

ABSTRACT
Osteoporosis and sarcopenia (osteosarcopenia (OS)) are twin-aging diseases. The biochemical crosstalk between muscle and bone seems to play a role in OS. We have previously shown that osteocytes produce soluble factors with beneficial effects on muscle and vice versa. Recently, enhanced FGF9 production was observed in the OmGFP66 osteogenic cell line. To test its role in myogenic differentiation, C2C12 myoblasts were treated with recombinant FGF9. FGF9 as low as 10 ng/mL inhibited myogenic differentiation, suggesting that FGF9 might be a potential inhibitory factor produced from bone cells with effects on muscle cells. FGF9 (10–50 ng/mL) significantly decreased mRNA expression of MyoG and Mhc while increasing the expression of Myostatin. Consistent with the phenotype, RT-qPCR array revealed that FGF9 (10 ng/mL) increased the expression of Icam1 while decreased the expression of Wnt1 and Wnt6 decreased, respectively. FGF9 decreased caffeine-induced Ca2+ release from the sarcoplasmic reticulum (SR) of C2C12 myotubes and reduced the expression of genes (i.e. Cacna1s, RyR2, Naftc3) directly associated with intracellular Ca2+ homeostasis. Myogenic differentiation in human skeletal muscle cells was similarly inhibited by FGF9 but required higher doses of 200 ng/mL FGF9. FGF9 was also shown to stimulate C2C12 myoblast proliferation. FGF2 and the FGF9 subfamily members FGF16 and FGF20 also inhibited C2C12 myoblast differentiation and enhanced proliferation. Intriguingly, the differentiation inhibition was independent of proliferation enhancement. These findings suggest that FGF9 may modulate myogenesis via a complex signaling mechanism.
KEYWORDS: FGF9, C2C12 myoblast, human skeletal muscle cells, differentiation, proliferation
Introduction
Bone and muscle are the two major integrated components in the musculoskeletal system. Osteoporosis/osteopenia (loss of bone mass) and sarcopenia (progressive muscle loss with a greater and disproportional loss of muscle force/strength) are consequences of the aging process and occur concurrently. Furthermore, musculoskeletal diseases are the most common cause of chronic disabilities worldwide, afflicting over 1.7 billion humans [1,2].
Bone and muscle tissues produce and secrete “hormone-like factors” called osteokines and myokines, respectively, demonstrating that bone and muscle can act as endocrine organs that can affect glucose, energy, and other metabolic functions by mutually influencing each other and other tissues [3,4].
Muscles secrete cytokines and growth factors able to act in a paracrine or endocrine manner on a variety of tissues [5,6]. Some factors secreted from muscle such as myostatin [7], osteonectin [8], irisin [9], and muscle-derived FGF-2 [10] regulate bone regeneration and metabolism. Our recent studies show that β-aminoisobutyric acid (BAIBA) secreted from muscle is a bone-protective factor that protected mice against the loss of bone induced by unloading [11]. This appears to be mediated by the ability of BAIBA to protect osteocytes against cell death induced by reactive oxygen species (ROS) [11].
Bones secrete a host of osteokines [12] that have clear paracrine and endocrine effects, such as IGF, TGFβ, BMP, FGF23, sclerostin, osteocalcin, WNTs, prostaglandin E2 (PGE2, etc. (reviewed in [13]). Some of these molecules have been or are being explored as therapeutic agents for bone diseases, showing the importance of understanding the cell biology of the effects of these factors not only in bone but also in other tissues [14]. We demonstrated that bone cell conditioned medium accelerates C2C12 cell differentiation, enhances calcium homeostasis of myotubes, and increases muscle contractile force, and that these effects could be attributed to WNT and PGE2 signaling pathways via WNT3a and PGE2 [3,15,16].
Fibroblast growth factors (FGFs) are secreted signaling proteins and are widely expressed in various tissues of the human body. In humans and mice, the FGF family comprises 22 members. FGFs are divided into seven subfamilies based on their evolutionary relationships [17]. Most FGFs play roles as paracrine or endocrine signals in embryonic development [18], organogenesis [19], angiogenesis [20], wound healing [21], metabolism [22], and cancer development [23]. They are involved in development, health, and disease in major organs including the liver, kidney, brain, and bone [24,25]. Impairment of FGFs involved in many diseases [26,27]. FGF ligands such as FGF2, 3, 4, 9, and 18 are involved in normal skeletal growth [28]. Various FGFs regulate skeletal muscle stem cells (satellite cells) and are essential for self-renewal of skeletal muscle stem cells and are required for the maintenance and repair of skeletal muscle [29], regulation of proliferation and differentiation (FGF13) [30], and modulation of skeletal muscle mass (FGF19) [31].
Specifically, the FGF9 subfamily consists of FGF9, FGF16, and FGF20 [32–34]. FGF9 and its subfamily members are expressed in bone [35–37]. The direction of the FGF9/16/20 signal polarizes the asymmetric division of mesenchyme/muscle blastomeres [38]. FGF9 signaling inhibits airway smooth muscle differentiation in mouse lung [39]. The specific role for FGF9 in skeletal muscle differentiation is elusive. Recent studies from our groups showed that FGF9 mRNA expression is highly enriched in osteocytes in the OmGFP66 osteogenic cell line as compared to the osteoblast population [40,41]. We have reasoned that this molecule could be functioning as an osteokine based on our observations (Huang & Brotto, Unpublished Results) that fibroblast-conditioned media can inhibit C2C12 differentiation. Thus, we hypothesized that FGF9 could exert an inhibitory effect on skeletal muscle cells, which may help maintain the fine balance required for skeletal muscle turnover and regeneration.
We examined the effects of FGF9 in the C2C12 myogenic cell line and in human skeletal muscle cells. FGF9 was found to inhibit the myogenic differentiation of both C2C12 muscle cells and human skeletal muscle cells (SkMC), via a complex signaling mechanism that involved myogenic regulatory factors and genes associated with calcium homeostasis.
Materials and methods
Materials
Alpha’s modification of minimal essential media (αMEM), Dulbecco’s modified Eagle’s media (DMEM)/high glucose, Dulbecco’s phosphate-buffered saline (DPBS), penicillin-streptomycin (P/S) 10,000 U/mL each, and trypsin-EDTA 1× solution were obtained from Mediatech Inc. (Manassas, VA, USA). Calf serum (CS), fetal bovine serum (FBS), horse serum (HS), and caffeine were obtained from Thermo Fischer Scientific Inc. (Waltham, MA, USA). Bovine serum albumin (BSA), diamidino-2-phenylindole (DAPI), and MTT assay kit were from Sigma-Aldrich (St Louis, MO, USA). TRI reagent was obtained from Molecular Research Center, Inc. (Cincinnati, OH, USA). High capacity cDNA reverse transcription kit was from Applied Biosystems. (Foster city, CA, USA). RT2 Real-TimeTM SYBR green/Rox PCR master mix and Mouse Signal Transduction PathwayFinder PCR Array ware from SABiosciences. (Valencia, CA, USA). Carboxyfluorescein (CFS)-conjugated mouse monoclonal anti-human Myosin Heavy Chain antibody (Catalog number: IC4470F) was from R&D Systems Inc. (Minneapolis, MN, USA); This antibody has also been previously validated for specificity in rodent muscle cells [42]. Anti-GDF8/Myostatin antibody (ab71808) was from Abcam (Cambridge, MA, USA). Previous studies validated this antibody specificity in rodent muscle cells [43]. 4x Laemmli Sample Buffer, TransBlot @TurboTM Mini-size PVDF membrane was from Bio-Rad (Hercules, CA, USA). Propidium iodide flow cytometry kit was obtained from Abcam (Cambridge, MA, USA). Fura-2/AM was obtained from Life Technologies (Grand Island, NY, USA). CellTiter 96® AQueous One kit and recombinant human basic FGF were obtained from Promega (Madison, WI, USA). Recombinant Mouse FGF-9, Recombinant Human FGF-9, Recombinant Mouse FGF-23, Recombinant Human FGF-16, and Recombinant Human FGF-20 were from R&D Systems Inc. (Minneapolis, MN, USA). C2C12 cells were obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). C2C12 cells were authenticated and tested for mycoplasma contamination by ATCC. Human Skeletal Muscle Cells (SkMC), Skeletal Muscle Cell Growth Medium and Skeletal Muscle Cell Differentiation Medium were obtained from ZenBio (Research Triangle Park, NC, USA). The SkMC were authenticated and tested for mycoplasma contamination by ZenBio.
Methods
No in vivo experiments were conducted in this study.
Animals
Six-month-old male C57BL/6 mice from Jackson Laboratory were used for isolation of intact extensor digitorum longus (EDL) and soleus (SOL) muscles for detection of FGF9 gene expression, following humane euthanasia via cervical dislocation. All animal procedures were performed according to an approved IACUC protocol at the University of Missouri–Kansas City (UMKC) and conformed to relevant federal guidelines. The UMKC animal facility is operated as a specific pathogen-free, AAALAC approved facility. Animal care and husbandry at UMKC meet the requirements in the Guide for the Care and use of Laboratory Animals (eighth edition), National Research Council. Animals were group housed and maintained on a 12-h light/dark cycle with ad libitum food and water at a constant temperature of 72°F and humidity of 45–55%. Daily health check inspections were performed by qualified veterinary staff and/or animal care technicians. To detect FGF9 gene expression, we lysed the tissue sample in 700 μl TRI Reagent, homogenized the tissue. Then, we followed the methods described in “RNA isolation and Real-time quantitative PCR (RT-qPCR)” below.
C2C12 and human skeletal muscle cell culture conditions
C2C12 myoblasts were cultured following our own previously published protocols [3,44]. Briefly, cells were grown at 37°C in a controlled humidified 5% CO2 atmosphere in growth medium (GM), DMEM/high glucose +10% FBS (100 U/mL P/S) and maintained at 40−70% cell density. Under these conditions, myoblasts proliferate but do not differentiate into myotubes. For experiments, cells were plated at 10 × 104 cells/well in six-well plates in GM and medium was changed every 48 h. To induce differentiation into myotubes, when the myoblasts reached about 75% confluence, GM was switched to differentiation medium (DM), DMEM/high glucose +2% horse serum (HS) (100 U/mL P/S). Fully differentiated, functional myotubes were formed within 5–7 days. During differentiation, medium was changed every 48 h.
