

ABSTRACT
TP53 mutations are found in 50% of all cancers and mutated TP53 status is considered poor for treatment. However, some TP53 mutations exhibit only partial loss-of-function (LOF), meaning they retain residual transcriptional and non-transcriptional activities that are potentially beneficial for therapy. Earlier we have characterized a knock-in mouse model for the partial LOF mutant Trp53E177R (p53RR). Reduced DNA binding cooperativity of this mutant led to the loss of p53-dependent apoptosis, while p53 functions in cell cycle control, senescence, metabolism, and antioxidant defense remained intact. Concomitantly, tumor suppression was evident but strongly compromised compared to wild-type mice. Here we used the Trp53E177R mouse as a model to investigate whether residual functions of mutant p53 can be engaged to induce cell death, which is considered the most desirable outcome of tumor therapy. We made use of Mdm2 knock-out in developing embryos as a sensitive tool for detecting remaining p53 activities. Genetic ablation of Mdm2 led to embryonic lethality in Trp53E177R/E177R homozygotes at days 9.5–11.5. This effect was not rescued by concomitant p21-knockout, indicating its independence of p21-mediated cell cycle arrest. Instead, immunohistochemical analysis showed widespread apoptosis in tissues of defective embryos accompanied by persistent accumulation of p53RR protein. This led to partial restoration of the mutant’s proficiency in transcriptional induction of the pro-apoptotic genes Bbc3 (Puma) and Bax. These data indicate that increased quantity can compensate for qualitative defects of p53 mutants and suggest that Mdm2-targeting (potentially in combination with other drugs) might be effective against cells bearing p53 partial LOF mutants.
KEYWORDS: Mutant p53, Mdm2, apoptosis
Introduction
Among all tumor suppressor genes, TP53 is the most recognized one. Also known as the “guardian of the genome”[1], it is activated upon DNA damage and other stresses and drives a plethora of cellular programs, ranging from temporary cell cycle arrest to apoptosis that either ensure repair and survival of damaged cells or promote their elimination – depending on the cell type, damage level and intensity of stress signal [2]. Loss of p53 activity makes cells vulnerable to malignant transformation, therefore a partial or complete inactivation of the p53-dependent network takes place in virtually all human cancers. Approximately 50% of tumors retain wild-type p53 but manage to blunt its functions by blocking up- or downstream pathways involved in p53-mediated responses; in another half of cancer cases p53 itself is hit by mutations [3]. Unlike other tumor suppressor genes, p53 is only rarely affected by nonsense or frame-shift mutations – more than 70% of all genetic alterations found in TP53 are missense mutations and most of them are located in the DNA-binding domain (DBD) [4]. The majority of such mutations result in functional inactivation of p53. Some codons are hit with an extraordinary frequency, like the positions R175, G245, R248, R249, R273, and R282 – so-called “hotspot” mutations [5]. Together they represent approximately 30% of all missense TP53 mutations found in tumors, but the frequency for each of them does not exceed 6%[6]. For some hotspot mutations, oncogenic “gain of function” (GOF) properties have been demonstrated, as they convert p53 into an oncogene: instead of suppressing tumorigenesis, mutant protein enhances metastasis, promotes genomic instability and supports survival of cancer cells under therapy [7]. However, the bulk of p53 mutants remains poorly characterized and the frequency of GOF variants is presently unclear. Nevertheless, several high-throughput screens demonstrated that the degree of functional inactivation and residual activities are very divergent between different mutants [8,9].
Missense mutations affecting the p53 DBD are typically classified as either DNA contact mutations affecting residues involved in binding DNA or structural mutations that thermodynamically destabilize the conformation of the DBD [10]. DNA binding cooperativity mutations represent a new third class of mutations that affect interactions between DBDs of four p53 monomers, bound to DNA. These interactions are maintained by ionic bonds between H1 helices located within the DBD, which are not involved in contact with DNA but play an essential role in stabilization of the p53-DNA complexes [11,12]. Double salt bridges established between two oppositely charged amino acids (Glu180 and Arg181 in human p53) provide the structural basis for DNA binding cooperativity, which determines the ability of p53 tetramers to recognize and bind specific DNA sequences, thus shaping p53’s transcriptional activity [13–15]. In contrast to structural and DNA-contact mutations, cooperativity mutations that affect the critical R180 and E181 residues have no or little effect on the DBD structure itself [13]. Cooperativity mutations are detected in spontaneous tumors and in hereditary cancer (Li-Fraumeni syndrome) and can lead to selective apoptosis defects [15]. Despite not being mutational hot-spots, cooperativity mutations are estimated to account for 34,000 cancer cases per year world-wide [3]. Recently we have described mouse models for the p53 cooperativity mutations E177R and R178E (p53RR and p53EE, corresponding to human E180 and R181 codons) [16,17]. The R178E mutation abolishes cooperative interactions between p53 monomers and therefore renders p53 entirely deficient in DNA binding, recapitulating mutations that lead to a severe loss of function. In contrast, E177R substitution leads to a reduced interaction between H1 helices and results in a complete deficiency in p53-dependent activation of pro-apoptotic genes, whereas regulation of genes involved in the control of the cell cycle, oxidative defense and metabolism remains intact or only slightly affected [18]. Importantly, tumor-suppressive functions of p53RR were dramatically impaired, making mice a suitable model for studying tumorigenic p53 mutations with partial loss of function.
Mdm2 and Mdm4 (MdmX) are major negative regulators of p53. These homologous proteins share C-terminal RING-finger domains that are necessary for the formation of Mdm2-Mdm4 heterodimers, but only Mdm2 possesses E3 ubiquitin ligase activity and can mono- and poly-ubiquitinate p53 protein thus controlling its intracellular localization and stability, respectively [19]. In addition, Mdm2 and Mdm4 can inhibit transcriptional activity of p53 via binding to its N-terminal transactivation domain [20]. In turn, Mdm2 transcription is regulated by p53, which represents a negative feedback loop mechanism that restrains p53 activity in normal cells [19]. Oncogenic stress mobilizes the ARF tumor suppressor protein which sequesters Mdm2 and thus leads to stabilization and activation of p53 [21]. When tumor cells retain wild-type p53, its level is usually kept low due to loss of ARF or overexpression of Mdm2, which provides a therapeutic window for using Mdm2 inhibitors in cancer treatment, aiming at the restoration of tumor-suppressive p53 functions in tumor cells. In the last two decades, multiple Mdm2 inhibitors were developed and tested in pre-clinical studies and early clinical trials [22]. Mutant p53 protein levels are typically high in tumor cells which indicates uncoupling from the ARF-Mdm2 regulatory circuit and suggests at least partial loss of p53’s tumor-suppressive functions. Therefore, the use of Mdm2 inhibitors is considered unbeneficial for treatment of tumors with mutant p53. Instead, alternative methods such as restoration of native conformation, disaggregation or destabilization are being tested [23]. Although specific accumulation of p53 in cancer cells makes the mutant protein a tempting target for therapy, insufficient knowledge of vulnerabilities created by different mutants hampers the development of such approaches.
Genetic ablation of Mdm2 in mice causes early embryonic lethality (3.5–5.5 dpc) due to massive apoptosis, whereas simultaneous disruption of the Trp53 gene completely rescues the lethal phenotype [24–26]. Moreover, inactivation of the pro-apoptotic p53 target Bax prolongs the development of Mdm2-null embryos up to day 6.5 [24], but knock-out of the cell cycle regulator Cdkn1a (p21) had no rescue effect [27]. These data underscore p53-dependent apoptosis as the primary lethal activity that must be inhibited by Mdm2 during embryogenesis. Importantly, typical p53 hotspot mutations R172H and R246S (corresponding to human R175H and R249S, respectively) and rare partial loss of function R172P (human R175P) mutant rescued embryonic lethality of Mdm2 knock-out mice [28,29]. In contrast, the hypomorphic p53neo allele that retained only about 16% of wild-type Trp53 gene expression did not [30], indicating that the Mdm2 knock-out model can be used for identification of even subtle residual lethal activities of mutant p53. Here we used the Trp53E177R (p53RR) mouse to test if Mdm2-targeting approaches can drive a therapy-relevant response in cells with partial LOF mutants that are defective for p53-dependent apoptosis.
