPCR and normalized to the housekeeping gene β-actin. The values are the mean ± SEM (n = 3) from a representative experiment. **P < 0.01 vs. controls. b and c Overexpression YFP-p53 in SW620 cell lines. d Chromatin immunoprecipitation (ChIP) was performed on HCT116 and SW620 after 24-h 5FU treatment. IgG was used as a control for the p53-specific antibody. e Molecular mechanism schematic represented that chemotherapy drugs did not induce apoptosis in cancer cells with mutp53.
Taken together, these data indicated that mutant p53 could not bind to PUMA’s promoter to start its transcription, probably due to the conformational change, therefore, lose the function on increasing PUMA expression and promoting apoptosis, which may be one of the reasons for chemotherapy resistance.
Discussion
Tumor-suppressor factors are the most important players in the development of cancer, an important example of which is p53. The research revealed Li-Fraumeni patients with p53 mutations that confer a higher risk of cancer [37,38]. P53 mutations found in 50% of the tumors left the growth out of control, promoting malignant development of the tumor [39,40], and were likely to be a clinical indicator of tumor [41,42]. For the therapy of cancer, including CRC, targeting mutp53 has become an increasingly attractive strategy to re-sensitize cancer cells to chemotherapies [43,44]. Therefore, it is significant to explore the role of mutp53 in cancer cells following chemotherapeutic agents.
The primary focus of our research was to study the effect of traditional drugs (5FU) on the proliferation of CRC cells with mutp53. Cell viability was analyzed by using Cell Counting Kit-8 and colony formation assay was conducted to estimate the effect of 5-FU on cell proliferation for a relatively long period (Figure 2), which suggested that mutant p53 makes SW620 cells arise resistance to 5-FU treatments. We wanted to know whether the tolerance of CRC cells with p53-R273H to 5FU was associated with apoptotic escape. Flow cytometry analysis and Hoechst 33,258 staining experiments make it clear that the p53 mutant plays an important role in the escape process of death (Figure 3(a,c)). The levels of c-caspase 3, typical indicators of mitochondria-mediated apoptosis, were significantly up-regulated by 5FU, in contrast, the activation of c-caspase 3 was not uniform, which did undetectable in SW620 and SW480 (Figure 3(d)). Pyroptosis (proinflammatory programmed cell death) is another type of programmed cell death besides apoptosis, which triggered by various pathological stimuli, such as stroke, heart attack or cancer. It is important for controlling microbial infections. Caspase 1 is the main activator of pyroptosis and a programmed process of cellular self-destruction mediated by caspases [35,45–47]. However, there was not cleaved in both wild-type and p53 mutant cells (Figure 3(e)). The results have an important implication that no caspase 1-dependent pyroptosis occurs in all of the CRC cells researched by us.
It is widely believed that tumor cell growth can be inhibited by p53-mediated cell cycle arrest, apoptotic cell death, and/or cellular senescence. In agreement with previous reports, lots of data indicate that the ability of p53 to regulate downstream signals that induce cell cycle arrest and apoptosis by upregulation of P21, BAX, PUMA and NOXA are essential for suppressing tumor proliferation in vivo and in vitro [13,15,48–50]. The tumor-suppressor protein of p53 up-regulates the expression of BAX that has been shown to be involved in p53-mediated apoptosis. Previous studies indicated that the crystal structure of the p53 core domain bound to naturally occurring RE located at the promoter of the Bcl-2-associated X protein (BAX) gene, which contains a one base-pair insertion between the two half-sites [51]. Upon induction of apoptosis, BAX becomes organelle membrane-associated, and in particular, mitochondrial membrane associated.