SkMC were cultured following the protocol from ZenBio. Briefly, cells were grown at 37°C and 5% CO2 atmosphere in Skeletal Muscle Cell Growth Medium and maintained at 40−70% cell density. Under these conditions, myoblasts proliferate but do not differentiate into myotubes. For experiments, cells were plated at 15 × 104 cells/well in 6-well plates in Skeletal Muscle Cell Growth Medium, medium was changed every 48 h. To induce SkMC differentiation into myotubes, when SkMC reached 80% confluence, Skeletal Muscle Cell Growth Medium was switched to Skeletal Muscle Cell Differentiation Medium. Fully differentiated, functional myotubes were formed within 2–3 days. During differentiation, medium was changed every 48 h.
C2C12 and SkMC cell morphometry and immunostaining
Cell Morphology
Phase-contrast images were taken with a LEICA DMI-4000B inverted microscope equipped with a 14-BIT CoolSNAP CCD camera (Photometrics), using the LEICA LAS imaging software for calibration (Leica microsystems) and Olympus IX73 inverted microscope equipped with a Hamamatsu digital camera C11440, using the CellSens Dimension software for calibration.
Immunostaining
Experiments were performed following our published protocols [15,42,44,45]. Briefly, cells were fixed with neutral buffered formalin and permeabilized with 0.1% Triton X-100 in PBS. Myosin heavy chain (MHC) was detected with Carboxyfluorescein (CFS)-conjugated mouse monoclonal anti-human Myosin Heavy Chain antibody (1:50) at room temperature for 30 min and counterstained with DAPI. Fluorescent images were taken using a 10X or 20X LEICA FLUO objective with the LEICA system and Olympus system described above or using a Nikon Eclipse TE300 Inverted Fluorescence Microscope.
Fusion index
To quantify myogenic differentiation of C2C12 and SkMC after treatments, the fusion index (FI) was calculated, where FI is defined as: (nuclei within myosin heavy chain-expressing myotubes/total number of myogenic nuclei) × 100 [46]. We conducted three independent experiments, with three areas per well randomly selected for the measurements. Approximately 2,000 nuclei of each area were analyzed.
Treatment of C2C12 cells with FGFs
C2C12 cells were plated in six-well plates, at 10 × 104 cells/well, and incubated overnight to allow the cells to attach and grow. The medium of C2C12 myoblasts was changed from GM to DM with various concentrations of FGF9, FGF2, FGF23, FGF16, and FGF20, respectively. Forty-eight hours later, medium was changed with fresh DM without test factors. At day 3 of differentiation, C2C12 cells were analyzed according to “C2C12 and SkMC Morphometry and Immunostaining” described above.
Pretreatment of C2C12 cell with differentiation media to reduce/stop proliferation
C2C12 cells were plated in six-well plates, at 10 × 104 cells/well, and incubated overnight to allow the cells to attach and grow. The medium of C2C12 myoblasts was changed from GM to DM for 48 h, then changed from DM to fresh DM with various concentrations of FGF9 2 ng-50 ng/mL. Forty-eight hours later, medium was changed with fresh DM without FGF9. At day 3 of differentiation, C2C12 cells were analyzed according to “C2C12 and SkMC Morphometry and Immunostaining” described above.
Treatment of SkMC cells with recombinant human FGF9
SkMC were plated in six-well plates, 40 × 104/well with Skeletal Muscle Cell Growth Medium and incubated overnight as previously described to allow the cells to attach and grow. Skeletal Muscle Cell Growth Medium was switched to Skeletal Muscle Cell Differentiation Medium with 50–200 ng/mL recombinant human FGF9. Forty-eight hours later, medium was changed to fresh Skeletal Muscle Cell Differentiation Medium without test factors. At day 3 of differentiation, SkMC cells were analyzed according to “C2C12 and SkMC Cell Morphometry and Immunostaining” described above.
RNA isolation and real-time quantitative PCR (RT-qPCR)
Total RNA was extracted from the cells with TRI reagent according to the manufacturer’s protocol and 1.0 μg complementary DNA (cDNA) was synthesized by reverse transcription using the high capacity cDNA reverse transcription kit. RT-qPCR was performed using the RT2 Real-TimeTM SYBR green/Rox PCR master mix. RT-qPCR primers used in this study are summarized in Supplemental Table 1. RT-qPCR was run in a 25 μl reaction volume on 96-well plates with the StepOnePlus instrument. Relative quantitation of target gene expression was calculated using the 2^-ΔΔCt method with normalization to Gadph (Glyceraldehyde-3-phosphate dehydrogenase) as a housekeeping gene. Data were then expressed as the fold or percentage change compared to the untreated controls. All experiments were repeated in triplicate.
RT-PCR gene arrays
We used the Mouse Signal Transduction Pathway Finder PCR Array that monitors 10 signaling pathways to detect gene expression changes in cells treated with FGF9. cDNA was synthesized using the RT2 First Strand Kit (this kit contains genomic DNA elimination buffer to eliminate genomic DNA) and the PCR Array was run according to the manufacturer’s instruction including a threshold of 0.25. As above, data were analyzed using the RT2 Profiler™ PCR Array Data Analysis Software; Ct values were normalized to six built-in reference housekeeping genes, genomic DNA control, reverse transcription control, and positive PCR control. We used this analytical software (http://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/) to set the significance of up/downregulation of all tested genes at twofold difference. Two independent experiments were performed.
Protein sample preparation and western blotting
C2C12 myoblasts cultured in six-well plates were washed three times with ice-cold Dulbecco’s phosphate-buffered saline before being lysed by RIPA buffer [1× Tris-buffered saline (TBS), 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.004% sodium azide] (Bio-Rad) with 1% cocktail of proteinase and phosphatase inhibitors (Sigma-Aldrich). Lysates were then collected and incubated in ice for 20 min, followed by centrifugation at 12,000 × g for 10 min at 4°C, and supernatants used for protein assay. Protein assay was performed using Micro BCA Protein Assay Kit (Thermo Scientific) according to the manufacturer’s instructions. Protein samples then were mixed with 4× Laemmle sample buffer (Bio-Rad) and denatured at 100°C for 5 min. For Western blots, 25 µg of total protein were fractionated by mini-protein TGX Gels (Bio-Rad), transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad), which were blocked in 5% nonfat dry milk in 1× TBS with 0.1% Tween 20 (TBST) for 1 h at room temperature (RT). After incubation with Anti-GDP8/Myostatin antibody (ab71808) (1:1000, Abcam) in 5% nonfat dry milk in TBST at 4°C overnight, horseradish peroxidase-conjugated secondary goat anti-rabbit Ab (1:5,000, Jackson immunoresearch) was applied to membranes for 1 h, RT. After three 10 min washes in TBST, membranes were incubated with ECL reagents (Bio-Rad), and signals were detected by Chemidoc MP Imaging System (Bio-Rad). Beta-Tubulin rabbit ab (2146S) (1:1000, cell signaling) was used as a loading control in the experiments. Experiments included three biological replicates.
Intracellular Ca2+ measurements
A Photon Technology International (PTI)-Horiba imaging system with an Eclipse Ti motorized with perfect focus and a SNAPCool 16-BIT CCD camera and automatic monochromators was used to measure intracellular Ca2+ transients in control and 10 ng/mL FGF9-treated C2C12 cells. These cells were treated for 48 h at differentiation days 1–2, and cells were tested at differentiation day 5 following our previously published method [45,47]. Briefly, each myotube imaged was loaded with 4 μM Fura-2/AM, a ratiometric calcium dye, for 30 min at 37°C, and followed by another 30 min incubation at room temperature for de-esterification. The intracellular Ca2+ transients (350/380 nm excitation ratio; 510 nm emission) were elicited by the stimulation of Ca2+ release from sarcoplasmic reticulum (SR) with 20 mM caffeine. Monitoring and analysis of the Ca2+ transients were performed using EasyRatioPro2 (Horiba Scientific). Experiments included three biological replicates, resulting in 6–10 cells analyzed per group [45,47].
Effects of FGFs on C2C12 cell proliferation
Proliferation was measured using various assays in undifferentiated C2C12 cells (Cell Counting, MTT, and CellTiter 96). In either MTT or cell counting assays, C2C12 cells were plated in 96-well or 24-well plates (1 × 104cell/cm2) in GM overnight. On the second day, cells were rinsed with DPBS, fed with GM supplemented with 2% FBS and treated with vehicle control or FGF9, FGF2, or FGF23 (2–50 ng/mL) for 3 days. MTT assay (N = 4, repeated twice) was performed as previously described [48]. Counting cells (N = 3, repeated twice) was performed using a hemocytometer: cells were rinsed with pre-warmed DPBS and incubated with 0.05% Trypsin/0.53 mM EDTA, cell number was counted manually. To test FGF9, FGF16, and FGF20 on C2C12 proliferation, 4000 cells/well C2C12 cells ware seeded in 96-well plates. Twenty-four hours later cells were treated with vehicle, FGF9 (10 ng/mL), FGF16 (100 ng/mL) or FGF20 (100 ng/mL) for 24 and 48 h, respectively. These concentrations were selected based on the results obtained in the differentiation experiments. Proliferation was detected by cell counting and by using the CellTiter 96® AQueous One kit according to manufacturer’s instructions (N = 3) and measured with SpetraMax i3x from Molecular Devices. All experiments were repeated in triplicate.