Materials and methods
Animals
The Trp53LSL-RR (p53LSL-RR) and Trp53RR (p53RR) mice with conditional expression of Trp53E177R mutant allele were described elsewhere [16]. For embryonic lethality studies, we used Mdm2Δ7−9 (129-Mdm2< tm1.2Mep>) mice that lack exons 7–9 [31]. These mice were obtained from the NCI Mouse Repository (Frederick, USA) and mated with Trp53LSL-RR/LSL-RR (not expressing p53 and used as the p53 knock-out control, further indicated as Trp53―/―) or Trp53RR/RR homozygote animals to generate double heterozygous Mdm2Δ7−9;Trp53+/RR and Mdm2Δ7−9;Trp53+/― mice for intercrossing. The p21/Cdkn1a knock-out mice (129S2-Cdkn1a<tm1Tyj>) were obtained from the Jackson Laboratory. All mouse experiments were approved by the local authorities and performed in accordance with the German Animal Welfare Act (Deutsches Tierschutzgesetz).
Cell culture and apoptosis assay
Mouse embryonic fibroblasts were isolated at 13.5 dpc using standard protocols and kept at low oxygen conditions (5% O2). MEF were immortalized using the E1A.12S adenoviral oncogene, infections and transfections were performed as described earlier [16]. Cells were cultured in DMEM (Gibco, Life Technologies) with 10% fetal calf serum (Sigma-Aldrich), supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (both from Gibco, Life Technologies). E1A MEFs were treated with 400 ng/ml doxorubicin for 16 h and collected for RNA isolation.
Isolation of embryos and whole-mount immunostaining
Whole-mount PECAM-staining was performed as described [32] with modifications. All solutions were filtered through 0.45 µM filters. Embryos were isolated, deposited in 12 well plates, fixed for 1.5 h in cold 4% buffered formalin, washed 3 × 10 minutes in 0.1% Tween-20 in PBS (PBST). All steps were performed upon gentle agitation. Catalase was inactivated by incubation in 50 mM NaN3 in PBST (20 min, RT). Endogenous peroxidase was blocked with 3% H2O2 + 50 mM NaN3 in PBST (20 min, RT). After washing with PBST (3X10 min, RT) embryos were blocked in PBST supplemented with 10% normal goat serum (NGS) and 0.5% bovine serum albumin (BSA) for 1.5 h (RT). Embryos were incubated in anti-PECAM antibodies (MEC 13.3, BD Pharmingen, 553370, 1:300 in PBST, 1% NGS, 0.5% BSA) for 24–48 h (+4°C). Embryos were washed (PBST, 0.5% BSA, 6 × 1 h, RT) and incubated with secondary antibodies (donkey anti-rat HRP-conjugate (Rockland, 612-703-120) 1:500 in PBST, 1% NGS, 0.5% BSA overnight (+4°C). After washing with PBST, 0.5% BSA (6x30 min, RT) embryos were incubated in TBTI buffer (10 mM imidazole, 0.2% BSA in PBST) for 1 h at RT. All further steps were performed in the dark. Solution was replaced with DyLight-488-tyramide amplification solution (1:100 in TBTI + 0.0015% H2O2) for 2 h at RT. Embryos were washed with PBST overnight (+4°C). Embryos were cleared as described [33]. Briefly, embryos were incubated in 25% formamide + 10% PEG 8000 solution (1 h), and then in 50% formamide + 20% PEG 8000 (two times). Embryos were imaged with a Zeiss stereo microscope. Synthesis of tyramide conjugate was performed as follows: stock solution A (10 mg/ml DyLight-488-NHS ester (Thermo Fisher Scientific, 46402 in dimethylformamide (DMF) and solution B (10 mg/ml tyramine-HCl (Sigma Aldrich, T2879 in DMF supplemented with 7.2 M trimethylamine (Sigma Aldrich, T0886) were prepared. Then 100 µl of solution A was mixed with 15.6 µl of solution B and incubated for 2 h in the dark at RT. Afterward, 884 µl of 100% ethanol was added to obtain a working tyramide amplification solution.
Immunohistochemistry
Embryos were fixed overnight in 4% buffered formalin, washed with PBS and stored until further processing in 70% ethanol. For embedding, embryos were sequentially treated as follows: 80% EtOH 5 min, 95% EtOH 2 × 5 min, 100% EtOH 1 × 5 min, 100% EtOH/Roticlear (Roth) 1:1 5 min, Roticlear – 5 min. Then embryos were incubated sequentially in 3 vessels with paraffin for 5 min (65°C) and embedded immediately after. 5 μm thick sections were mounted to SuperFrost glass slides (Thermo Fischer Scientific) and processed as described earlier[16]. The following antibodies and dilutions were used: anti-p53 (NCL-p53-505, Leica Microsystems, 1:1000), anti-cleaved caspase-3 (#9661, Cell Signaling, 1:100), anti-PCNA (sc-56, clone PC10, Santa Cruz Biotech, 1:100), anti-mouse CD31/PECAM-1 (#553370, clone MEK 13.3, BD Pharmingen). Apoptosis was detected with the DeadEnd TM colorimetric TUNEL System (Promega).
IHC quantification
Staining intensity was quantified using Aperio ImageScope software and a positive pixel count algorithm v9. For each embryo at least 10 regions, covering in total > 50% of the whole section area were randomly selected from several sections at 10x magnification. 3 embryos of each genotype were quantified for every staining. The positivity index was calculated as the ratio of positive pixels over the total number of pixels in regions used for quantification. Statistical significance was assessed using ANOVA with Holm-Sidak multiple comparison test.
Cellular fractionation and western blotting
Nuclear and cytosolic fractions were prepared as follows: cells were collected and resuspended in 2–3 volumes of Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM β-mercaptoethanol) supplemented with protease inhibitors, and incubated for 10 minutes on ice. Then 10% NP-40 was added to a final concentration of 0.25%. Cells were passed through a 27G needle 5–10 times, nuclei were pelleted by centrifugation (500xg, 10 min). The cytoplasmic fraction was collected and re-centrifuged for 10–15 minutes. Nuclear fractions were washed 3 times in 5–10 volumes of Buffer A and pelleted by centrifugation. Nuclei were lysed with 1–2 volumes of Buffer C (20 mM HEPES pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM β-mercaptoethanol) and soluble fraction was collected by centrifugation (10000xg, 10 min). Mitochondrial fractions were isolated using Mitochondria Isolation Kit for Cultured Cells (Thermo Scientific) as described by the manufacturer. For Western Blotting 50 µg of total protein were resolved on 4-12% NuPAGE polyacrylamide gels (Invitrogen). After wet transfer to Hybond-P nitrocellulose membrane (GE Healthcare) antigens were detected using the following antibodies: anti-p53 (NCL-p53-505, Leica Microsystems, 1:2000), anti-PCNA (PC10, #sc-56, 1:1000), anti-Bak (At8B4, Abcam, 1:250), anti-Tom20 (FL-145, #sc-11415, 1:100). Specific immunocomplexes were detected with secondary anti-mouse or anti-rabbit IgG-HRP (GE Healthcare, 1:5,000) and SuperSignal ECL kit (Thermo Fisher).