Lower expression of BAX results in better survival in cells with mutant p53. Interestingly, low BAX expression was also observed in the presence of wild-type p53, which leads to worse results [52]. In fact, all tumor-derived p53 mutants failed to activate BAX transcription, because of the lack of DNA binding and transactivation capabilities. However, some mutants retain the ability to activate a subset of the p53 target gene, like p21 [53]. The majority of BAX is found in the cytosol, but upon initiation of apoptotic signaling, BAX undergoes a conformational shift. BAX is believed to interact with, and induce the opening of the mitochondrial voltage-dependent anion channel, VDAC. The functional consequence of pro-apoptotic signaling is mitochondrial membrane perturbation and release of cytochrome c in the cytoplasm, where it forms a complex or apoptosome with apoptotic protease activating factor 1 (APAF1) and the inactive form of caspase-9.
When the WT p53 transformed into a mutant p53, it may affect the intrinsic apoptosis via three ways by regulating BAX expression and activity. Firstly, it will lose the function of up-regulating BAX transcription, which directly leads to the decrease of BAX protein level and inhibits apoptosis. Secondly, it will also lose the function of up-regulating PUMA, which is a critical upstream activator of BAX, thus decreasing BAX activity and apoptosis, as we demonstrated in this present study. Thirdly, previous studies have shown that WT p53 can directly activate BAX by their interaction and changing BAX conformation [54]. Mutant p53 may fail to do this and block intrinsic apoptosis, which should be further verified in future. Of course, there may exist some new targets that are involved in mutant p53 inhibited apoptosis, waiting to be discovered.
How p53’s use as a diagnostic marker in view of the findings reported. The clinical data analysis of 95 patients with chronic lymphocytic leukemia shows that 65% of patients showed TP53 changes at the time of diagnosis, with a median follow-up of 7 years from the initial diagnosis of chronic lymphocytic leukemia [55]. Mutations of the TP53 tumor-suppressor gene are the most frequent of all somatic genomic alterations in head and neck squamous cell carcinomas (HNSCCs), with a mutation frequency in non-human papilloma virus-associated HNSCC cases ranging from 75% to 85% [56].
However, this marker should be used with caution. It is reported that about 60% allele show concomitant deletion among the missense mutation of p53. The remaining group (40%) does not undergo LOH, retaining a wild-type p53 allele, in which wild-type p53 expression and function may be inhibited through a dominant-negative mechanism. Furthermore, a smaller but considerable fraction of mutant p53 (around 10%) are nonsense mutations that give rise to truncated p53 proteins with impaired wild-type function [57]. We need to be approached with caution because its mutation is not completely free of wild-type function. In addition, some studies have found that most tumor-associated p53 mutations occur in the core region responsible for sequence-specific DNA binding highlights the importance of its transcriptional activity. The mutant p53 can activate Rac1 (a small GTPase) through that inhibited the interaction of SENP1 (an important posttranslational protein modification to regulate protein stability and/or activity) with Rac1. It demonstrates that mutant p53 is a novel regulator for Rac1 and reveal that Rac1 activation by mutant p53 is a novel and critical mechanism in tumorigenesis [25]. Moreover, clinical patient records of CRC indicated mutant p53 cells escape chemotherapy-induced cell death by upgrading c-Yes and YAP transcript levels [58]. The expression of p53 is not rigorous for the diagnosis of cancer because that it may be regulated by other signaling pathways.
Generally, mutp53 regulates some genes that are also regulated by WT p53 but the outcome is diametrically opposed [59]. Taking into account these findings, we focus on the expressions of the PUMA and p21, which accumulated in WT p53 cancer cells with time- and dose dependent after 5FU treatment, yet both proteins were inactivated in the presence of mutant p53 (Figures 4 and 5). Consequently, we can make a preliminary judgment that cancer patients with mutant p53 may appear more resistance to chemotherapeutics by not regulate its downstream targets in the wild-type CRC cells.
