

ABSTRACT
Hepatocellular carcinoma appears as an extremely angiogenic solid tumor marked by apoptosis evasion, dysregulated cell cycle and low sensitivity to chemotherapy. TGF-β, a multifunctional cytokine, plays a pleiotropic role in the tumor microenvironment and has implications in cancer drug resistance. The current study provides novel evidence that TGF-β signaling contributes to drug resistance in liver cancer cells by inducing the expression of xenobiotic nuclear receptor PXR. We observed that PXR increases the expression of drug efflux transporters; therefore, accounting for exacerbated drug resistance. Additionally, anti-apoptotic nature of PXR contributes to TGF-β mediated chemoresistance as seen by procaspase-3 and Mcl-1 cellular levels. TGF-β binding to the TGF-β receptor triggers a complex downstream signaling cascade through a non-canonical SMAD-independent ERK pathway that leads to increased PXR expression. Activated ERK activates ETS1 transcription factor which is a critical regulator of endogenous PXR expression in hepatic cells. Loss of function of ETS1 abrogates the TGF-β induced PXR expression. Together these findings indicate that PXR modulates TGF-β induced resistance to chemotherapy in liver cancer cells. This underscores the need for combinatorial approaches with focus on PXR antagonism to improve drug effectiveness in hepatocellular carcinoma.
Abbreviations: HCC: Hepatocellular Carcinoma; FDA: Food and Drug Administration; TGF-β: Transforming growth factor-β; PXR: Pregnane X receptor; CAR: Constitutive androstane receptor; P-gp/ABCB1: P-glycoproteins/ATP-binding cassette transporter subfamily B member 1; MRP1/ABCC1 and MRP2/ABCC2: Multidrug-resistance associated proteins; BCRP/ABCG2: Breast cancer resistant protein; DMEs: Drug-metabolizing enzymes; CFDA: 5,6-carboxyfluorescein diacetate; ETS1: Transcription factor E26 transformation specific sequence 1.
KEYWORDS: Hepatocellular carcinoma, transforming growth factor-β, chemoresistance, nuclear receptors, pregnane X receptor, extracellular‐signal‐regulated kinase, sorafenib
Introduction
Hepatocellular Carcinoma (HCC) is the fifth most common malignancy and the fourth leading cause of cancer mortality worldwide [1]. According to American Cancer Society, an estimated 42,030 new cases of liver cancer would be diagnosed in the US during 2019 and an approximate three-fourths of which would be HCC. Due to poor prognosis and late detection, the 5-y survival rate is merely 18% [2]. Although several risk factors for HCC are known, the major ones are liver cirrhosis and fibrosis [3]. Sorafenib, a multikinase inhibitor, is the FDA approved drug for advanced HCC and inhibits the survival and growth of HCC cells by targeting various raf kinases in RAS/RAF/MEK/ERK signaling cascade as well as tyrosine kinases such as Flt3, c-Kit, VEGFR-2/3 and PDGFR-β [4–6]. However, there are numerous reports that suggest decreased overall survival of HCC patients treated with sorafenib as they develop resistance against this drug at later stages [7]. HCC is an extremely malignant tumor with low sensitivity to chemotherapy and various mechanisms have been reported for chemoresistance in HCC including aberrations in DNA repair pathways [8], impairment of apoptotic machinery [9,10], activation of survival signaling pathways [11], reduced drug uptake, drug efflux, drug metabolism [12,13] and contribution of tumor microenvironment [14], etc. Identification of appropriate targets to increase the sensitivity of cancer cells toward chemotherapy is the need of the hour.
Transforming growth factor-β (TGF-β) is one of the most important pleiotropic cytokine and acts as a two-edged sword in tumorigenesis. In normal cells and early carcinomas, it suppresses tumor by virtue of induction of cell cycle arrest and apoptosis. Interestingly, during tumor progression, there are aberrations in TGF-β signaling pathways causing loss of growth inhibitory property of TGF-β. These modifications in the TGF-β signaling shift the function of TGF-β from inhibitor of tumor growth to their promoter, leading to penetration of cancer cells in neighboring tissues and metastasis. This dual character of TGF-β during carcinogenesis is known as the “TGF-β Paradox” [15,16]. Furthermore, there are recent reports, which indicate the emanating role of TGF-β signaling in cancer drug resistance including colorectal, HCC and squamous cell carcinoma [17–21]. Although substantial amount of studies have dissected the role of TGF-β in resistance against anticancer drugs; however, the underlying genomic mechanisms still remain obscure. Thus, there arises a need to search for cleaner genomic targets.