PCR and RTqPCR
Genotypes of mice, isolated embryos and embryonic tissues were identified by PCR using following primers: 5ʹTCTTTGTGAAGGAACCTTACT3ʹ; 5ʹCATTCATCAGTTCCATAGGTT3ʹ; 5ʹCCCTGAGAAGAGCAAGGC3ʹ, 5ʹAACCAGATCAGGAGGGTCAC3ʹ for genotyping of p53RR; 5ʹCGCCACCAGAAGAGAAACCT3ʹ; 5ʹTGTCCCTATGTACCTGTCTCACT3ʹ; 5ʹGTATTGGGCATGTGTTAGACTGG3ʹ; 5ʹCCTGGATTTAATCTGCAGCACTC3ʹ for genotyping of Mdm2; 5ʹATCAGCAGCCTCTGTTCCAC3ʹ; 5ʹGTCTAGCTCCGGCATTCTCG3ʹ; 5ʹACTCCATGTCTCCAGCCTCT3ʹ for genotyping of p21. For PCR genotyping, tail tips or tissues were lysed overnight at 55°C in PBND buffer (10 mM Tris-HCl pH 8.3, 50 mM KCl, 2.5 mM MgCl2, 0.45% NP-40, 0.45% Tween-20) supplemented with 8 U/ml proteinase K (AppliChem). Proteinase was heat-inactivated at 95°C for 10 min. For reverse transcription quantitative PCR (RTqPCR), RNA was isolated from cells or whole embryos using the RNeasy Mini kit (Qiagen) according to the manufacturer’s protocols. Isolated embryos were controlled microscopically to ensure the absence of resorption and intactness of tissues. cDNA was synthesized from 0.5–1 μg of total RNA with the SuperScript VILO cDNA Synthesis Kit (Invitrogen). Gene expression was assessed on a LightCycler-480 (Roche) using the ABsolute QPCR SYBR Green Mix (Thermo Scientific). Data were analyzed with the ΔΔCt method using β-actin as a reference gene for normalization. Sequences of primers used for RTqPCR were described earlier[16].
Flow cytometry
Flow cytometry analysis was performed on the Accuri C6 flow cytometer (BD Biosciences). For analysis of apoptosis Annexin V-APC (MabTag) kit was used according to the manufacturer’s protocol. Cell viability was determined by staining of freshly collected cells with 0.2μg/ml propidium iodide (PI) in PBS in non-permeable conditions.
Results
P53RR does not rescue embryonic lethality caused by deletion of Mdm2
We used heterozygous mice with a deletion of exons 7–9 of the Mdm2 locus (Mdm2Δ7−9)[31] which were bred with homozygous Trp53E177R/E177R (RR) mice to generate double heterozygous Mdm2+/Δ7−9; Trp53+/RR animals. The double heterozygous progenies were viable and we used them for further breeding. When we genotyped newborn mice obtained from intercrossing of double heterozygous Mdm2+/Δ7−9; Trp53+/RR animals, we observed a clear deviation from the expected Mendelian distribution: notably, no double homozygous Mdm2Δ7−9/Δ7−9; Trp53RR/RR mice were born alive (Figure 1(a)). Similar results were obtained upon breeding of Mdm2+/Δ7−9; Trp53RR/RR animals to each other (Figure 1(c)), indicating that the double homozygous Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos die in utero. In contrast, upon crossing of double heterozygous Mdm2+/Δ7−9; Trp53+/― mice used as the control, double homozygous offspring was obtained with a frequency only insignificantly deviating from the expected one (Figure 1(b)). This data clearly indicated that the cooperativity mutant p53RR, similar to wild-type p53 and different from other partially inactive or cancer-associated mutants such as R172P, R172H, and R246S, exhibits lethal activity in the absence of Mdm2.
Figure 1.
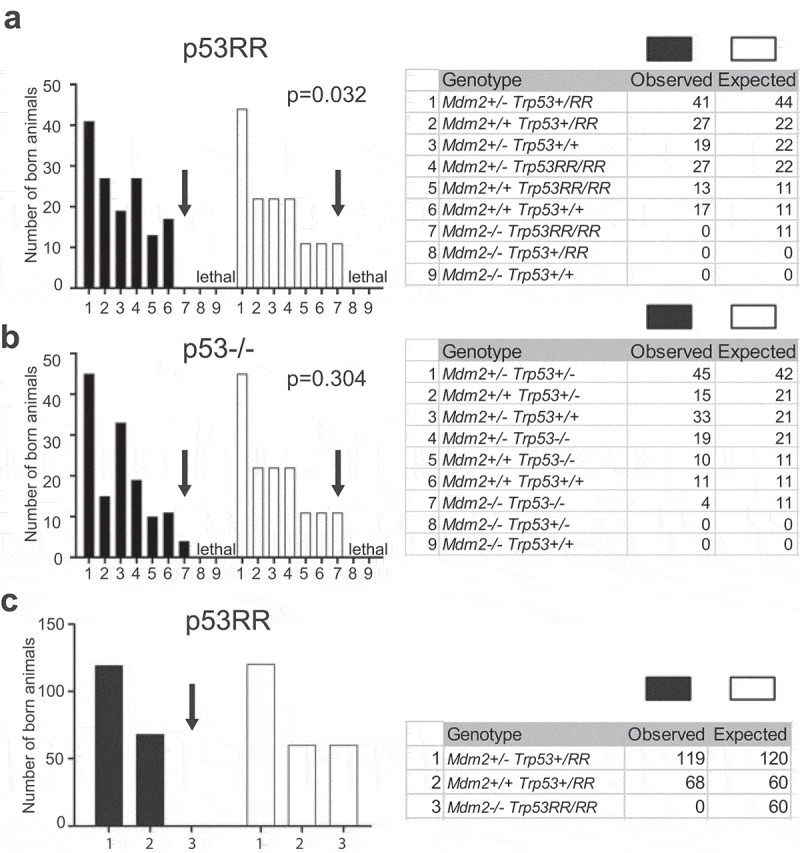
p53RR does not rescue embryonic lethality caused by deletion of Mdm2. (a) Observed and expected genotype distribution of newborn offspring from matings of Mdm2+/Δ7−9;Trp53+/RR mice (total number of pups n = 144; contingency test P = 0.032). (b) Observed and expected genotype distribution of newborn offspring from matings Mdm2+/Δ7−9; Trp53+/− mice (total number of pups n = 137; contingency test P = 0.3044). (c) Observed and expected genotype distribution of newborn offspring from matings Mdm2+/Δ7−9; Trp53RR/RR mice (total number of pups n = 187; contingency test P < 0.0001).
Double homozygous mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos show severe developmental defects
We have previously demonstrated that p53RR is deficient for induction of apoptosis and transcriptional activation of apoptosis-related genes such as Puma, Noxa, and Bax [16] in response to DNA damage. We, therefore, expected survival of double homozygous Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos beyond the terms observed in p53 wild-type background (3.5–5.5 dpc). We started collecting embryos from 6.5 dpc and did not observe any abnormalities until day 7.5–8.5 (Figure 2(a) and not shown). Strikingly, at 9–9.5 dpc the majority of double homozygous embryos displayed severe developmental defects and after day 11 no normal embryos with this genotype were recovered (Figure 2(b,c), Suppl. Figure 1(a)). Also, no living double homozygous embryos were found at later developmental stages. At day 9 dpc (TS 14) pronounced defects in neural tube closure and formation of brain vesicles of Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos were evident (Suppl. Figure 1(a)), which became further aggravated at day 9.5–10.5 and were accompanied with growth retardation (Figure 2(c)). Hemizygous Mdm2Δ7−9/Δ7−9; Trp53RR/― embryos displayed the same morphological abnormalities (Suppl. Figure 1(b)). Notably, the double homozygous embryos that were found alive (heart beating) at day 10.5–11.5 looked pale and bloodless (Figure 2(c,d)). The yolk sac of those embryos had a clearly reduced number of large blood vessels and abnormal structure of the capillary network (Figure 2(e)). Similar defects were detected in whole-mount Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos stained with antibodies to the endothelial marker Pecam-1 (Figure 2(f)).