Based on our results, we believe that the mutant p53 motif may alter its activity through the following ways. The core region of p53 shows as a tetramer that binds to DNA from two different rotation angles according to the cBioportal database, the TP53 website and the International Agency for Research on Cancer (IARC) TP53 database [50]. The structure of mutant p53 undergoes a certain degree of change that the tetrameric structure is likely to refold. Therefore, some p53 related active sites furtherly exposing and affecting its original activity. In addition, the p53’s cocrystal structure with DNA suggests direct binding of positively charged Arg273 with the negatively charged phosphate backbone of DNA [51]. Recent work has found that the lowering pHi attenuated the tumorigenic effects of p53-R273H. The mutant p53 (p53-R273H) decreased its transcriptional activity and attenuated the DNA damage response with high pHi in fibroblasts and breast cancer cells [60]. We can speculate that the activity of mutant p53 is associated with dynamic changes in pHi or dynamic micro-environmental stress in cancer.
Besides, mutant p53 possibly decreased PHLPP2 mRNA level [61], through which activating AKT, promoting cell cycle progression and inhibiting apoptosis [62,63]. Recent reports show that mutant p53 regulates PD-L1 expression by either transcriptional or posttranscriptional mechanisms mediated by PI3K/AKT [64]. One of the hypotheses is mutant p53 may up-regulate c-myc that binding to the promoter regions of the genes coding for PD-L1 [65,66]. In conclusion, the mutant site of p53 may lead to its conformational change, which blocks the active area that is responsible for binding to target DNA, like PUMA’s promoter. However, this conformational change may also sensitize other areas, which results in mutant p53 binding to some new target DNAs, like PHLPP2, c-myc or PD-L1.
Until now, our results demonstrated that PUMA expression in all analyzed cancer cells is strongly related to p53 status, as well as in mRNA levels. 5-FU stimuli enhanced the transcriptional level of PUMA in HCT116 cells, but not in p53 mutant cells (SW620) (Figure 6(a)). Furthermore, PUMA upregulation was clearly observed by overexpression of WT p53 (YFP-p53) in SW620 (Figure 6(b,c)), which strongly supports the previous results that WT p53 can induce PUMA expression to initiate apoptosis but not mutp53 in CRC cells after exposure to chemotherapeutic agents. WT p53 had a direct interaction with PUMA’s promoter in HCT116 cells, which was expected. Notably, mutant p53 was no longer translocate into the nucleus and bound to the promoter of PUMA to stimulate its transcription (Figure 6(d)).
In summary, our data testified that cancer cells with mutp53 are more resistant to chemotherapeutic agents than those of WT, which is related to PUMA-induced apoptosis and p21-mediated cell cycle arrest. Here, ChIP assay results showed that mutant p53 could not bind to the promoter of PUMA, which inactivates PUMA transcription. Furthermore, overexpression of p53 in mutant p53 cell could induce PUMA. Our results have important implications for further exploring the mechanism of mutant p53 in human cancer cells that promote chemotherapeutic resistance, which provides a theoretical basis for clinical trials.
Funding Statement
This work was supported by the National Natural Science Foundation of China [31801140 and 31701132]; Fundamental Research Funds for Central Universities of the Central South University (CN) [Nos. 531118040098 and 14700-502044001]; the Basic Research Program of Shenzhen Municipal Science and Technology Innovation Committee [JCYJ20160530192802733].