The cardinal roles of nuclear receptors in different biological spheres such as infection and immunity have already been recognized in the scientific world [22–25]. Xenobiotic nuclear receptors such as Constitutive androstane receptor (CAR) and Pregnane X receptor (PXR) are activated by many small molecules including commonly used chemotherapeutic agents or clinical drugs and are also involved in various cellular signaling pathways important in carcinogenesis [26–28]. Additionally, in our recent study, we have shown that activation of PXR in macrophages leads to compromised efficacy of the anti-TB drug rifampicin leading to drug non-responsiveness [29] and is the major transcription factor behind modulating the expression of drug efflux transporters. One of the mechanisms of acquired resistance against anticancer drugs is the decreased intracellular drug accumulation resulting from elevated expression of drug efflux transporters such as multidrug resistance associated proteins (MRP1/ABCC1 and MRP2/ABCC2), P-glycoproteins/ATP-binding cassette transporter subfamily B member 1 (p-gp/ABCB1) and breast cancer resistance protein (ABCG2/BCRP) [11,30,31]. Therefore, this intrigued us to examine the role of PXR in regulating the expression of drug efflux transporters and associated signaling pathways in cancer cells.
The current study is undertaken to demonstrate the contribution of TGF-β and the underlying mechanism in chemoresistance in HCC. It has been known previously that TGF-β signaling contributes to the pathogenesis of HCC in particular liver fibrosis and cirrhosis and also plays a major role in cancer drug resistance. We have demonstrated that recombinant TGF-β treatment induces drug resistance in HepG2 cells by enhancing efflux potential of cells, which was lowered in the presence of TGF-β receptor inhibitor LY2157299. Further, we have discovered a novel crosstalk between TGF-β signaling and PXR. TGF-β treatment led to increased expression of PXR and its target genes including drug efflux transporters conferring to TGF-β mediated drug resistance in HCC. Mechanistically, the ERK-ETS1 axis is involved in the regulation of PXR expression by TGF-β. Downregulation of PXR resulted in sensitization toward anticancer drug sorafenib. Our studies suggest the predominant role of TGF-β induced PXR in determining the response of tumor cells toward chemotherapy.
Materials and methods
Cell culture
HepG2, Huh7 and PLC/PRF/5 cells (National Center for Cell Science, India) were grown in DMEM (Gibco, Invitrogen) with 100 U/ml penicillin-streptomycin (Gibco, Invitrogen) and 10% FBS (Gibco, Invitrogen) at 37 °C and 5% CO2.
Cell cytotoxic assay
1 × 104 HepG2 cells/well were seeded into 96-well plates and treated with 10 ng/ml TGF-β plus anticancerous drugs in the presence or absence of TGF-β inhibitor LY2157299 for 72 h. After 72 h, the plate was incubated with MTT dye (5 mg/ml) at 37 °C for 4 h and later the formazan crystals were dissolved by adding lysis solution. Absorbance was quantified by using ELISA plate reader at 570 nm.
Clonogenic assay
For the clonogenic assay, 1.5 × 103 HepG2 cells per well were seeded in a six-well plate and allowed to adhere overnight. Cells were then treated with TGF-β with or without sorafenib (5 µM) and doxorubicin (0.5 µM) and cells were allowed to grow for 14 d. When cells exhibited colonies, fixation was done using 4% paraformaldehyde followed by staining with 0.5% crystal violet. The colonies were counted manually and surviving fraction was calculated as the ratio of plating efficiency of treated and control cells.
Efflux assay
For efflux assay, HepG2 cells were plated in 12 well plates at a density of 5 × 105 cells/well and left for overnight adherence. Cells were then subjected to treatments according to experimental need. At the end of the experiment, cells were detached from the plate using BD acutase followed by washing and incubation in 10 μM rhodamine123 and 10 μM of mitoxantrone for 15 min at 37ᵒC. Washing was done with cold PBS and cells were resuspended in fresh media and incubated for 90 min at 37°C. After incubation, cells were washed two times with PBS followed by resuspension in cold PBS. Fluorescence was measured by using BD accuriC6 flow cytometry.