Figure 2.
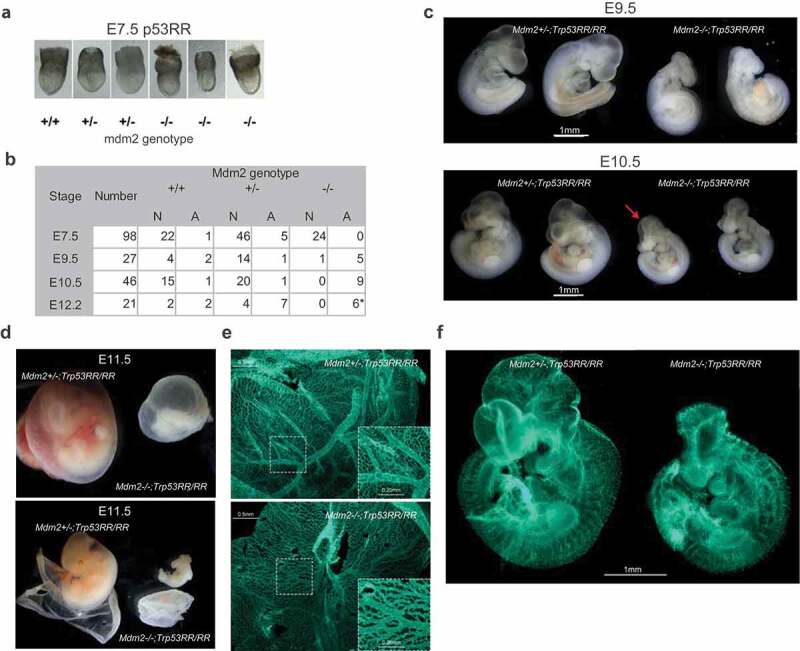
Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos show severe developmental defects. (a) Representative images of E7.5 embryos demonstrating absence of developmental abnormalities in Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos at this stage. (b) Phenotype analysis of Trp53RR/RR embryos with different Mdm2 genotypes reveals developmental defects in Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos starting from E9.5. N – normal, A – abnormal morphology. Asterisk – embryos were partially resorbed. (c) Representative images of Trp53RR/RR embryos with hetero- and homozygous Mdm2 deletion at stages E9.5 (TS15, upper panel) and E10.5–11 (TS18, bottom panel) show progressive growth retardation, abnormal head morphology and neural tube closure defects (arrow) in Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos. (d) Representative image of Mdm2+/Δ7−9 and Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos at stage E11.5, (TS19) with the intact (upper panel) and opened (bottom panel) embryonic envelope demonstrates a “bloodless” phenotype of the Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos. (e) Representative picture of PECAM-1 (endothelial marker) staining in whole mount yolk sac samples shows a reduced number of large blood vessels and decreased branching of the capillary network (zoom-in) in homozygous Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos (bottom panel). (f) Representative picture of Pecam-1 staining in whole mount embryos shows a reduced branching in blood vessel network and abnormal head morphology of homozygous Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos.
Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos die with signs of apoptosis
To investigate the pathological phenotype of double heterozygous embryos in more detail, we used intact embryos with no signs of resorption for immunohistochemical staining. First, we analyzed p53 protein levels. As expected, in all analyzed samples collected at 9–10.5 dpc (TS 14–17) we observed a strong accumulation of p53RR protein, particularly in the neuroepithelium, limb buds, and somites, whereas in the cardiac tissues the p53 level was markedly lower (Figure 3(a,b) and Suppl. Figure 2(b). The p53RR protein retains the ability to induce expression of the Cdkn1a gene and therefore is proficient in mounting cell cycle arrest [16]. The abnormal phenotype of Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos was reminiscent of that published for Mdm4 deficient mice which die at 7.5–10.5 dpc because of cessation of proliferation by p21-mediated cell cycle arrest [34]. We hypothesized that accumulation of p53RR may lead to high expression of p21 and loss of proliferative potential resulting in developmental aberrations and death of Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos. However, quantitative analysis of PCNA staining that is commonly used as a marker for proliferating cells revealed no difference between normal Mdm2+/Δ7−9; Trp53RR/RR and abnormal Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos (Figure 3(c,d)). Abnormalities in development of embryonic vasculature can result in insufficient blood supply and acute ischemia, which in turn can lead to necrotic cell death. However, we have not seen typical microscopic signs of necrosis such as swelling of cells and membrane rupture in tissues of double homozygous embryos. Instead we observed widespread apoptosis, especially in the neuroepithelial tissues, as suggested by chromatin condensation, nuclear DNA fragmentation (TUNEL) and caspase-3 cleavage (CC3), which started approximately at 9 dpc and progressively increased at later stages (Figure 3(e,g) and Suppl. Figure 2(c,f)). Importantly, tissues with weak staining for p53 protein also demonstrated low levels of apoptosis (Figure 3(a,e,g)), indicating that high levels of p53 protein may be responsible for apoptosis. This was entirely unexpected, given that all our previous experiments had demonstrated a complete loss of p53-dependent apoptosis in human and mouse p53RR cells that was explained by a defect of p53RR in binding and transactivating pro-apoptotic target genes [1,5,16]. To analyze this surprising finding in more detail, we examined different tissues at 9.5 and 10.5 dpc (magnification panels in Figure 3(a,e,g) and Suppl. Figure 2(c)) of intact embryos and quantified p53, TUNEL and CC3 staining at these stages. As shown in Figure 3(f,h) and Suppl. Figure 2(f), we consistently observed a strong accumulation of p53 and increase of apoptosis in Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos. Moreover, we detected a direct correlation between p53 protein and apoptosis levels (Suppl. Figure 2d,e). Collectively, these data pointed to p53RR as the cause of cell death.
Figure 3.