Flow cytometry (FCM) analysis of cell cycle
C2C12 myoblasts were plated at 2.5 × 105 cells/well in six-well plates in GM overnight, followed by induction of cell cycle arrest in G0/G1 phase by switching the medium to DMEM/high glucose with 1% FBS for 24 h. For cell cycle analysis, cells were then treated with GM, FGF9 (10 ng/mL) in GM. At 24 and 36 h. Cells were harvested with trypsin and fixed in 66% ethanol for cell cycle analysis. After fixing, cells were incubated with propidium iodide (PI)/RNase staining buffer (Abcam) for 30 min for cell cycle analysis using LSRII Multi-Color Flow Cytometer (BD Biosciences, San Jose, CA, USA), PI was excited with the 488 nm blue laser and imaged with the 575 ± 26 nm band–pass filter. Cell cycle population analyses were performed using FlowJo® software (Tree Star). Experiments included three biological replicates.
Statistical analysis
IBM SPSS Statistics v.23 was used for Statistical Analyses. Data are presented with all individual data points and a horizontal line indicating the average. Comparisons were made using one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. For comparisons of differences between two groups, the t-test was used. P value <0.05 was considered as being significantly different. We measured the effect size when performing our experiments and all differences reported range from 30% to 800%. The experiments for the determination of FI were conducted blindly by the operator.
Results
FGF9 is expressed in C2C12 cells, EDL, and SOL muscles of adult mouse
To detect the expression of FGF9 gene in C2C12 cells and mouse muscle, we performed RT-qPCR. The expression of FGF9 was detected in C2C12 myoblasts and myotubes (Figure 1(a)). In C2C12 myoblasts at the proliferation stage, FGF9 was expressed and its expression increased significantly by 48 h of proliferation. The expression of FGF9 further increased significantly at days 3 and 5 of differentiation, as much as eightfold higher than the expression level in 24 h myoblasts. FGF9 mRNA expression was also detected in EDL and soleus (SOL) muscles of young adult (6-month-old male) wild type mice. The relative expression of FGF9 was significantly higher in SOL compared to EDL muscles (Figure 1(b)). In these experiments, the Ct of the housekeeping gene Gapdh was ~13, and did not change in any condition by more than 0.3 cycles.
Figure 1.
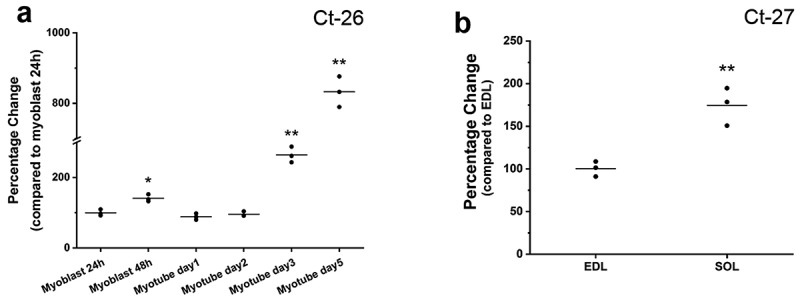
FGF9 is expressed in C2C12 cells, EDL, and soleus muscles of the adult mouse. (a) Summary data of RT-qPCR results of FGF9 expression at 24 and 48 h of myoblast proliferation, and at day 1, day 2, day 3, and day 5 of myotube differentiation. Compared to proliferation at 24 h, FGF9 expression was significantly increased at 48 h, day 3 and day 5 of differentiation. (n = 3, 48 h proliferation, p < 0.05; day 3 and day 5, p< 0.01), while there is no significant difference between day 1 and day 2. (b) FGF9 expression was detected in EDL and SOL of 6-month-old young adult male mice. FGF9 expression is significantly higher in SOL compared with EDL (n = 3, p< 0.01). Ct numbers for the highest level of gene expression are indicated in each graph.
FGF9 inhibits the myogenic differentiation of C2C12 cells
To determine the effect of FGF9 on myogenic differentiation, C2C12 cells were treated with recombinant FGF9 (2, 10, and 50 ng/mL), respectively. At day 3 of differentiation, control and treatment groups were stained with Anti-Human Myosin Heavy Chain-CFS antibody, which only stains maturing myocytes/myotubes not myoblasts, and with DAPI, which stains only the nuclei (Figure 2(a)). Compared to control, no obvious difference was found in myotubes treated with 2 ng/mL FGF9, while smaller and fewer myotubes were observed in the 10 and 50 ng/mL FGF9-treated groups. To confirm these observations, the specific fusion index (FI) of each experimental group was measured. Figure 2(b), showing that compared to control, FI significantly decreased in the 10 and 50 ng/mL FGF9-treatment groups, while no difference was observed in the 2 ng/mL FGF9-treatment group.
Figure 2.
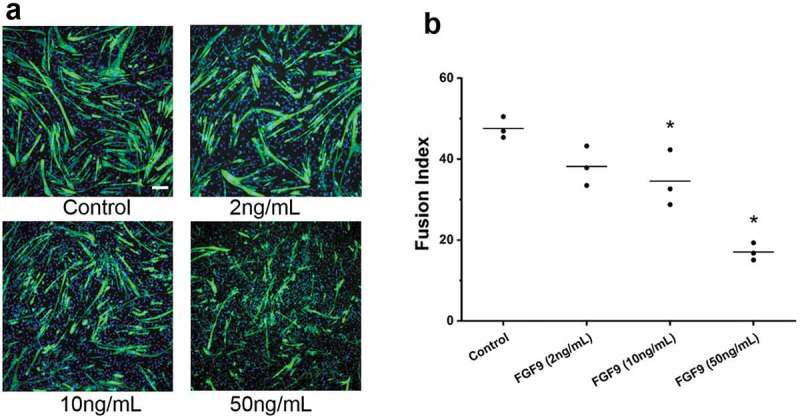
FGF9 inhibits C2C12 myoblast differentiation. (a) Representative fluorescence images of DAPI and myosin heavy chain antibody stained myocytes/myotubes of C2C12 cells at differentiation day 3 after FGF9 treatment. (b) Summary data for FI for treatments of 2, 10, and 50 ng/mL FGF9. FI decreased significantly in the 10 ng/mL and 50 ng/mL FGF9 groups (n = 3, compared to control, 10 ng/mL FGF9, p < 0.05; 50ng/mL FGF9, p< 0.01), while there is no significant difference between 2 ng/mL FGF9 and control. Calibration bar = 100 µm.
Modulation of gene expression by FGF9
RT-qPCR was used to detect the expression of four key regulatory genes of myogenesis (Mhc, MyoD, MyoG, and Myostatin) in C2C12 cells after treatment with FGF9 (2, 10, and 50 ng/mL). At day 3 of differentiation, compared to control, the expression of MyoD was not changed significantly in any of the FGF9-treated groups (Figure 3(a)). Expression of MyoG decreased by ~25% at 10 and 50 ng/mL FGF9, with no effect at 2 ng/mL. FGF9 also dose-dependently inhibited Mhc expression by as much as 60%, with significance at 10 and 50 ng/mL. In contrast to the downregulation of genes associated with myogenesis, expression of Myostatin, an inhibitor of myogenesis, increased in a dose-dependent manner, with significance at 10 and 50 ng/mL and a maximal stimulation close to twofold (Figure 3(a)). As shown in Figure 3(b-c), Western blot demonstrated that Myostatin protein content increased after FGF9 treatment. Completed Western blot images are shown in Supplemental Figure 5. To determine the potential mechanisms underlying the inhibitory effects of FGF9 on C2C12 myoblast differentiation, the Mouse Signal Transduction PathwayFinder PCR Array was used. This protein-validated array monitors differences in the expression of 10 signaling pathways and was used to compare control and FGF9 (10 ng/mL) treated C2C12 cells. Twenty-four hours of FGF9 treatment reduced the expression of Wnt1 and Wnt6, compared to control, by 3.46 and 4.32 fold, respectively, while the expression of Icam1 increased by 2.79 fold (Table 1).
Figure 3.
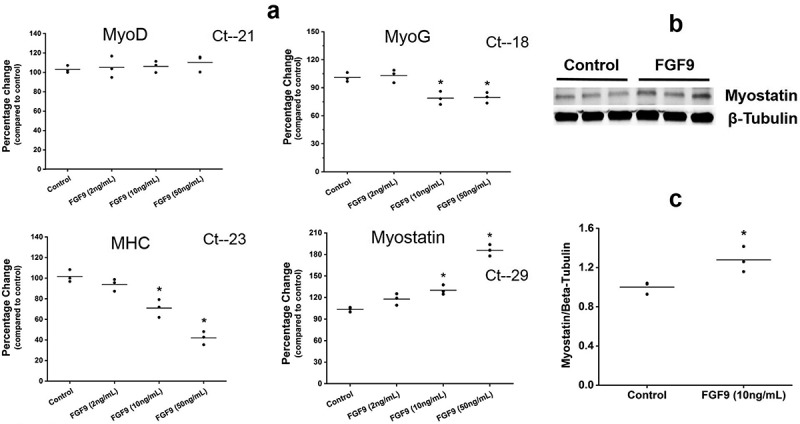
FGF9 modulates the expression of muscle regulatory factors. (a) Summary data of RT-qPCR detection of gene expression changes for MyoD, MyoG, Mhc, and Myostatin at differentiation day 3 after 2, 10, and 50 ng/mL FGF9 treatment. In the 2 ng/mL FGF9-treatment group, no significant difference was detected for any of the genes evaluated (n = 3, p > 0.05). In the 10ng/mL and 50ng/mL FGF9-treatment groups the expression of Mhc and MyoG was significantly decreased (n = 3, p < 0.05), while the expression of Myostatin increased significantly (n = 3, p < 0.05). Expression of MyoD was not affected by FGF9 at any of the doses tested (n = 3, p > 0.05). Ct numbers for the highest level of gene expression are indicated in each graph. (b) Western blot illustrating that Myostatin protein content increased after FGF9 treatment. (c) Quantification of the data presented in b (n = 3, p < 0.05).
Table 1.