Annexin V and PI staining
5 × 105 HepG2 cells per well were seeded on 12-well plate and they were permitted to adhere overnight. Transfection was done with either control scrambled siRNA or PXR siRNA for 48 h and then treated with sorafenib (5 µM) with or without TGF-β for 24 h. Cells were detached from the plate and were washed with 1X PBS twice followed by Annexin V and PI staining using kit according to the manufacturer’s instructions. Acquisition was done on BD accuriC6 flow cytometer and data was analyzed by FlowJo software.
Immunoblot analysis
For western blotting, 40 μg of protein was separated by 10% SDS-PAGE and transferred to a PVDF membrane (Millipore, Immobilon, IPVH00010). This was followed by membrane blocking for 1 h at RT with 5% BSA in Tris-buffered saline (pH 7.4). The immunoblots were incubated overnight at 4°C with primary antibodies to Caspase-3 (BD Bioscience, 610322), MCL1 (BD Bioscience, 559027), ETS1 (Santa Cruz, sc-55581), phospho ETS1 (Sigma, SAB4301235), PXR (Santa Cruz, sc25381) and CAR (Santa Cruz, sc-373791) and washed three times with Tris-buffered saline with 0.1% Tween 20 (TBST, pH 7.4), then incubated with appropriate HRP-conjugated secondary antibody for 60 min at RT and visualized by chemiluminescent HRP substrate (Millipore, WBLUF0500). β-actin (Santa Cruz, sc47778) was used as loading control for whole-cell extract.
RNA isolation and quantitative real time PCR (qRT-PCR)
TRIzol reagent (Ambion, Life Technologies, 15596018) was used to isolate total RNA from HepG2 cells. Reverse transcription was carried out using 1 μg of RNA by the verso c-DNA synthesis kit (Thermo scientific, AB1453/B) according to the manufacturer’s instructions. By using gene-specific primers expression of various genes was examined by qRT-PCR and normalized by β-actin gene expression.
siRNA knock down
For transient silencing, cells at 70% confluence were transfected with 60 nM final concentration of scrambled control small interfering RNA (siRNA) or gene-specific siRNA using lipofectamine RNAi Max solution diluted according to manufacturer’s instructions.
Chromatin immunoprecipitation (ChiP) assay
ChiP analysis was performed to determine the invivo binding of ETS1 to PXR promoter region. 1 × 106 cells were subjected to formaldehyde fixation followed by quenching of crosslinking reaction with 125 mM glycine. Further, cells were washed with 1X ice-cold PBS twice and the pellet was dissolved in 300 µl warm SDS lysis buffer. Chromatin was digested by sonication to yield DNA fragments of desired length ranging from 500 to 1000 bp. Before subjecting the lysate to immunoprecipitation, preclearing was done with Protein G agarose beads and salmon sperm DNA and simultaneously beads were incubated with 2 µg of ETS1 antibody for 4 h at 4ºC. The beads-antibody complex was then incubated with lysate overnight at 4ºC. Precleared lysates were subjected to immunoprecipitation with the anti-ETS1 antibody. Precipitated DNA was purified by ethanol precipitation followed by PCR amplification with PXR promoter-specific primers.
Statistical analysis
Sigma Plot was used for statistical analysis and the results are presented as the mean ± S.D unless otherwise mentioned. P values were obtained using two-tailed Student’s t tests at p < 0.05 (*), p < 0.005 (**), p < 0.0005 (***).
Results
TGF-β signaling is associated with chemoresistance in HepG2 cells
TGF-β signaling is known to induce resistance to standard anticancer drugs in various cancer models [32]. To unravel the role of TGF-β in resistance against chemotherapeutic agents in HCC, we studied the effect of recombinant TGF-β on the cytotoxic action of anticancer drugs in HepG2 cells. HepG2 cells were treated with increasing concentrations of sorafenib or doxorubicin with or without recombinant TGF-β and cell viability was determined by MTT assay. Interestingly, recombinant TGF-β treatment rescued HepG2 cells from chemotherapy-induced cell death and this rescue effect was abrogated in the presence of TGF-β receptor inhibitor LY2157299 (Figure 1(a)). To further confirm the cell viability clonogenic assay was performed. Cells were treated with sorafenib or doxorubicin in the presence or absence of TGF-β and LY2157299. Drug treatment leads to a decrease in survival fraction and the colony-forming ability of HepG2 cells which was rescued in the presence of TGF-β but was negated when cells were co-treated with TGF-β and LY2157299 (Figure 1(b)). These data clearly suggest that TGF-β signaling helps in the induction of chemotherapy resistance in HepG2 cells.
Figure 1.