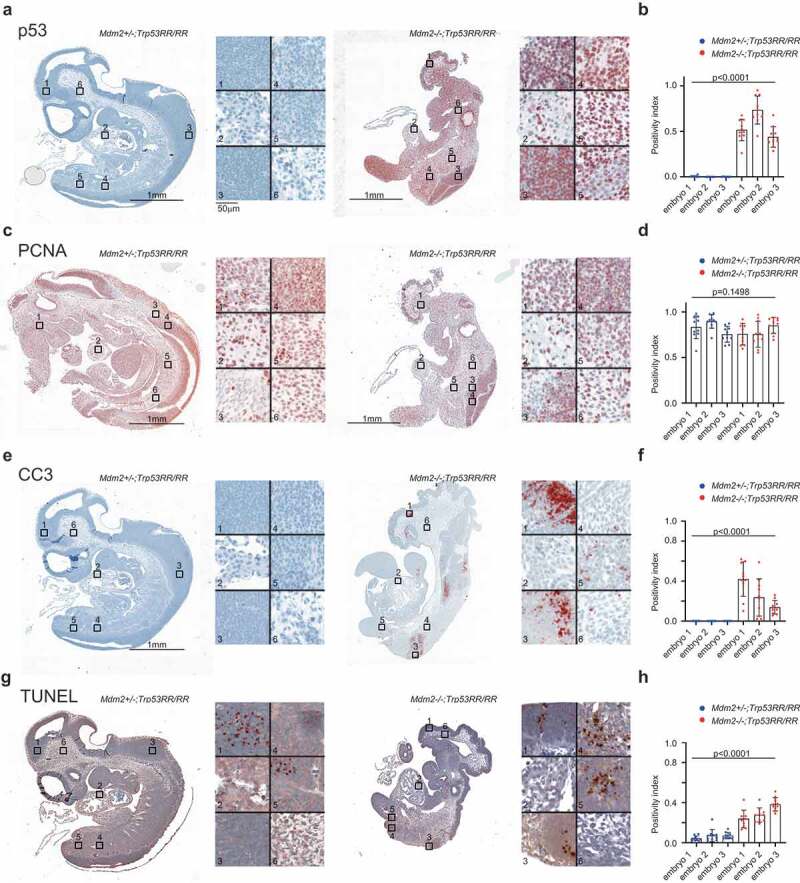
Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos die with signs of apoptosis. (a,b) Representative immunohistochemical (IHC) staining of Mdm2+/Δ7−9 and Mdm2Δ7−9/Δ7−9;Trp53RR/RR E10.5 embryos using anti-p53 antibodies. Right panel: different tissues shown at high magnification (1,3 – neuroepithelial, 2 – heart, 4,5 – somites, 6 – mesenchyme). Strong accumulation of p53 protein is evident in all tissues except the heart in Mdm2Δ7−9/Δ7−9;Trp53RR/RR samples. (b) – quantification of IHC staining in 3 embryos for each genotype, 10 random fields of view covering in total >50% of the section area, ANOVA test. (c,d) IHC staining of proliferating cells nuclear antigen (PCNA) in a serial section from the same samples as in (a). Right panel: different tissues shown at high magnification (1 – mesenchyme, 2 – heart, 3,4 – neuroepithelial, 5,6 – somites). (d)- quantification of PCNA staining, done as in (b), shows a slight reduction in PCNA levels in Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos, ANOVA test. (e,f) IHC staining of apoptosis marker cleaved caspase 3 (CC3) in a serial section from the same samples as in (a). Right panel: different tissues shown at high magnification (1,3 – neuroepithelial, 2 – heart, 4,5 – somites/limb bud, 6 – mesenchyme). Note apoptosis in neural tissues and limb bud of Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryo. (f) – quantification of IHC staining in 3 embryos for each genotype, done as in (b), ANOVA test. (g,h) Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) assay in a serial section from the same samples as in (a). Right panel shows the same sites as in (e). Note only a small foci of apoptosis in neural tissue of the normal Mdm2+/Δ7−9;Trp53RR/RR embryo, whereas massive apoptosis is observed in multiple tissues of the abnormal double homozygous Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryo. (h) – quantification of IHC staining, done as in (b), ANOVA test.
Genetic ablation of Cdkn1a does not provide even partial rescue of embryonic lethality
Apart from transactivating tumor-suppressive target genes, p53 also activates the p21-DREAM pathway and thereby indirectly represses dozens of genes involved in cell proliferation and survival, which might lead to developmental defects, embryonic death, and secondary apoptosis [35]. Importantly, p53RR retains the ability to induce p21 and therefore engages the p21-DREAM pathway for gene repression [18]. We reasoned that if embryonic lethality was caused by activation of the p53-p21-DREAM pathway, knock-out of p21/Cdnk1a should provide at least partial rescue of developmental defects. We, therefore, generated triple transgenic Mdm2Δ7−9/Δ7−9;Cdkn1a–/–;Trp53RR/RR mice and bred them to obtain triple-homozygous offspring. However, we detected no triple homozygotes upon genotyping of newborn pups obtained from such matings (Figure 4(a)). In fact, genetic inactivation of p21 did not provide even partial rescue of the lethal Mdm2-null phenotype – Mdm2Δ7−9/Δ7−9;Cdkn1a–/―;Trp53RR/RR embryos died at E9.5–10.5 dpc with the same morphological abnormalities as observed in Mdm2Δ7−9/Δ7−9;Trp53RR/RR mice – developmental retardation, impaired blood supply, neural tube closure defects (Figure 4(b,c,d). It suggested that the observed abnormal phenotype was caused primarily by a mechanism distinct from the lack of proliferation or p21-DREAM mediated gene repression. Like Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos, triple homozygous embryos expressed very high levels of p53 protein (Figure 4(e)). Moreover, also in Mdm2Δ7−9/Δ7−9;Cdkn1a–/―;Trp53RR/RR embryos we detected high levels of apoptosis in neuroepithelial tissues and somites as indicated by TUNEL assay, similar to double homozygous samples (compare Figures 4(f) and 3(g)). Although it is unclear, whether p53-induced apoptosis or developmental defects caused the embryonic lethality, these data show that the apoptosis defect of p53RR, previously reported for newborn and adult tissues following DNA damage [16], was rescued in embryonic cells by loss of Mdm2.
Figure 4.
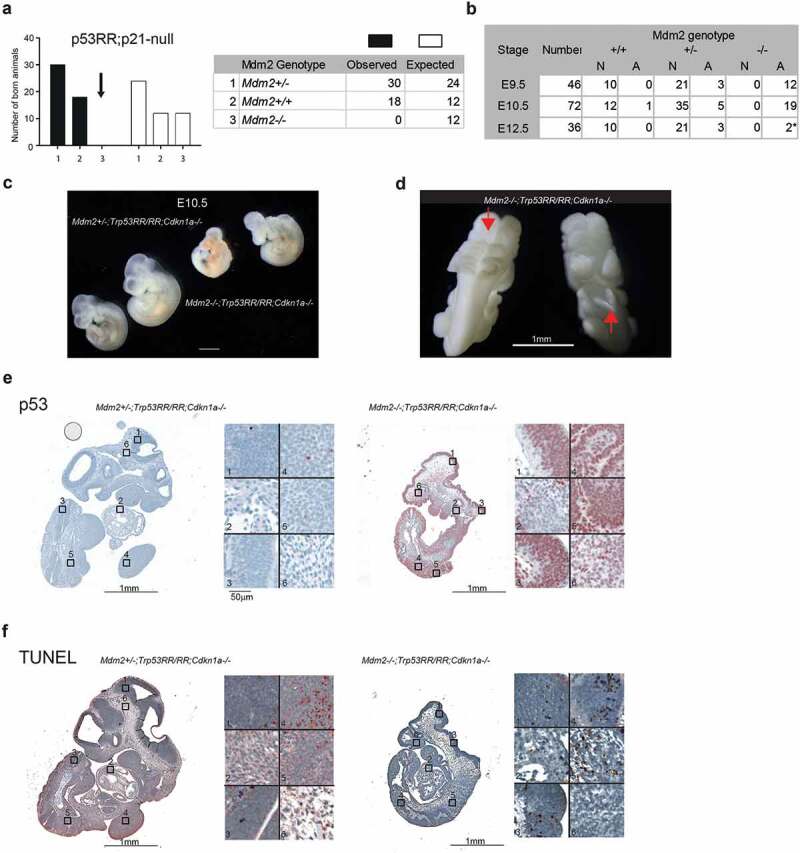
Genetic ablation of Cdkn1a fails to rescue embryonic lethality. (a) Observed and expected genotype distribution of newborn offspring from matings of Mdm2Δ7−9/Δ7−9;Cdkn1a–/–;Trp53RR/RR mice (total number of pups n = 48; contingency test P = 0.001). (b) Phenotype analysis of Cdkn1a–/–;Trp53RR/RR embryos with different Mdm2 genotypes shows the onset of development defects at the same stage (E9.5 – E10.5) as in Mdm2Δ7−9/Δ7−9; Trp53RR/RR embryos. N – normal, A – abnormal morphology. Asterisk – embryos were partially resorbed. (c) Representative images of Cdkn1a–/–;Trp53RR/RR embryos with hetero- and homozygous Mdm2 deletion at stage E10.5 (TS17-18) show same phenotypic abnormalities as in Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos. (d) Representative image of an Mdm2Δ7−9/Δ7−9;Cdkn1a–/–;Trp53RR/RR embryo shows neural tube closure defects (arrows) in cranial (left) and caudal (right) parts of the embryo. (e) Representative IHC staining of E10.5 embryos for p53 shows strong accumulation of p53 protein in Mdm2Δ7−9/Δ7−9;Cdkn1a–/–;Trp53RR/RR samples. Right panel: different tissues shown at high magnification (1,3 – neuroepithelial, 2 – heart, 4,5 – somites/limb bud, 6 – mesenchyme). (e) TUNEL assay in a serial section from the same samples as in (E). Note the enhanced apoptosis in neuroepithelial tissues and somites of Mdm2Δ7−9/Δ7−9;Cdkn1a–/–;Trp53RR/RR embryos.