Gene expression alterations detected by the Mouse Signal Transduction PathwayFinder PCR.
| Array in C2C12 cells treated with FGF9 (10 ng/mL) |
|
|---|---|
| Altered genes | 10 ng/mL FGF9 |
| Wnt1 | −3.46 ± 0.54 |
| Wnt6 | −4.32 ± 0.59 |
| Icam1 | 2.79 ± 0.49 |
Data are expressed as Fold-Change of treatment groups compared to control groups.
Summary data from two independent experiments for the Mouse Signal Transduction Pathway Finder PCR.
Array. Twenty-four hours after 10 ng/mL FGF9 treatment compared to control, the expression of Wnt1 and Wnt6 decreased by 3.46 and 4.32 fold, respectively, while the expression of Icam1 increased by 2.79 fold.
FGF9 inhibits caffeine-induced Ca2+ release from the sarcoplasmic reticulum (SR)
Ca2+ homeostasis plays important roles in myoblast differentiation [49], and is important to skeletal muscle function [45,49]. To determine if Ca2+ release from SR of myotubes was influenced by FGF9 treatment, we tested the direct effects of caffeine’s ability to release Ca2+ from the SR in Fura-2 loaded myotubes. In FGF9-treated myotubes at day 5 of differentiation, the amplitude peak of Ca2+ response to caffeine significantly decreased by 29.8% in FGF9-treated cells (1.01 ± 0.04 vs. 1.44 ± 0.05, respectively, mean ± SD, n = 3, p< 0.05). The relaxation phase of the transients (i.e. time to return from peak to baseline) was 10.1% shorter (392 ± 7.2 s vs. 436 ± 9.1 s, respectively, mean ± SD, n = 3, p < 0.05) (Figure 4(a)).
Figure 4.
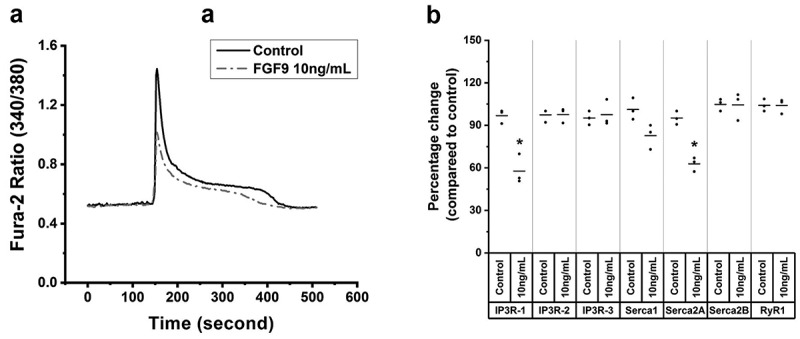
FGF9 decreases Ca2+ release from the sarcoplasmic reticulum (SR). (a) Ca2+ transient tracing that represents the average response of C2C12 myotubes loaded with Fura-2/AM exposed to 20 mM caffeine (arrows) in control and FGF9-treated cells. Compared to control, in 10 ng/mL FGF9-treated C2C12 myotubes, the average amplitude peak Ca2+ response to caffeine, which is a measure of SR Ca2+ release was significantly decreased, and the relaxation phase of the transient was shorter (n = 3, p < 0.05). (b) Summary data of the RT-qPCR results for the expression levels of IP3R-1, IP3R-2, IP3R-3, Serca1, Serca2A, Serca2B and RyR1 at day 3 of myotube differentiation. Compared to control, IP3R-1 and Serca2A expression was significantly decreased (n = 3, p < 0.05), while the expression of IP3R-2, IP3R-3, Serca1, Serca2B, and RyR1 was not altered (n = 3, p > 0.05).
FGF9-induced downregulation of calcium homeostasis, muscle regeneration and mitochondrial biogenesis genes and upregulation of a muscle dystrophy gene
Expression of important genes related to calcium homeostasis, muscle regeneration, mitochondrial biogenesis, and muscle dystrophy/cellular repair in C2C12 cells at day 3 of differentiation after FGF9 treatments was detected by a custom-built RT-PCR gene array using the same technology that the PathwayFinder was used. Furthermore, the expression of calcium homeostasis-associated genes IP3R-1, IP3R-2, IP3R-3, Serca1, Serca2A, Serca2B, and RyR1 was confirmed by RT-qPCR. In agreement with the reduced SR Ca2+ release by FGF9, the expression of genes associated with calcium homeostasis, muscle regeneration, and mitochondrial biogenesis genes was downregulated while a muscle dystrophy/cellular repair gene was upregulated (Tables 2–3) and the expression of genes that regulate SR Ca2+ release/uptake (IP3R-1 and Serca2A) was significantly downregulated (Figure 4(b)).
Table 2.
Gene expression alterations detected by the Custom-built RT-PCR gene array in C2C12 cells treated with FGF9 (10 ng/mL).
| Altered genes | 10 ng/mL FGF9 |
|---|---|
| Cacna1s | −2.50 ± 0.25 |
| RyR2 | −2.77 ± 0.23 |
| PGC1α | −2.17 ± 0.16 |
| Hspa1a | −2.21 ± 0.20 |
| Btk | −2.28 ± 0.16 |
| Nfatc3 | −3.30 ± 0.33 |
| Trim72 | 3.80 ± 0.36 |
Data are expressed as Fold-Change of treatment groups compared to control groups.
Summary data from two independent experiments for the Custom-built RT-PCR gene array.
Three days after 10 ng/mL FGF9 treatment compared to control, the expression of Cacna1s, RyR2, PGC1α, Hspa1a, Btk, and Nfatc3 decreased by 2.50, 2,77, 2.17, 2.21, 2.28 and 3.30 fold, respectively, while the expression of Trim72 increased by 3.80 fold.
Table 3.
Altered pathways and genes detected by the Custom-built RT-PCR gene array.
| Signaling pathways | Genes |
|---|---|
| Ca2+ homeostasis | Cacna1s, RyR2, Btk, Nfatc3, Trim72 |
| Muscle regeneration/cellular repair | Hspa1a, Trim72 |
| Mitochondrial biogenesis | PGC1α |
FGF9 subfamily members: FGF16 and FGF20 inhibit the differentiation of C2C12 cells
To determine whether other FGF9 family members had similar effects on myotube differentiation, FGF16 and FGF20 were examined as they are from the same subfamily as FGF9. C2C12 cells were treated with FGF16 and FGF20 (10–100 ng/mL). At day 3 of differentiation no obvious difference was found in 10 ng/mL and 50 ng/mL FGF16-treated and FGF20-treated myotubes compared to controls, while smaller and fewer myotubes were observed at 100 ng/mL of FGF16 (Figure 5(a)) and FGF20 (Figure 5(b)). FI quantification confirmed that 100 ng/mL FGF16 or FGF20 significantly decreased myoblast fusion to form myotubes (Figure 5(c-d)).
Figure 5.
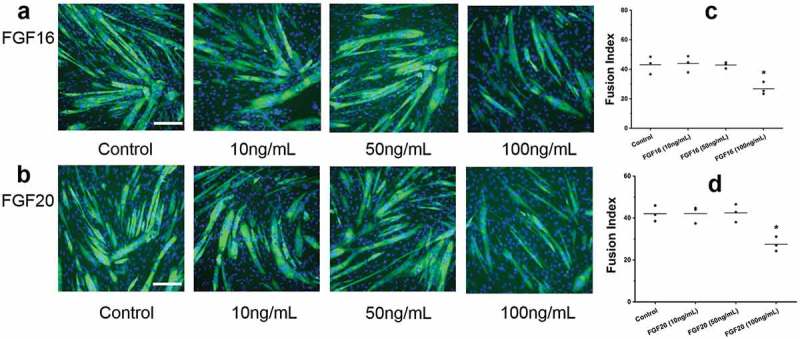
Two other members (FGF16/20) of the FGF9 subfamily also inhibit the differentiation of C2C12 cells. Fluorescence images of C2C12 cells at differentiation day 3 after FGF16 (a) and FGF20 (b) treatment. Summary data for FI for control and FGF16 (c) and FGF20 (d) treatment groups. Compared to control, the FI for FGF16 and FGF20 (10 ng/mL and 50 ng/mL) did not change significantly, while FI for FGF16 and FGF20 (100ng/mL) decreased significantly (n = 3; compared to control, p < 0.05). Calibration bar = 100 µm.
FGF9 inhibits human skeletal muscle cell differentiation
To determine whether FGF9 has similar effects on human myoblasts, primary human skeletal muscle cells (SkMC) were treated with FGF9 at a dose range from 50 to 200 ng/mL. At day 3 of differentiation, no obvious difference was found with FGF9 treatment at 50 and 100 ng/mL compared to controls, but in the 200 ng/mL FGF9-treated group, smaller myotubes were observed (Figure 6(a)). Quantitation confirmed that the FI was significantly decreased in the 200 ng/mL FGF9-treated group, compared to the control group (Figure 6(b)).
Figure 6.
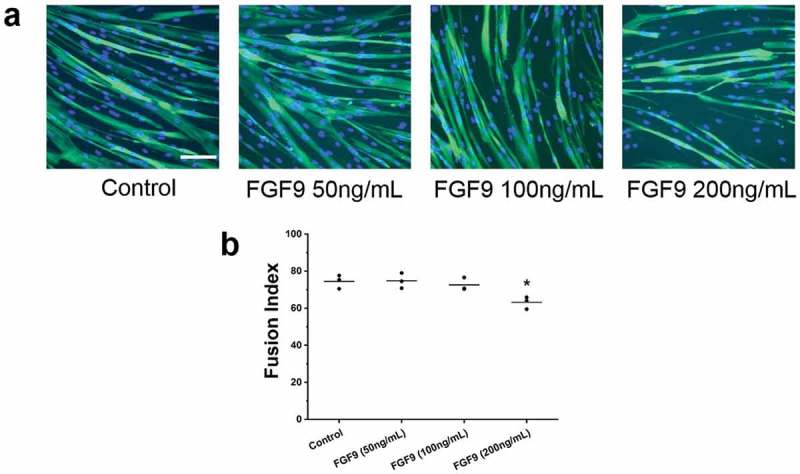
FGF9 inhibits human skeletal muscle cell differentiation. (a) Fluorescence images of DAPI and myosin heavy chain antibody stained myocytes/myotubes of human skeletal muscle cells at differentiation day 3 after FGF9 treatment (50–200 ng/mL). (b) Summary data for FI for control and FGF9-treatment group. FI decreased significantly for FGF9 compared to control (n = 3; compared to control, 200 ng/mL FGF9, p < 0.05). Calibration bar = 100 µm.