Acknowledgments
We would like to thank the support of the National Natural Science Foundation of China (31801140 and 31701132), the Basic Research Program of Shenzhen Municipal Science and Technology Innovation Committee (JCYJ20160530192802733), the Fundamental Research Funds for the Central South Universities (Nos. 531118040098 and 14700-502044001), and the start funds from College of Biology, Hunan University.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Vega P, Valentín F, Cubiella J.. Colorectal cancer diagnosis: Pitfalls and opportunities. World J Gastrointest Oncol. 2015;7:422––33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Arnold M, Sierra MS, Laversanne M, et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683–691. [DOI] [PubMed] [Google Scholar]
- [3].Sullivan KD, Galbraith MD, Andrysik Z, et al. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018;25:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Krause K, Wasner M, Reinhard W, et al. The tumour suppressor protein p53 can repress transcription of cyclin B. Nucleic Acids Res. 2000;28:4410–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Taylor WR, Schonthal AH, Galante J. Stark GR. p130/E2F4 binds to and represses the cdc2 promoter in response to p53. J Biol Chem. 2001;276:1998–2006. [DOI] [PubMed] [Google Scholar]
- [7].Wang H, Yan C. A small-molecule p53 activator induces apoptosis through inhibiting MDMX expression in breast cancer cells. Neoplasia. 2011;13:611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683. [DOI] [PubMed] [Google Scholar]
- [9].Yu J, Zhang L, Hwang PM, et al. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673. [DOI] [PubMed] [Google Scholar]
- [10].Shamas-Din A, Kale J, Leber B, et al. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yu J, Wang Z, Kinzler KW, et al. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100:1931–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Huanan Wang LZ. PUMA mediates the combinational therapy of 5-FU and NVP-BEZ235 in colon cancer. Oncotarget. 2015;6:14385–14398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang S, Zhu Z, Zhang X, et al. Idelalisib induces PUMA-dependent apoptosis in colon cancer cells. Oncotarget. 2017;8:6102–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fruman DA, Cantley LC. Idelalisib – a PI3Kdelta inhibitor for B-cell cancers. N Engl J Med. 2014;370:1061–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang L, Huang X, Li W. Pazopanib, a novel multi-kinase inhibitor, shows potent antitumor activity in colon cancer through PUMA-mediated apoptosis. Oncotarget. 2017;8:3289–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Paoluzzi L, Cacavio A, Ghesani M, et al. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin Sarcoma Res. 2016;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sun L, Huang Y, Liu Y, et al. Ipatasertib, a novel Akt inhibitor, induces transcription factor FoxO3a and NF-kappaB directly regulates PUMA-dependent apoptosis. Cell Death Dis. 2018;9:911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim SB, Dent R, Im SA, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18:1360–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol Cb. 1994;4:1–7. [DOI] [PubMed] [Google Scholar]
- [20].Rodrigues NR, Rowan A, Smith ME, et al. p53 mutations in colorectal cancer. Proc Natl Acad Sci U S A. 1990;87:7555–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Muller PAJ, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2. [DOI] [PubMed] [Google Scholar]
- [22].Levine AJ, Wu MC, Chang A, et al. The spectrum of mutations at the p53 locus. Evidence for tissue-specific mutagenesis, selection of mutant alleles, and a “gain of function” phenotype. Ann N Y Acad Sci. 1995;768:111–128. [DOI] [PubMed] [Google Scholar]
- [23].Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang Y, Coillie SV, Fang JY, et al. Gain of function of mutant p53: R282W on the peak? Oncogenesis. 2016;5:e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yue X, Zhang C, Zhao Y, et al. Gain-of-function mutant p53 activates small GTPase Rac1 through SUMOylation to promote tumor progression. Genes Dev. 2017;31:1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schulz-Heddergott R, Stark N, Edmunds SJ, et al. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits stat3-mediated tumor growth and invasion. Cancer Cell. 2018;34:298–314 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Walerych D, Lisek K, Sommaggio R, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol. 2016;18:897–909. [DOI] [PubMed] [Google Scholar]
- [28].Weissmueller S, Manchado E, Saborowski M, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell. 2014;157:382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tan BS, Tiong KH, Choo HL, et al. Mutant p53-R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 2015;6:e1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wiegering A, Matthes N, Mühling B, et al. Reactivating p53 and inducing tumor apoptosis (RITA) enhances the response of RITA-sensitive colorectal cancer cells to chemotherapeutic agents 5-fluorouracil and oxaliplatin. Neoplasia. 2017;19:301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Raguz S, Yague E. Resistance to chemotherapy: new treatments and novel insights into an old problem. Br J Cancer. 2008;99:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- [33].Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33:125–36 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Audrey P, Ewy M, Shunsuke K, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2010;28:622–629. [DOI] [PubMed] [Google Scholar]