High levels of mutant p53RR protein can partially compensate for the cooperativity defect
The apoptosis deficiency of p53RR is caused by weakened interactions between p53 monomers that result in a compromised ability to bind promoters of many pro-apoptotic target genes [15,18,36]. To analyze expression of p53-dependent genes in embryos, we bred Mdm2+/Δ7−9; Trp53RR/RR mice and collected embryos at 10.5 dpc. All embryos were genotyped for Mdm2 and used for isolation of RNA. As expected, quantitative PCR analysis of Mdm2-deficient embryos showed a strongly elevated expression of Cdkn1a, Ccng1, Aldh4a, Gls2, and Sesn2 genes that have been demonstrated earlier as p53RR targets in human and mouse cells [16,18] (Figure 5(a), left panel). Surprisingly, we also detected significant upregulation of pro-apoptotic Bbc3 (Puma) and Bax transcripts, which was comparable to activation levels observed in p53 wild-type MEFs after treatment with doxorubicin, used as a positive control (compare Figure 5(a), right panel and Figure 5(b)). This finding was unexpected because in our earlier experiments we did not detect any increase in expression of Puma or Bax in p53RR cells after irradiation or doxorubicin treatment [16]. The validity of our previous findings was confirmed by the lack of Bbc3 and Bax induction in doxorubicin-treated p53RR MEFs (Figure 5(b)). Because of the very limited amount of embryonic tissues, we decided to use readily available mouse fibroblasts (MEFs) to further analyze the mechanisms of apoptosis triggered by p53RR. Recently, the combination of Mdm2 inhibitor Nutlin-3a and doxorubicin was shown to evoke apoptotic activity, mediated by direct mitochondrial functions, of the entirely DNA binding-deficient cooperativity mutant R178E [17]. To check if p53RR MEFs are sensitive to such a combination of drugs, we analyzed apoptosis and overall cell death in treated cells using flow cytometry and found p53RR cells to be highly apoptotic in these conditions (Figure 5(c)), whereas treatment with Nutlin-3a or doxorubicin alone had no effect. Importantly, apoptosis was associated with transcriptional activation of proapoptotic target genes Bbc3 and Bax, similarly as observed in Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos (Figure 5(d)). Of note, combination treatment also induced Bbc3 and Bax in p53-null MEFs, but to significantly lesser extent (Figure 5(d)). It is possible that the apoptotic activity of p53RR mutant was also supported by transcription-independent mechanisms because we detected p53RR protein in mitochondrial fractions (Figure 5(e)). Taken together, these data suggest that massive stabilization of p53RR protein – either due to complete genetic inactivation of Mdm2 or upon its pharmacological inhibition complemented with cytotoxic stress can partially compensate for the defect in DNA binding cooperativity and restore apoptotic proficiency of the mutant.
Figure 5.
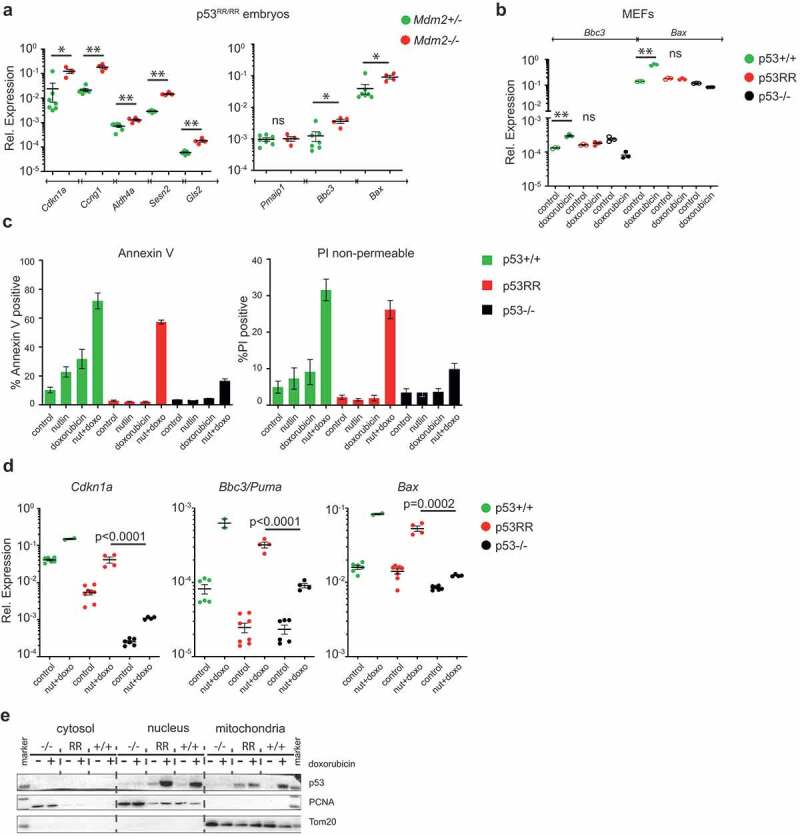
High levels of mutant p53RR protein can partially compensate for the cooperativity defect. (a) mRNA expression analysis (RTqPCR) of p53 target genes in Trp53RR/RR embryos with indicated Mdm2 genotypes. Shown are expression values normalized to β-actin, normal n = 7, abnormal n = 4. (b) RTqPCR analysis of the expression of pro-apoptotic p53 target genes Bbc3 (Puma) and Bax in primary Trp53+/+, Trp53RR/RR and Trp53–/ – MEFs after 16 h treatment with 400 ng/ml doxorubicin. Shown are technical triplicates. In (a) and (b) * P < 0.05, ** P < 0.01, ns – not significant, error bars represent SD, two-tailed Mann Whitney test. (c) Quantification of apoptosis detected by flow cytometry with Annexin-V and non-permeable PI staining in Trp53+/+, Trp53RR/RR and Trp53–/– E1A-imortalized MEFs upon 16 h treatment with 10 μM Nutlin-3a, 400 ng/ml doxorubicin or a combination of both. (d) RTqPCR quantification of p53 target genes in E1A-MEFs from (c) upon treatment with Nutlin-3a and doxorubicin. Each dot indicates one biological replicate, ANOVA test. (e) Representative western blot of cytosolic, nuclear and mitochondrial fractions from primary MEFs (untreated and treated with 400 ng/ml doxorubicin), probed with anti-p53 antibodies. PCNA and Tom20 antibodies were used as fractionation controls.