FGF9, FGF16, and FGF20 promote C2C12 myoblast proliferation
To determine whether FGF9 has effect on C2C12 myoblast proliferation, cell counting, and MTT assay were used. C2C12 cells were treated with (2–50 ng/mL) FGF9. Cell counting was performed at day 1, day 2, and day 3 after FGF9 treatment. FGF9 dose-dependently stimulated the proliferation of C2C12 cells at all three time points, with the 50 ng/mL dose giving the maximal response (Figure 7(a-b)). The increase was as highest (~3.5-fold) at 50 ng/mL FGF9 at the 3-day time point. In the MTT assay, similar results were observed. We further confirmed these results using the CellTiter 96® Aqueous One kit to detect the effects of FGF9 (10 ng/mL) (as positive control), FGF16 (100 ng/mL) and FGF20 (100 ng/mL) on C2C12 myoblast proliferation. At 48 h after treatment FGF9, FGF16, and FGF20 significantly increased the number of C2C12 cells compared to controls (Figure 7(c-d)). Next, we determined whether the changes in proliferation are associated with modulation of cell cycle. After treatment with FGF9 (10 ng/mL) for 24 and 36 h, cells in the exponential growth phase were stained with propidium iodide (PI) and analyzed by flow cytometry. Twenty-four hours after FGF9 treatment, the population of cells in the S-phase significantly increased, while the population in the G1 phase significantly decreased in the FGF9-treated cells (Figure 8(a)). At 36 h, FGF9 treatment increased the number of cells in S phase and G2/M phase and decreased G1 phase cells (Figure 8(b)). These data suggested that FGF9 signaling promoted the G1-S phase transition in C2C12 myoblasts, providing further evidence for the mechanism of increased proliferation.
Figure 7.
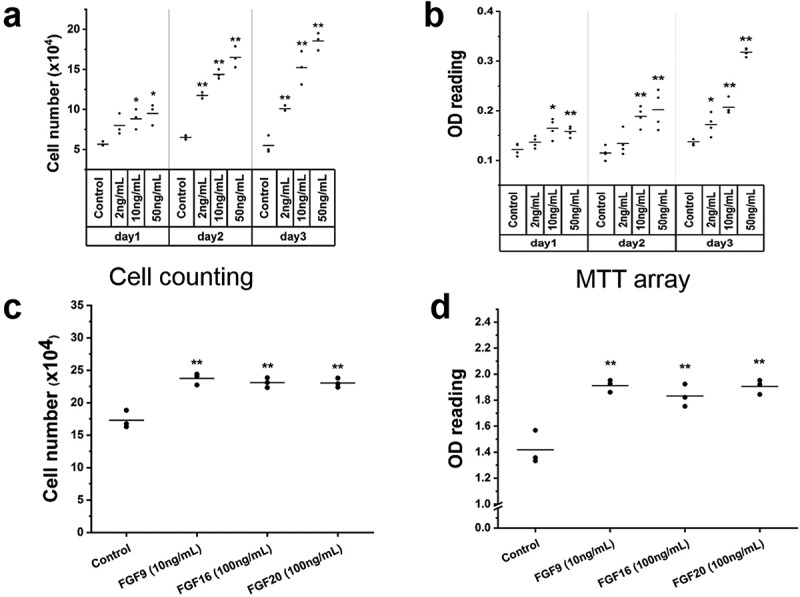
FGFs enhance C2C12 cell proliferation.
Results of cell counting, MTT assay and CellTiter 96® AQueous One kit in C2C12 cells treated with FGF9 (2–50 ng/mL), FGF16 (100 ng/mL) and FGF20 (100 ng/mL). (a) With FGF9 treatment, compared to control, no significant difference of cell number was observed with 2 ng/mL at day 1. A significant increase in cell number was detected in the 2 ng/mL group at days 2 and 3. At days 1–3, in the 10 ng/mL and 50ng/mL groups a significant increase in cell number was observed (n = 3, *p< 0.05, **p< 0.01). (b) In the MTT assay, similar results were observed, except that no significant difference was noted with 2 ng/mL FGF9 at day 2 (n = 4, *p< 0.05, **p< 0.01). The effect of FGF16 and FGF20 on C2C12 myoblast proliferation was determined by cell counting and CellTiter 96® AQueous One kit. (c) In the cell counting assay, compared to control, FGF9 (10 ng/mL) (as positive control), FGF16 (100 ng/mL) and FGF20 (100 ng/mL) significantly increased the number of C2C12 cells. (d) Similar results were observed with CellTiter 96® AQueous One kit (n = 3, *p< 0.05, **p< 0.01).
Figure 8.
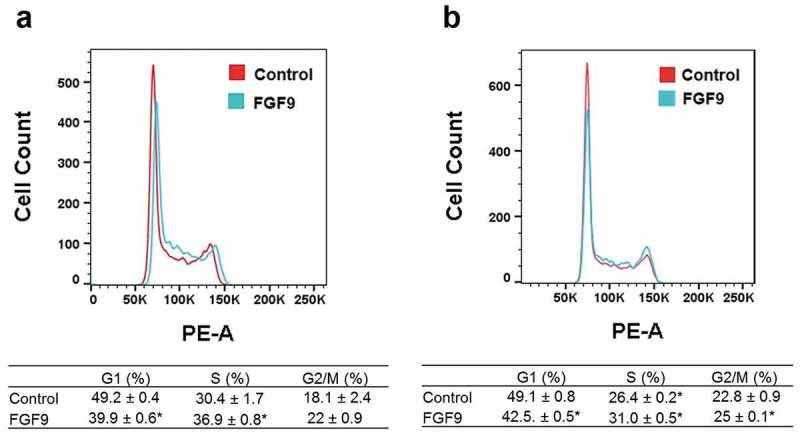
FGF9 alters the G1-S phase cell cycle transition in C2C12 myoblasts, consistent with enhanced proliferation. Representative cell cycle profile at 24 h (a) and 36 h (b), with respective quantification of cell distribution in cell cycle. n = 3, *: P < 0.05 compared with control.
FGF2 promotes C2C12 cell proliferation and inhibits C2C12 cell differentiation
FGF2, another FGF family member known to be produced by bone cells, was next evaluated. C2C12 cells were treated with (2–50 ng/mL) FGF2. Using the cell counting assay, a dose-dependent increase in proliferation was observed with FGF2 treatment (Supplemental Figure 1(a)), akin to the response seen with FGF9 treatment. These results were confirmed using the MTT assay, which gave similar results, showing a dose-dependent stimulation of C2C12 cell proliferation by FGF9 (Supplemental Figure 1(b)). Similar to FGF9, FGF2 was also found to dose-dependently decrease myogenic differentiation (Supplemental Figure 1c-d) suggesting that this FGF family member functions similarly to FGF9.
FGF23 does not alter C2C12 cell proliferation and differentiation
FGF23 is another member of the FGF family that is highly expressed in osteocytes and is released into the circulation to act as an endocrine regulator of phosphate homeostasis. We, therefore, determined whether this osteocyte-produced factor has effects on C2C12 cell proliferation and differentiation. Experiments were carried out similar to the FGF2 and FGF9 experiments. C2C12 cells were treated with FGF23 at a dose range from 2 to 50ng/mL. No significant difference was observed in either cell proliferation (Supplemental Figure 2a-b) or FI (Supplemental Figure 2c) in C2C12 cells treated with FGF23 compared to controls.
FGF9 inhibits C2C12 cell differentiation independently of its proliferation effects
Because FGF9 has dual effect on C2C12 cell proliferation and differentiation, to clarify whether the differentiation phenotype is dependent or independent of the proliferation phenotype, we treated C2C12 cells with DM for 48 h to reduce/stop cell proliferation. After this treatment, cells were treated with FGF9 (2, 10, and 50 ng/mL), respectively. At day 3 of differentiation, compared to control, no obvious difference was found in myotubes treated with 2 ng/mL FGF9, while smaller and fewer myotubes were observed in the 10 and 50 ng/mL FGF9-treated groups (Supplemental Figure 3(a)). FI of each experimental group Supplemental Figure 3(b) showed that compared to control, FI was significantly decreased in the 10 and 50 ng/mL FGF9-treatment groups, while no difference was observed in the 2 ng/mL FGF9-treatment group.
Discussion
Bone (osteoblasts and osteocytes) functions as an endocrine organ, by producing and secreting circulating factors such as FGF23 and osteocalcin [13]. Osteocytes also secrete large amounts of PGE2, which enhances of C2C12 myoblast myogenesis [3] and proliferation [16]. Furthermore, bone-produced WNT3a enhances myogenesis [15]. BAIBA secreted from muscle prevents unloading-induced bone-loss apparently by reducing osteocyte cell death induced by oxidative stress [11]. These data support the concept of a biochemical crosstalk between bone-muscle, which could be important for the pathophysiology of OS.
Fine-tuning of muscle mass and function is essential for optimal health and organism survival. Therefore, it is conceivable that inhibitory factors (i.e. osteokines or others) are also produced by bone cells.
Our previous study has shown that that Fgf9 expression increases with osteocyte differentiation in the OmGFP66 and SW3 osteogenic cell lines [40]. We, therefore, examined whether it might have a potential role in the regulation of muscle cell function. In this study, we also found that Fgf9 is expressed in C2C12 myoblasts and myotubes and in mouse SOL and EDL muscles, suggesting the potential for an autoregulatory feedback mechanism for control of myogenesis and muscle size.