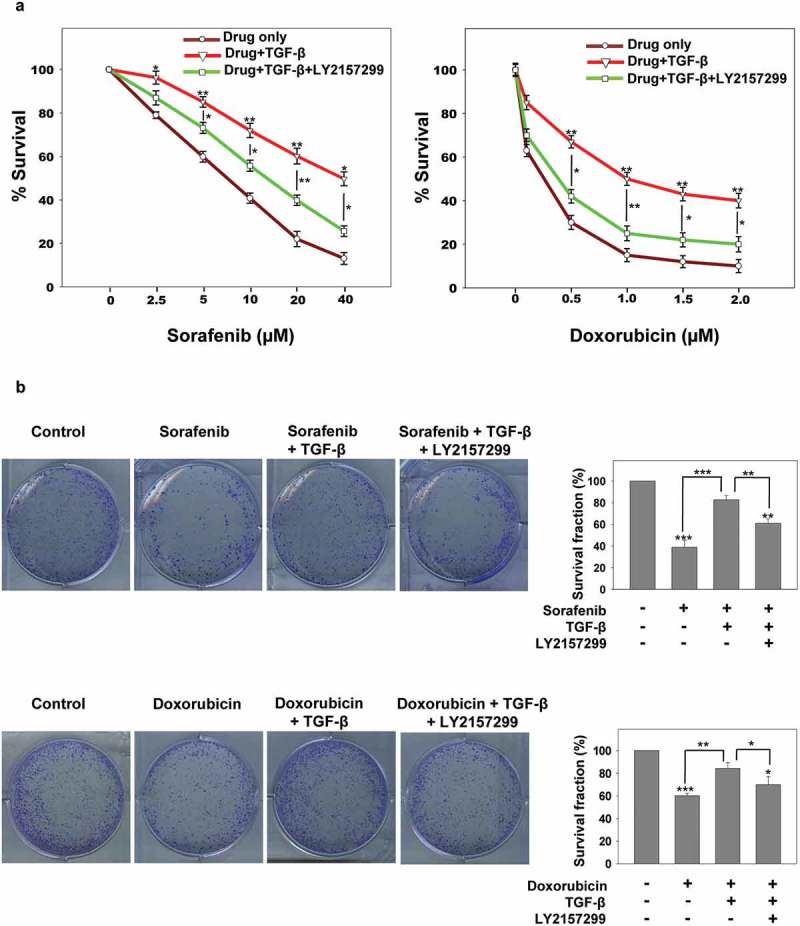
TGF-β induces chemoresistance in HepG2 cells. (a and b) MTT and clonogenic assay of HepG2 cells sensitized with recombinant TGF-β alone or in combination with LY2157299 in presence of Sorafenib or doxorubicin. Asterisks represent significant differences (p< 0.05 (*), p < 0.005 (**), p < 0.0005 (***)). Data are mean ± SD or representative of three independent experiments performed in triplicate.
TGF-β signaling induces the expression of xenobiotic nuclear receptor PXR
The explicit mechanisms by which TGF-β signaling leads to resistance toward chemotherapy are of high interest and understanding these mechanisms are crucial for the development of new therapeutic strategies. One credible explanation could be the induction of host innate cellular drug detoxifying mechanisms. Drug-metabolizing enzymes (DMEs) and transporters are major players in disposition and detoxification of various xeno- and endobiotic chemicals. The expression of these DMEs and transporters is critically controlled by ligand-activated transcription factors called xenobiotic sensing nuclear receptors. PXR and CAR are prototype of xenobiotic nuclear receptors. Together, these two receptors control the expression of an overlapping array of genes encoding DMEs, and drug transporters [33,34]. To elucidate the effect of TGF-β on host drug detoxification mechanisms, we measured the expression of xenobiotic nuclear receptors PXR and CAR in control and TGF-β treated HepG2 cells both at mRNA and protein level (Figure 2(a,b)). Interestingly, we found that TGF-β treatment increases the expression of PXR but not CAR in HepG2 cells. Moreover, TGF-β induced PXR expression was abolished in the presence of TGF-β receptor inhibitor LY2157299 (Figure 2(a,b)).
Figure 2.