- [35].Lv Y, Zhang Y, Rubinsky B. Molecular and histological study on the effects of electrolytic electroporation on the liver. Bioelectrochemistry. 2018;125:79–89. [DOI] [PubMed] [Google Scholar]
- [36].Wang P, Yu J, Zhang L The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci USA. 2007;104(10):4054–4059. DOI: 10.1073/pnas.0700020104. Epub 2007 Feb 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–1256. [DOI] [PubMed] [Google Scholar]
- [38].Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. [DOI] [PubMed] [Google Scholar]
- [39].Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. [DOI] [PubMed] [Google Scholar]
- [40].Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9:95–107. [DOI] [PubMed] [Google Scholar]
- [41].Punt CJA, Koopman M, Vermeulen L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat Rev Clin Oncol. 2016;14:235. [DOI] [PubMed] [Google Scholar]
- [42].Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. 2018;15:13–30. [DOI] [PubMed] [Google Scholar]
- [43].Alam SK, Yadav VK, Bajaj S, et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 2016;23:707–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].He C, Li L, Guan X, et al. Mutant p53 gain of function and chemoresistance: the role of mutant p53 in response to clinical chemotherapy. Chemotherapy. 2017;62:43–53. [DOI] [PubMed] [Google Scholar]
- [45].Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 2017;38:261–271. [DOI] [PubMed] [Google Scholar]
- [46].Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li T, Kon N, Jiang L, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25:114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bykov VJN, Eriksson SE, Bianchi J, et al. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18:89–102. [DOI] [PubMed] [Google Scholar]
- [51].Chen Y, Zhang X, Dantas Machado AC, et al. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013;41:8368–8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Farooqi AA, de la Roche M, Djamgoz MBA, et al. Overview of the oncogenic signaling pathways in colorectal cancer: mechanistic insights. Semin Cancer Biol. 2019;58:65–79. [DOI] [PubMed] [Google Scholar]
- [53].Campomenosi P, Monti P, Aprile A, et al. p53 mutants can often transactivate promoters containing a p21 but not Bax or PIG3 responsive elements. Oncogene. 2001;20:3573–3579. [DOI] [PubMed] [Google Scholar]
- [54].Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. [DOI] [PubMed] [Google Scholar]
- [55].Liu YC, Margolskee E, Allan JN, et al. Chronic lymphocytic leukemia with TP53 gene alterations: a detailed clinicopathologic analysis. Mod Pathol. 2019. DOI:10.1038/s41379-019-0356-z [DOI] [PubMed] [Google Scholar]
- [56].Tanaka N, Zhao M, Tang L, et al. Gain-of-function mutant p53 promotes the oncogenic potential of head and neck squamous cell carcinoma cells by targeting the transcription factors FOXO3a and FOXM1. Oncogene. 2018;37:1279–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Blandino G, Di Agostino S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J Exp Clin Cancer Res. 2018;37:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Touil Y, Igoudjil W, Corvaisier M, et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin Cancer Res off J Am Assoc Cancer Res. 2014;20:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Stambolsky P, Tabach Y, Fontemaggi G, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell. 2010;17:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].White KA, Ruiz DG, Szpiech ZA, et al. Cancer-associated arginine-to-histidine mutations confer a gain in pH sensing to mutant proteins. Sci Signal. 2017;10(495). pii: eaam9931. DOI:10.1126/scisignal.aam9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nowak DG, Katsenelson KC, Watrud KE, et al. The PHLPP2 phosphatase is a druggable driver of prostate cancer progression. J Cell Biol. 2019;218:1943–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Brognard J, Sierecki E, Gao T, et al. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–931. [DOI] [PubMed] [Google Scholar]
- [63].Newton AC, Trotman LC. Turning off AKT: PHLPP as a drug target. Annu Rev Pharmacol Toxicol. 2014;54:537–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chen J, Jiang CC, Jin L, et al. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016;27:409–416. [DOI] [PubMed] [Google Scholar]
- [65].Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gutgemann I, Eilers M, et al.MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Atsaves V, Tsesmetzis N, Chioureas D, et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia. 2017;31:1633–1637. [DOI] [PubMed] [Google Scholar]