It was observed previously that exogenously administered FGF1 and FGF2 suppress differentiation of myoblasts in culture [50,51]. In this study, we confirmed that FGF2 at concentrations as low as 2 ng/mL inhibited C2C12 cell differentiation (Supplemental Fig. 1c-d). FGF9 overexpression prevented myofibroblastic differentiation of mesothelial cells [52] and repressed the differentiation of progenitor cells into airway smooth muscle cells [53], but a specific role for FGF9 in skeletal muscle differentiation has not yet been reported. Our results showed that FGF9 dose-dependently inhibited C2C12 myogenic differentiation (Figure 2). This inhibition was associated with decreased expression of Myogenin [54], an important myogenic regulatory factor, as well as a strong downregulation of Mhc, an indicator of terminal myogenic differentiation. While genes that promote myogenesis were downregulated, expression of the negative myogenic regulator, myostatin [55] mRNA and protein content increased with FGF9 treatment. Myostatin belongs to the TGF‐β (transforming growth factor β) superfamily and is a negative regulator of muscle differentiation [55–57]. The increase of Myostatin coupled with the downregulation of Myogenin offers a feasible mechanism to explain both the inhibition of myogenesis and the increased proliferation (Figure 3). Moreover, our findings with FGF9 suggest a mechanism similar to other FGFs, FGF1, and FGF2, which have been shown to suppress MyoG expression in BC3H-1 myoblasts [58], with no effect on MyoD [58].
Calcium homeostasis is very important in myoblast differentiation [59], and is a major surrogate of skeletal muscle function [47,59,60]. To determine if SR Ca2+ release was influenced by FGF9, we performed Fura-2 imaging of intracellular Ca2+ transients in response to caffeine-induced SR Ca2+ release. Our data showed that SR Ca2+ release was significantly decreased by FGF9 treatment (Figure 4(a)).
To search for molecular insights into these effects of FGF9, the Mouse Signal Transduction PathwayFinder PCR Array was used. Treatment with 10 ng/mL FGF9 decreased expression of Wnt1 and Wnt6 and increased expression of ICAM-1(Table 1). Wnt1 is directly involved in myogenesis [61,62], exogenous Wnt1 enhances C2C12 cell differentiation [15]. The decreased expression of Wnt1 after FGF9 treatment may, therefore, play a role in decreased C2C12 cell differentiation. Exogenous Wnt6 inhibits satellite cell proliferation by promoting muscle cell differentiation [63]; therefore, its inhibition is expected to decrease differentiation as in our studies. Based on this, we postulate that the key molecular mechanism for the FGF9-induced differentiation results from the downregulation of the Wnt (Wnt1, 6). In contrast, ICAM-1 is a member of the immunoglobulin superfamily of adhesion molecules that has been associated with muscle overload-induced hypertrophy [64] and enhanced myogenesis [65]. We interpreted the increased expression of ICAM-1 in our experiments as a likely compensatory mechanism to the other changes leading to inhibition of myogenesis and enhanced proliferation.
Our custom-built RT-PCR gene array results allowed us to demonstrate that FGF9 downregulated genes associated to pathways linked to calcium homeostasis, muscle regeneration, mitochondrial biogenesis, and upregulated a gene associated with muscular dystrophy. In support of our observations of reduced Ca2+ released induced by FGF9, we found down expression of specific genes associated with calcium homeostasis (Cacna1s [66], RyR2 [67,68], Nfatc3[69], BTK [70]), and up expression of Trim72, a gene associated with muscle dystrophy/cellular repair [71–73]. The down-regulation of these calcium homeostasis genes could help explain the reduced Ca2+ capacity in muscle cells after FGF9 treatment. A gene that is critical for muscle regeneration (Hspa1a [74]) as well as a gene associated with mitochondrial biogenesis (PGC1α [75]) were also downregulated (Tables 2–3). It is feasible to postulate that as cells arrested in the cell cycle and do not progress to become myotubes, they require less mitochondria and membrane fusion. RT-qRCR revealed that consistent with the decreased Ca2+ release and uptake, the expression of IP3R-1 and Serca2A is decreased (Figure 4(b)). IP3R-1 is associated and modulates SR Ca2+ release [76,77] and SERCA2A is an integral membrane protein that regulates myofibrillar Ca2+ removal by pumping Ca2+ ions back in the SR under ATP usage [78,79]. It is predicted that reduced levels of IP3R-1 and SERCA will lead into a combination of reduced availability of SR Ca2+ and reduced SR Ca2+ release.
Our results are also in agreement with previous reports that suggested the maintenance of normal intracellular Ca2+ concentration is vital to myocyte gene expression and differentiation [49]. It is also intriguing that FGF9 modulates intracellular Ca2+ in mesenchymal stem cells [80], suggesting that this might be a common mechanism of FGF9 action. We believe that the modulation in intracellular Ca2+ might work as the biophysical linker between the molecular-genetic adaptations and the changes (phenotypes) induced by FGF9 in muscle cells. This result is also consistent with the decreased differentiation of myotubes treated with FGF9, although this phenotype could arise from underdeveloped myotubes with a less developed capacity to control calcium homeostasis.
The FGF9 subfamily consists of FGF9, FGF16, and FGF20, sharing 62–73% amino acid sequence similarity [32,33], and has sequence homology between human and mouse [81]. FGF9 subfamily members have similar receptor specificity [82]. Fgfr1 to −4 and α-Klotho are known to be expressed in skeletal muscle and C2C12 cells [48]. Most importantly, it is well established that signaling by FGF9 subfamily members regulates mesenchymal development and regulates the morphogenesis of multiple organs. Impaired development of multiple organs was seen in FGF9 knockout mice that died soon after birth due to early embryonic lung hypoplasia [83]. Osteoblast-derived FGF9 positively regulates skeletal homeostasis [84] and FGF9 het mice have impaired long bone repair [85]. Evidence also suggests that FGF9/16/20 members have similar biological effects in neurological development [38], heart specification [86], and cancer progression [87]. An important question raised by our experiments with FGF9 was whether other FGF9 subfamily members might exert similar effects in muscle. As there is very little data on FGF16 and FGF20 function in skeletal muscle, we tested this possibility by treating C2C12 myoblasts with FGF16 and FGF20. Interestingly, FGF16 and FGF20 inhibited C2C12 cell differentiation, although higher concentrations were required compared to FGF9. This suggests that they are less potent than FGF9. However, an alternative explanation may be that the decreased potency occurs because the FGF16 and FGF20 used in this study are human recombinant proteins, as the mouse recombinant proteins were not commercially available. Overall, our data showed that FGF9 subfamily members have a similar function of inhibition of muscle cell differentiation.
We also tested an FGF member outside of this subfamily, FGF23, which is expressed at high levels by osteocytes and is secreted into the circulation. We recently demonstrated that FGF23 did not alter myogenic gene expression, calcium homeostasis, or ex vivo skeletal muscle function [48]. Here, we also found that unlike FGF9, 16, 20, and FGF2, which inhibited myogenic differentiation and stimulated proliferation, FGF23 had no effect on either myogenic differentiation or proliferation (Supplemental Fig. 2). This data suggests that inhibition of myogenesis is not a general property of all members of the FGF family. The difference in response may be due to the different structures and actions of the subfamilies of FGFs or the action of these molecules on specific receptors and co-receptors. In this regard, it is notable that FGFs 2, 9, 16, and 20 are all heparin-binding FGFs, while FGF23 does not bind to heparin. It is truly intriguing since heparin is known to inhibit SR Ca2+ release.
In addition to its functions in regulating cell differentiation, it is intriguing that FGF9 significantly increased the proliferation of lung mesenchyme [88] and increased cell proliferation in mouse Leydig tumor cells [89]. FGF9 is required for smooth muscle cell proliferation [90], and cardiomyocyte proliferation [91]. FGF16 was also required for cardiomyocyte proliferation in the mouse embryonic heart [92], but there is very little data on the role of FGF9 and FGF9 subfamily members in regulating proliferation of skeletal muscle cells. Our data showed that FGF9, FGF16, and FGF20 enhanced the proliferation of C2C12 myoblasts. This is consistent with their function in stimulating cell proliferation in other cell types and suggests that this might be a common function of FGF9 subfamily members. In our study, FGF2 at concentrations as low as 2 ng/mL also promoted the proliferation of C2C12 cells (Supplemental Fig. 1a-b), consistent with its known effects to promote satellite cell proliferation [93], and previous reports showing stimulation of C2C12 cell proliferation [48]. In contrast, FGF23 (2–50 ng/mL) had no significant effect on C2C12 myoblast proliferation (Supplemental Figure 2(a-b)). A potential mechanism for the proliferation-related effects of FGF9 is our discovery that it modulates the G1-S phase transition in C2C12 myoblasts, indicating that the enhancement of FGF9 on proliferation associated with modulation of cell cycle (Figure 8(a-b)).
To clarify whether the differentiation phenotype is dependent or independent of the proliferation phenotype, C2C12 cells were pretreated with DM for 48 h to reduce/stop cell proliferation. After this treatment, cells were treated with FGF9. The result showed that the phenotype of differentiation inhibition (Supplemental Fig. 3a-b) was essentially identical to the phenotype without the DM pre-treatment (Figure 2(a-b)), indicating the differentiation phenotype is independent of the proliferation phenotype. We also detected the expression levels of a caspase 3, a key apoptosis regulatory gene [94], and two key autophagy regulatory genes (LC3B and P62) [95] and found that FGF9 treatment did not alter their expression levels. This result indicated that FGF9 treatment did not alter C2C12 cell apoptosis and autophagy (Supplemental Figure 4).
Limitations of this study: The aim of this study was to investigate the functional consequences of treating muscle cells with FGF9 in vitro. Our rationale was based on our studies showing that osteokines secreted from bone cells can influence muscle cell function and myogenesis. These studies are a necessary step before in vivo studies to demonstrate that FGF9 might function in bone-muscle crosstalk. Therefore, one important limitation is that we did not investigate the functions of FGF9 in vivo.