Discussion
Cooperativity mutations belong to a third and distinct class of p53 missense mutations affecting the DBD. They are found as sporadic cancer mutations and also as germ-line mutations giving rise to hereditary cancer susceptibility [15]. They are estimated to account for approximately 34,000 cancer cases per year world-wide [3]. In contrast to p53 “hotspot” mutations, cooperativity mutations commonly do not result in a complete loss of DNA binding but rather cause a partial loss of function. In particular, reduction in cooperative interactions between p53 monomers severely impairs the ability of p53RR to bind and transactivate pro-apoptotic target genes, whereas induction of cell cycle arrest and senescence along with other homeostatic p53 functions are only weakly affected [15,18,16]. Such a selective loss of p53-induced apoptosis has also been observed for many other non-hotspot p53 mutants [8,37–41], making p53 cooperativity mutants a valuable model to study the consequences of a partial loss of p53 activity for tumorigenesis or cancer therapy.
Here we report the surprising observation that the apoptosis deficiency of the cooperativity mutation p53RR was overcome by the loss of Mdm2. Although it is unclear whether the enhanced apoptosis is the main reason for embryonic lethality caused by p53RR, this genetic model demonstrated that a p53 mutant, deficient for apoptosis in other settings, can exert lethal activity. Recently, also using the Mdm2 knock-out model, we have shown that another cooperativity mutant Trp53E177R (p53EE) can induce embryonic lethality [17]. The Trp53E177R (p53RR) and Trp53R178E (p53EE) knock-in mice with cooperativity mutations have different phenotypes as the DNA binding and transcriptional activity of p53 is affected to a different degree: whereas p53EE is fully deficient in DNA binding and devoid of direct transcriptional activity, p53RR is only partially compromised regarding DNA binding and transactivation resulting in a selective loss of transcription-dependent apoptosis. As a consequence, substantial differences between these mutants in p53-dependent response and tumor suppression were observed [16,17]. Interestingly, although both mutations strongly affect transactivation of p53-dependent apoptotic genes and render cells apoptosis-deficient, our data show that this deficiency is not absolute and can be compensated, yet by different mechanisms: while p53EE mutant induces apoptosis independently of regular transcriptional activity, p53RR seems to engage both mitochondrial and transcription-mediated pathways, as suggested by a restored ability to induce proapoptotic Puma/Bbc3 and Bax expression. One reasonable explanation for this unexpected finding could be that in developing embryos an abnormal accumulation of p53RR triggered by the loss of Mdm2 can compensate for the cooperativity defect and in combination with endogenous stress signals mobilize accumulated p53 to transactivate pro-apoptotic targets. The ability of wild-type p53 to trigger apoptosis is known to be dependent on at least three factors: the expression level of p53, the duration of increased p53 expression and the intrinsic apoptotic sensitivity of cells, i.e. its apoptosis threshold [42–45]. Embryonic cells have an exceptionally low apoptosis threshold, explained at least in part by high levels of mitochondrial priming [46]. In the case of Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos, p53RR protein is massively and constitutively stabilized because of the lack of Mdm2 as the major negative regulator of p53. We speculate that the sustained high-level expression of p53RR restores binding to pro-apoptotic gene promoters by simple mass action: the more protein is available, the higher is its probability to bind DNA. The elevated expression of Puma/Bbc3 and Bax in Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos and MEFs treated with Mdm2 inhibitor Nutlin-3a combined with doxorubicin also supports this hypothesis. We, therefore, assume that the combination of these factors in Mdm2-deficient embryos suffices to rescue the transcriptional apoptosis defect of p53RR cells.
Furthermore, p53 possesses transcription-independent cytotoxic activity [47]. Several studies have shown that p53 drives mitochondrial apoptosis via direct interaction with Bcl2 family proteins: it inhibits anti-apoptotic members (Bcl2, BclxL, Mcl1) and activates pro-apoptotic Bak and Bax, leading to mitochondrial outer membrane permeabilization [48–50]. Interestingly, it has been reported that the Mdm2 inhibitor Nutlin-3a can enhance the non-transcriptional pro-apoptotic functions of p53 and treatment of chronic lymphocytic leukemia cells with nutlin induces a more robust non-transcriptional than transcription-dependent apoptotic response [51,52]. Thus, activation of non-transcriptional apoptosis by targeting Mdm2 may contribute to and support the lethal activity of p53 mutants with impaired transcriptional functions. Whereas hotspot mutations that severely affect the DNA-interaction interface or global structure of p53 were shown to reduce both transcriptional and non-transcriptional apoptosis [53], non-hotspot mutants may retain some of these functions. In support of this, the Trp53R178E (p53EE) cooperativity mutant is capable of inducing mitochondrial apoptosis in the absence of target gene activation [17]. It is therefore conceivable also for p53RR that residual non-transcriptional cell death activities further contribute to the apoptosis observed in Mdm2Δ7−9/Δ7−9;Trp53RR/RR embryos and E1A MEFs treated with nutlin and doxorubicin. In line with this hypothesis we detected p53RR in mitochondrial fractions (Figure 5(e)).
It has been shown for wild-type p53 that the amount and dynamics of p53 protein accumulation determine the shape and outcome of the stress response [43–45,54,55]. Intriguingly, our results identify the well-established apoptosis deficiency of cooperativity mutants as a relative, context-dependent defect that, in principle, can be overcome by interventions which increase the level and duration of mutant p53 expression or lower the cell-intrinsic apoptosis threshold. It is tempting to speculate that the residual transcriptional activities described for numerous other p53 mutants could be boosted therapeutically, for example with Mdm2 inhibitors, to drive pro-apoptotic target gene expression beyond the apoptosis threshold. Such efforts might be supported with compounds such as BH3 mimetics which lower the apoptosis threshold. As many cancer-associated p53 mutants, even those with residual transcriptional activity, are highly accumulated in tumor cells, it might be possible to identify proper stimuli to limit the apoptotic response to cancer cells while sparing normal cells.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [TRR81 A10, TI 1028/2-1]; Deutsche Krebshilfe [111250, 70112623, 111444]; José Carreras Leukämie-Stiftung [R13/08, R09/2018]; German Center for Lung Research (DZL).
Acknowledgments
Authors thank Sigrid Bischofsberger, Antje Grzeschiczek, Angela Mühling, Björn Geissert for technical assistance and Dr. Sabrina Elmshäuser and employees of the animal facility of Marburg University for help in mouse experiments. We acknowledge Dr. Deckelbaum and Julia Lerner (Regeneron Pharmaceuticals, NY, USA) for sharing a protocol of whole-mount PECAM staining and preparation of tyramide conjugates; we thank Dr. Gergana Dobreva (Heidelberg University) for help with microscopy.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemented data of this article can be accessed here.