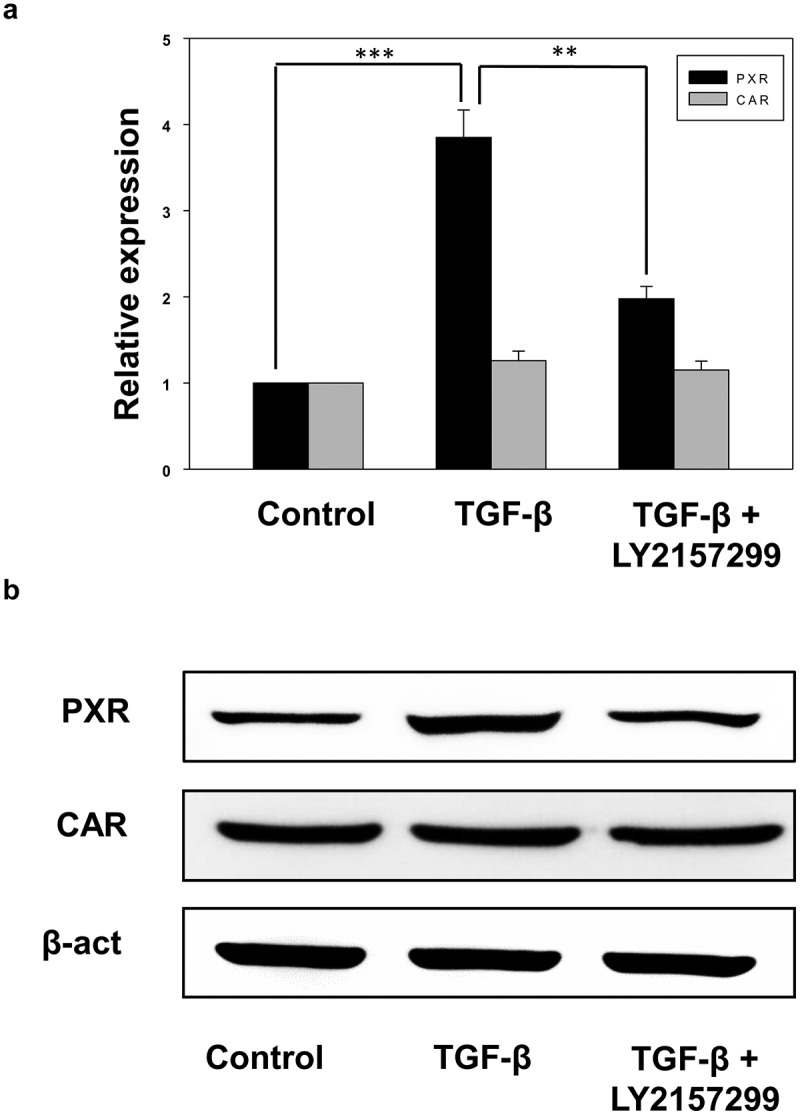
Recombinant TGF-β induces the expression of xenobiotic nuclear receptor PXR. (a and b) qRT-PCR (a) and immunoblot analysis (b) of PXR and CAR in control, TGF-β and TGF-β+ LY2157299 treated HepG2 cells. Asterisks represent significant differences (p< 0.05 (*), **p < 0.005, ***p < 0.0005). Data are mean ± SD or representative of three independent experiments performed in triplicate.
Effect of TGF-β on the drug efflux mechanisms in HepG2 cells
As TGF-β treatment induces PXR expression in HepG2 cells, it was of interest to investigate the effect of TGF-β on drug efflux transporters, expressions of which are modulated by PXR activation. Drug efflux transporters like MRP1/ABCC1, MRP2/ABCC2, P-gp/ABCB1 and BCRP/ABCG2 modulates the pharmacokinetics and effectiveness of numerous drugs. Thus, to evaluate the effect of TGF-β on the expression of drug transporters upon PXR activation by SR12813 (a pharmacological activator of PXR), HepG2 cells were treated with vehicle and TGF-β in presence or absence of PXR agonist SR12813. The results revealed that TGF-β treatment leads to an increase in efflux transporters ABCB1, ABCC1 and ABCC2 whereas no change was observed in the expression of ABCG2 when compared to control. However, increased expression of all four transporters was observed in SR12813 treated cells. When the cells were co-treated with TGF-β and SR12813, we observed a synergistic effect leading to a significant increase in the expression of drug efflux transporters (Figure 3(a)). In addition, we also monitored the efflux potential of HepG2 and observed that HepG2 cells co-treated with TGF-β and SR12813 are more efficient, compared to TGF-β or SR12813 treated cells in the efflux of drugs like fluorescent molecule such as rhodamine 123, 5,6-carboxyfluorescein diacetate (CFDA) and mitoxantrone. This effect was abrogated in the presence of TGF-β receptor inhibitor LY2157299 (Figure 3(b,c)). This enhanced efflux potential could be due to enhanced expression of PXR and its target genes (ABC transporters). These data clearly suggest that TGF-β signaling induces chemoresistance in HepG2 cells by modulating the expression of xenobiotic nuclear receptor PXR and its target genes.