Conclusions
In conclusion, our data suggest that FGF9 has the intrinsic capacity to inhibit myogenic differentiation and promote myoblast proliferation. The inhibitory effect on C2C12 cell differentiation is modulated by a complex signaling mechanism that appears to involve MyoG and Myostatin and genes associated with the WNT signaling pathway leading to modulation of the cell cycle and genetic changes in the cells that while enhancing proliferation, reduce their capacity to progress into fully matured myotubes. We postulate that FGF9 could act as a modulatory osteokine from bone cells to maintain an optimal balance of muscle growth, but additional in vivo studies will be required to confirm this concept.
Funding Statement
This study was directly supported by NIH-NIA P01 AG039355 (SLD, LFB, MB), the George and Hazel Jay Endowment Fund (MB), and the UT System Science and Technology Acquisition and Retention Program (STARS) (MB). JH and LB were partially supported by NIH-NIA R01AG056504 and R01 AG060341 (MB).
Acknowledgments
We thank Dr. Kimberly Bowles, Life Sciences Core Facility, Department of Biology at UTA for her expert assistance with the Flow Cytometry studies. We thank Mr. Julian Vallejo and Mr. Derek Wang for performing the myogenic assay for the FGF23 series of experiments.
Authors’ roles
Study design: JH, KW, SLD, and MB. Study conduct: JH, KW. Data collection: JH, KW, LAS, and LB. Data analysis: JH, KW, MJW, SLD, and MB. Data interpretation: JH, KW, LFB, SLD, and MB. Drafting manuscript: JH, KW, LFB, SLD, MJW, and MB. Revising manuscript: JH, KW, LAS, SLD, LFB, and MB. Approving final version of manuscript: all co-authors. MB takes responsibility for the integrity of the data analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005-2025. J Bone Miner Res. 2007;22(3):465–475. [DOI] [PubMed] [Google Scholar]
- [2].Kaji H. Linkage between muscle and bone: common catabolic signals resulting in osteoporosis and sarcopenia. Curr Opin Clin Nutr Metab Care. 2013;16(3):272–277. [DOI] [PubMed] [Google Scholar]
- [3].Mo C, Romero-Suarez S, Bonewald L, et al. Prostaglandin E2: from clinical applications to its potential role in bone- muscle crosstalk and myogenic differentiation. Recent Pat Biotechnol. 2012;6(3):223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hamrick MW. A role for myokines in muscle-bone interactions. Exerc Sport Sci Rev. 2011;39(1):43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pedersen BK. Muscles and their myokines. J Exp Biol. 2011;214(2):337–346. [DOI] [PubMed] [Google Scholar]
- [6].Norheim F, Raastad T, Thiede B, et al. Proteomic identification of secreted proteins from human skeletal muscle cells and expression in response to strength training. Am J Physiol Endocrinol Metab. 2011;301(5):E1013–21. [DOI] [PubMed] [Google Scholar]
- [7].Elkasrawy MN, Hamrick MW. Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interact. 2010;10(1):56–63. [PMC free article] [PubMed] [Google Scholar]
- [8].Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19(8):815–827. [DOI] [PubMed] [Google Scholar]
- [9].Colaianni G, Cuscito C, Mongelli T, et al. The myokine irisin increases cortical bone mass. Proc Natl Acad Sci U S A. 2015;112(39):12157–12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hamrick MW, McNeil PL, Patterson SL. Role of muscle-derived growth factors in bone formation. J Musculoskelet Neuronal Interact. 2010;10(1):64–70. [PMC free article] [PubMed] [Google Scholar]
- [11].Kitase Y, Vallejo JA, Gutheil W, et al. beta-aminoisobutyric acid, l-BAIBA, is a muscle-derived osteocyte survival factor. Cell Rep. 2018;22(6):1531–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Karsenty G, Oury F. Biology without walls: the novel endocrinology of bone. Annu Rev Physiol. 2012;74:87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell … and more. Endocr Rev. 2013;34(5):658–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63(5):561–571. [DOI] [PubMed] [Google Scholar]
- [15].Huang J, Romero-Suarez S, Lara N, et al. Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/β-Catenin pathway. JBMR Plus. 2017;1(2):86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mo C, Zhao R, Vallejo J, et al. Prostaglandin E2 promotes proliferation of skeletal muscle myoblasts via EP4 receptor activation. Cell Cycle (Georgetown, tex). 2015;14(10):1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn. 2008;237(1):18–27. [DOI] [PubMed] [Google Scholar]
- [18].Yamanaka Y, Lanner F, Rossant J. FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development. 2010;137(5):715–724. [DOI] [PubMed] [Google Scholar]
- [19].Jung J, Zheng M, Goldfarb M, et al. Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science (New York, NY). 1999;284(5422):1998–2003. [DOI] [PubMed] [Google Scholar]
- [20].Presta M, Dell’Era P, Mitola S, et al. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16(2):159–178. [DOI] [PubMed] [Google Scholar]
- [21].Barrientos S, Stojadinovic O, Golinko MS, et al. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16(5):585–601. [DOI] [PubMed] [Google Scholar]
- [22].Kharitonenkov A, Shiyanova TL, Koester A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115(6):1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. [DOI] [PubMed] [Google Scholar]
- [24].Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. WIRES Dev Biol. 2015;4(3):215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Brewer JR, Mazot P, Soriano P. Genetic insights into the mechanisms of FGF signaling. Genes Dev. 2016;30(7):751–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Krejci P, Prochazkova J, Bryja V, et al. Molecular pathology of the fibroblast growth factor family. Hum Mutat. 2009;30(9):1245–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Itoh N, Nakayama Y, Konishi M. Roles of FGFs as paracrine or endocrine signals in liver development, health, and disease. Front Cell Dev Biol. 2016;4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Javed A, Chen H, Ghori FY. Genetic and transcriptional control of bone formation. Oral Maxillofac Surg Clin North Am. 2010;22(3):283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pawlikowski B, Vogler TO, Gadek K, et al. Regulation of skeletal muscle stem cells by fibroblast growth factors. Dev Dyn. 2017;246(5):359–367. [DOI] [PubMed] [Google Scholar]
- [30].Lu H, Shi X, Wu G, et al. FGF13 regulates proliferation and differentiation of skeletal muscle by down-regulating spry1. Cell Prolif. 2015;48(5):550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Benoit B, Meugnier E, Castelli M, et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat Med. 2017;23(8):990–996. [DOI] [PubMed] [Google Scholar]
- [32].Miyake A, Konishi M, Martin FH, et al. Structure and expression of a novel member, FGF-16, on the fibroblast growth factor family. Biochem Biophys Res Commun. 1998;243(1):148–152. [DOI] [PubMed] [Google Scholar]
- [33].Ohmachi S, Watanabe Y, Mikami T, et al. FGF-20, a novel neurotrophic factor, preferentially expressed in the substantia nigra pars compacta of rat brain. Biochem Biophys Res Commun. 2000;277(2):355–360. [DOI] [PubMed] [Google Scholar]
- [34].Wang S, Li Y, Jiang C, et al. Fibroblast growth factor 9 subfamily and the heart. Appl Microbiol Biotechnol. 2018;102(2):605–613. [DOI] [PubMed] [Google Scholar]
- [35].Schmid GJ, Kobayashi C, Sandell LJ, et al. Fibroblast growth factor expression during skeletal fracture healing in mice. Dev Dyn. 2009;238(3):766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Charoenlarp P, Rajendran AK, Iseki S. Role of fibroblast growth factors in bone regeneration. Inflamm Regen. 2017;37:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hajihosseini MK, Heath JK. Expression patterns of fibroblast growth factors-18 and −20 in mouse embryos is suggestive of novel roles in calvarial and limb development. Mech Dev. 2002;113(1):79–83. [DOI] [PubMed] [Google Scholar]
- [38].Kim GJ, Kumano G, Nishida H. Cell fate polarization in ascidian mesenchyme/muscle precursors by directed FGF signaling and role for an additional ectodermal FGF antagonizing signal in notochord/nerve cord precursors. Development. 2007;134(8):1509–1518. [DOI] [PubMed] [Google Scholar]
- [39].Yi L, Domyan ET, Lewandoski M, et al. Fibroblast growth factor 9 signaling inhibits airway smooth muscle differentiation in mouse lung. Dev Dyn. 2009;238(1):123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].McCormick LA, Wang K, Tiede-Lewis LM, et al. Role of FGF9 in promotion of early osteocyte differentiation and as a potent inducer of fgf23 expression in osteocytes. J Bone Miner Res. 2016;31(Suppl 1):S39. [Google Scholar]
- [41].Wang K, Le L, Chun BM, et al. A novel osteogenic cell line that differentiates into GFP-tagged osteocytes and forms mineral with a bone-like lacunocanalicular structure. J Bone Miner Res. 2019;34(6):979–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Huang J, Hsu YH, Mo C, et al. METTL21C is a potential pleiotropic gene for osteoporosis and sarcopenia acting through the modulation of the nf-kappa B signaling pathway. J Bone Miner Res. 2014;29(7):1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ono Y, Sakamoto K. Lipopolysaccharide inhibits myogenic differentiation of C2C12 myoblasts through the Toll-like receptor 4-nuclear factor-kappaB signaling pathway and myoblast-derived tumor necrosis factor-alpha. PloS One. 2017;12(7):e0182040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jahn K, Lara-Castillo N, Brotto L, et al. Skeletal muscle secreted factors prevent glucocorticoid-induced osteocyte apoptosis through activation of beta-catenin. Eur Cell Mater. 2012;24:197–209, discussion −10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shen J, Yu WM, Brotto M, et al. Deficiency of MIP/MTMR14 phosphatase induces a muscle disorder by disrupting Ca(2+) homeostasis. Nat Cell Biol. 2009;11(6):769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Filigheddu N, Gnocchi VF, Coscia M, et al. Ghrelin and des-acyl ghrelin promote differentiation and fusion of C2C12 skeletal muscle cells. Mol Biol Cell. 2007;18(3):986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhao X, Weisleder N, Thornton A, et al. Compromised store-operated Ca2+ entry in aged skeletal muscle. Aging Cell. 2008;7(4):561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Avin KG, Vallejo JA, Chen NX, et al. Fibroblast growth factor 23 does not directly influence skeletal muscle cell proliferation and differentiation or ex vivo muscle contractility. Am J Physiol Endocrinol Metab. 2018;315:E594-E604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Porter GA Jr., Makuck RF, Rivkees SA. Reduction in intracellular calcium levels inhibits myoblast differentiation. J Biol Chem. 2002;277(32):28942–28947. [DOI] [PubMed] [Google Scholar]