References
- [1].Lane DP. 1992. Cancer p53 guardian of the genome. Nature 358 (6381):15-6. [DOI] [PubMed] [Google Scholar]
- [2].Kastenhuber ER, Lowe SW.. Putting p53 in context. Cell. 2017;170:1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Leroy B, Girard L, Hollestelle A, et al. Analysis of TP53 mutation status in human cancer cell lines: A reassessment. Hum Mutat. 2014;35(6):756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer [Internet] 2009; 9:701–713. [DOI] [PubMed] [Google Scholar]
- [5].Muller PAJ, Vousden KH. P53 mutations in cancer. Nat Cell Biol. [Internet] 2013; 15:2–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23263379 [DOI] [PubMed] [Google Scholar]
- [6].Stiewe T, Haran TE. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist Updat. 2018;38:27–43. [DOI] [PubMed] [Google Scholar]
- [7].Muller PAJ, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shibata H, Kato S, Han S-Y, et al. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci. 2003;100(14):8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kotler E, Shani O, Goldfeld G, et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell. 2018;71:178–190.e8. [DOI] [PubMed] [Google Scholar]
- [10].Joerger AC, Fersht AR. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu Rev Biochem. [Internet] 2016; 85:375–404. Available from: http://www.annualreviews.org/doi/10.1146/annurev-biochem-060815-014710 [DOI] [PubMed] [Google Scholar]
- [11].Kitayner M, Rozenberg H, Kessler N, et al. Structural basis of DNA recognition by p53 tetramers. Mol Cell. 2006;22:741–753. [DOI] [PubMed] [Google Scholar]
- [12].Klein C, Planker E, Diercks T, et al. NMR spectroscopy reveals the solution dimerization interface of p53 core domains bound to their consensus DNA. J Biol Chem. 2001;276:49020–49027. [DOI] [PubMed] [Google Scholar]
- [13].Dehner A, Klein C, Hansen S, et al. Cooperative binding of p53 to DNA: regulation by protein-protein interactions through a double salt bridge. Angew Chemie - Int Ed. 2005;44:5247–5251. [DOI] [PubMed] [Google Scholar]
- [14].Kitayner M, Rozenberg H, Rohs R, et al. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat Struct Mol Biol. [Internet]. 2010;17:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schlereth K, Beinoraviciute-Kellner R, Zeitlinger MK, et al. DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2010;38:356–368. [DOI] [PubMed] [Google Scholar]
- [16].Timofeev O, Schlereth K, Wanzel M, et al. P53 DNA binding cooperativity is essential for apoptosis and tumor suppression invivo. Cell Rep. 2013;3:1512–1525. [DOI] [PubMed] [Google Scholar]
- [17].Timofeev O, Klimovich B, Schneikert J, et al. Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses. Embo J. 2019;38(20):e102096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schlereth K, Heyl C, Krampitz AM, et al. Characterization of the p53 cistrome - DNA binding cooperativity dissects p53’s tumor suppressor functions. PLoS Genet. 2013;9:e1003726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Marine J-C, Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. Internet. 2010;17:93–102. [DOI] [PubMed] [Google Scholar]
- [20].Marine JC, Francoz S, Maetens M, et al. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. [Internet] 2006; 13:927–934. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16543935 [DOI] [PubMed] [Google Scholar]
- [21].Weber JD, Taylor LJ, Roussel MF, et al. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. [DOI] [PubMed] [Google Scholar]
- [22].Burgess A, Chia KM, Haupt S, et al. Clinical overview of MDM2/X-targeted therapies. Front Oncol. 2016;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sabapathy K, Lane DP, Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. [Internet] 2017; Available from: http://www.nature.com/doifinder/10.1038/nrclinonc.2017.151 [DOI] [PubMed] [Google Scholar]
- [24].Chavez-Reyes A, Parant JM, Amelse LL, et al. Switching mechanisms of cell death in mdm2- and mdm4-null mice by deletion of p53 downstream targets. Cancer Res. 2003;63:8664–8669. [PubMed] [Google Scholar]
- [25].Jones SN, Roe AE, Donehower LA, et al. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. [DOI] [PubMed] [Google Scholar]
- [26].Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. [DOI] [PubMed] [Google Scholar]
- [27].Oca Luna RM, De, Amelse LL, Chavez-Reyes A, et al. Deletion of p21 cannot substitute for p53 loss in rescue of mdm2 null lethality. Nat Genet. 1997;16(4):336–337. [DOI] [PubMed] [Google Scholar]
- [28].Lee MK, Teoh WW, Phang BH, et al. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell. Internet. 2012;22:751–764. [DOI] [PubMed] [Google Scholar]
- [29].Abbas HA, MacCio DR, Coskun S, et al. Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ros-induced p53 activity. Cell Stem Cell. 2010;7:606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang Y, Suh YA, Fuller MY, et al. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mendrysa SM, McElwee MK, Michalowski J, et al. Mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. 2003;23:462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lazarus A, Del-Moral PM, Ilovich O, et al. A perfusion-independent role of blood vessels in determining branching stereotypy of lung airways. Development. 2011;138:2359–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kuwajima T, Sitko AA, Bhansali P, et al. ClearT: a detergent- and solvent-free clearing method for neuronal and non-neuronal tissue. Development. 2013;140:1364–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Migliorini D, Lazzerini, Denchi E, et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. [Internet] 2002; 22:5527–5538. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12101245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018;25:114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schlereth K, Charles JP, Bretz AC, et al. Life or death: p53-induced apoptosis requires DNA binding cooperativity. Cell Cycle. 2010;9:4068–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rowan S, Ludwig RL, Haupt Y, et al. Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. Embo J. 1996; 15(4):827–838. [PMC free article] [PubMed] [Google Scholar]
- [38].Ludwig RL, Bates S, Vousden KH. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol Cell Biol. 1996;16:4952–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Campomenosi P, Monti P, Aprile A, et al. P53 mutants can often transactivate promoters containing a p21 but not Bax or PIG3 responsive elements. Oncogene. 2001;20:3573–3579. [DOI] [PubMed] [Google Scholar]
- [40].Jordan JJ, Inga A, Conway K, et al. Altered-function p53 missense mutations identified in breast cancers can have subtle effects on transactivation. Mol Cancer Res. 2010;8:701–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Menendez D, Inga A, Resnick MA. The biological impact of the human master regulator p53 can be altered by mutations that change the spectrum and expression of its target genes. Mol Cell Biol. 2006;26:2297–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Le Pen J, Laurent M, Sarosiek K, et al. Constitutive p53 heightens mitochondrial apoptotic priming and favors cell death induction by BH3 mimetic inhibitors of BCL-xL. Cell Death Dis. [Internet] 2016. [cited 2016 June24]; 7:e2083. Available from: http://www.nature.com/doifinder/10.1038/cddis.2015.400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Speidel D, Helmbold H, Deppert W. Dissection of transcriptional and non-transcriptional p53 activities in the response to genotoxic stress. Oncogene. [Internet] 2006; 25:940–953. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16247471 [DOI] [PubMed] [Google Scholar]
- [44].Kracikova M, Akiri G, George A, et al. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013; 20(4):576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Purvis JE, Karhohs KW, Mock C, et al. p53 dynamics control cell fate. Science. 2012;336:1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sarosiek KA, Fraser C, Muthalagu N, et al. Developmental regulation of mitochondrial apoptosis by c-myc governs age- and tissue-specific sensitivity to cancer therapeutics. Cancer Cell. 2017;31:142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Marchenko ND, Moll UM. Mitochondrial death functions of p53. Mol Cell Oncol. [Internet] 2014. [cited 2017 December8]; 1:e955995. Available from: https://www.tandfonline.com/doi/full/10.1080/23723548.2014.955995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. J Biol Chem. 2000;275(21):16202–16212. [DOI] [PubMed] [Google Scholar]
- [49].Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53~{m}ediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. [DOI] [PubMed] [Google Scholar]
- [50].Leu JIJ, Dumont P, Hafey M, et al. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–450. [DOI] [PubMed] [Google Scholar]
- [51].Kojima K, Konopleva M, McQueen T, et al. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108:993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Steele AJ, Prentice AG, Hoffbrand AV, et al. P53-mediated apoptosis of CLL cells: evidence for a transcription- independent mechanism. Blood. 2008;112:3827–3834. [DOI] [PubMed] [Google Scholar]
- [53].Tomita Y, Marchenko N, Erster S, et al. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. [Internet] 2006. [cited 2018 March13]; 281:8600–8606. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16443602 [DOI] [PubMed] [Google Scholar]
- [54].Chen X, Ko LJ, Jayaraman L, et al. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 1996;10:2438–2451. [DOI] [PubMed] [Google Scholar]
- [55].Paek AL, Liu JC, Loewer A, et al. Cell-to-cell variation in p53 dynamics leads to fractional killing. Cell. 2016;165:631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.