- [50].Clegg CH, Linkhart TA, Olwin BB, et al. Growth factor control of skeletal muscle differentiation: commitment to terminal differentiation occurs in G1 phase and is repressed by fibroblast growth factor. J Cell Biol. 1987;105(2):949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Spizz G, Roman D, Strauss A, et al. Serum and fibroblast growth factor inhibit myogenic differentiation through a mechanism dependent on protein synthesis and independent of cell proliferation. J Biol Chem. 1986;261(20):9483–9488. [PubMed] [Google Scholar]
- [52].Justet A, Joannes A, Besnard V, et al. FGF9 prevents pleural fibrosis induced by intrapleural adenovirus injection in mice. Am J Physiol Lung Cell Mol Physiol. 2017;313(5):L781–l95. [DOI] [PubMed] [Google Scholar]
- [53].El Agha E, Kheirollahi V, Moiseenko A, et al. Ex vivo analysis of the contribution of FGF10(+) cells to airway smooth muscle cell formation during early lung development. Dev Dyn. 2017;246(7):531–538. [DOI] [PubMed] [Google Scholar]
- [54].Ridgeway AG, Petropoulos H, Wilton S, et al. Wnt signaling regulates the function of MYoD and myogenin. J Biol Chem. 2000;275(42):32398–32405. [DOI] [PubMed] [Google Scholar]
- [55].McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387(6628):83–90. [DOI] [PubMed] [Google Scholar]
- [56].Langley B, Thomas M, Bishop A, et al. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277(51):49831–49840. [DOI] [PubMed] [Google Scholar]
- [57].Rios R, Carneiro I, Arce VM, et al. Myostatin is an inhibitor of myogenic differentiation. Am J Physiol Cell Physiol. 2002;282(5):C993–9. [DOI] [PubMed] [Google Scholar]
- [58].Brunetti A, Goldfine ID. Role of myogenin in myoblast differentiation and its regulation by fibroblast growth factor. J Biol Chem. 1990;265(11):5960–5963. [PubMed] [Google Scholar]
- [59].Thornton AM, Zhao X, Weisleder N, et al. Store-operated Ca(2+) entry (SOCE) contributes to normal skeletal muscle contractility in young but not in aged skeletal muscle. Aging (Albany NY). 2011;3(6):621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Park KH, Brotto L, Lehoang O, et al. Ex vivo assessment of contractility, fatigability and alternans in isolated skeletal muscles. J Vis Exp. 2012;(69):e4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Stern HM, Brown AM, Hauschka SD. Myogenesis in paraxial mesoderm: preferential induction by dorsal neural tube and by cells expressing Wnt-1. Development. 1995;121(11):3675–3686. [DOI] [PubMed] [Google Scholar]
- [62].Porter JD, Baker RS. Absence of oculomotor and trochlear motoneurons leads to altered extraocular muscle development in the Wnt-1 null mutant mouse. Brain Res Dev Brain Res. 1997;100(1):121–126. [DOI] [PubMed] [Google Scholar]
- [63].Hitchins L, Fletcher F, Allen S, et al. Role of Sulf1A in Wnt1- and Wnt6-induced growth regulation and myoblast hyper-elongation. FEBS Open Bio. 2013;3:30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Dearth CL, Goh Q, Marino JS, et al. Skeletal muscle cells express ICAM-1 after muscle overload and ICAM-1 contributes to the ensuing hypertrophic response. PloS One. 2013;8(3):e58486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Goh Q, Dearth CL, Corbett JT, et al. Intercellular adhesion molecule-1 expression by skeletal muscle cells augments myogenesis. Exp Cell Res. 2015;331(2):292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Schartner V, Romero NB, Donkervoort S, et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 2017;133(4):517–533. [DOI] [PubMed] [Google Scholar]
- [67].Waddell HMM, Zhang JZ, Hoeksema KJ, et al. Oxidation of ryr2 has a biphasic effect on the threshold for store overload-induced calcium release. Biophys J. 2016;110(11):2386–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jiang D, Xiao B, Yang D, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A. 2004;101(35):13062–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Phuong TT, Yun YH, Kim SJ, et al. Positive feedback control between STIM1 and NFATc3 is required for C2C12 myoblast differentiation. Biochem Biophys Res Commun. 2013;430(2):722–728. [DOI] [PubMed] [Google Scholar]
- [70].Scharenberg AM, Kinet JP. PtdIns-3,4,5-P3: a regulatory nexus between tyrosine kinases and sustained calcium signals. Cell. 1998;94(1):5–8. [DOI] [PubMed] [Google Scholar]
- [71].Yi JS, Park JS, Ham YM, et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun. 2013;4:2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Weisleder N, Takizawa N, Lin P, et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci Transl Med. 2012;4(139):139ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Weisleder N, Takeshima H, Ma J. Immuno-proteomic approach to excitation–contraction coupling in skeletal and cardiac muscle: molecular insights revealed by the mitsugumins. Cell Calcium. 2008;43(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Brzeszczynska J, Meyer A, McGregor R, et al. Alterations in the in vitro and in vivo regulation of muscle regeneration in healthy ageing and the influence of sarcopenia. J Cachexia Sarcopenia Muscle. 2018;9(1):93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Vina J, Gomez-Cabrera MC, Borras C, et al. Mitochondrial biogenesis in exercise and in ageing. Adv Drug Deliv Rev. 2009;61(14):1369–1374. [DOI] [PubMed] [Google Scholar]
- [76].Powell JA, Carrasco MA, Adams DS, et al. IP(3) receptor function and localization in myotubes: an unexplored Ca(2+) signaling pathway in skeletal muscle. J Cell Sci. 2001;114(Pt 20):3673–3683. [DOI] [PubMed] [Google Scholar]
- [77].Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004;2004(215):re1. [DOI] [PubMed] [Google Scholar]
- [78].Berchtold MW, Brinkmeier H, Muntener M. Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol Rev. 2000;80(3):1215–1265. [DOI] [PubMed] [Google Scholar]
- [79].Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35(4):430–442. [DOI] [PubMed] [Google Scholar]
- [80].Kizhner T, Ben-David D, Rom E, et al. Effects of FGF2 and FGF9 on osteogenic differentiation of bone marrow-derived progenitors. In Vitro Cell Dev Biol Anim. 2011;47(4):294–301. [DOI] [PubMed] [Google Scholar]
- [81].Sontag DP, Cattini PA. Cloning and bacterial expression of postnatal mouse heart FGF-16. Mol Cell Biochem. 2003;242(1–2):65–70. [PubMed] [Google Scholar]
- [82].Zhang X, Ibrahimi OA, Olsen SK, et al. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281(23):15694–15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Colvin JS, White AC, Pratt SJ, et al. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development. 2001;128(11):2095–2106. [DOI] [PubMed] [Google Scholar]
- [84].Wang L, Roth T, Abbott M, et al. Osteoblast-derived FGF9 regulates skeletal homeostasis. Bone. 2017;98:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Behr B, Leucht P, Longaker MT, et al. Fgf-9 is required for angiogenesis and osteogenesis in long bone repair. Proc Natl Acad Sci U S A. 2010;107(26):11853–11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Davidson B, Shi W, Beh J, et al. FGF signaling delineates the cardiac progenitor field in the simple chordate, Ciona intestinalis. Genes Dev. 2006;20(19):2728–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang S, Lin H, Zhao T, et al. Expression and purification of an FGF9 fusion protein in E. coli, and the effects of the FGF9 subfamily on human hepatocellular carcinoma cell proliferation and migration. Appl Microbiol Biotechnol. 2017;101(21):7823–7835. [DOI] [PubMed] [Google Scholar]
- [88].White AC, Xu J, Yin Y, et al. FGF9 and SHH signaling coordinate lung growth and development through regulation of distinct mesenchymal domains. Development. 2006;133(8):1507–1517. [DOI] [PubMed] [Google Scholar]
- [89].Chang MM, Lai MS, Hong SY, et al. FGF9/FGFR2 increase cell proliferation by activating ERK1/2, Rb/E2F1 and cell cycle pathways in mouse Leydig tumor cells. Cancer science. 2018;109(11):3503–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Agrotis A, Kanellakis P, Kostolias G, et al. Proliferation of neointimal smooth muscle cells after arterial injury. J Biol Chem. 2004;279(40):42221–42229. [DOI] [PubMed] [Google Scholar]
- [91].Lavine KJ, Yu K, White AC, et al. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8(1):85–95. [DOI] [PubMed] [Google Scholar]
- [92].Hotta Y, Sasaki S, Konishi M, et al. Fgf16 is required for cardiomyocyte proliferation in the mouse embryonic heart. Dev Dyn. 2008;237(10):2947–2954. [DOI] [PubMed] [Google Scholar]
- [93].Lefaucheur JP, Sebille A. Basic fibroblast growth factor promotes in vivo muscle regeneration in murine muscular dystrophy. Neurosci Lett. 1995;202(1–2):121–124. [DOI] [PubMed] [Google Scholar]
- [94].Thomas CN, Berry M, Logan A, et al. Caspases in retinal ganglion cell death and axon regeneration. Cell Death Discov. 2017;3:17032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Gomez-Sanchez R, Yakhine-Diop SM, Rodriguez-Arribas M, et al. mRNA and protein dataset of autophagy markers (LC3 and p62) in several cell lines. Data Brief. 2016;7:641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.