Figure 3.
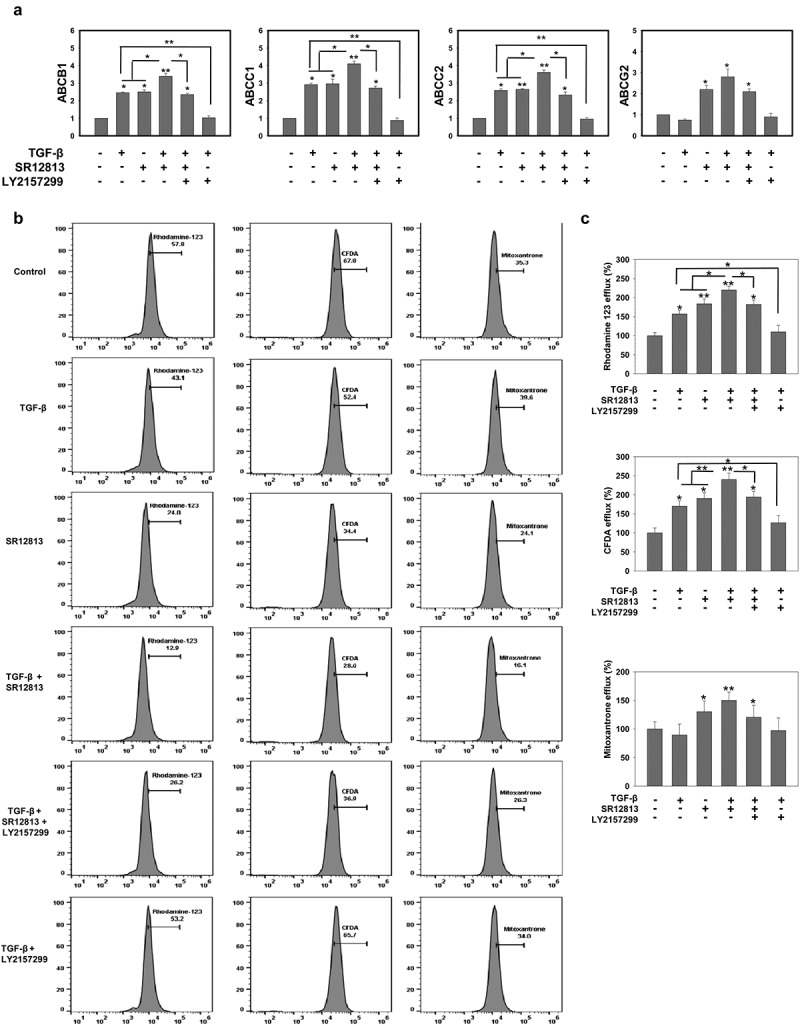
TGF-β rescues HepG2 cell from cytotoxic effects of anticancer drugs by increasing drug efflux potential. (a) qRT-PCR analysis of drug efflux transporters ABCB1, ABCC2, ABCC1 and ABCG2 in HepG2 cells treated with TGF-β in the presence or absence of PXR agonist SR12813 and TGF-β inhibitor LY2157299. (b and c) Representative images (b) and Analysis (c) of rhodamine 123, CFDA and mitoxantrone efflux potential of HepG2 cells. Asterisks represent significant differences (*p < 0.05, **p < 0.005, ***p < 0.0005). Data are mean ± SD or representative of three independent experiments performed in triplicate.
TGF-β inhibits chemotherapy-induced apoptosis
In addition to the induction of drug efflux transporter, PXR is known to act as an anti-apoptotic in many cancers [35,36] and this could additionally contribute to TGF-β mediated chemoresistance. To explore the effect of anticancer drugs on apoptosis in TGF-β treated control and PXR knockdown cells, we incubated HepG2 cells with sorafenib in the presence or absence of TGF-β in control and PXR knockdown cells. Percentage of apoptotic cells were examined by flow cytometry using Annexin V and PI staining (Figure 4(a)). Strikingly, TGF-β treated cells undergo significantly less apoptosis compared to the cells treated with drug alone in control HepG2 cells. However, in PXR knockdown cells this effect was abrogated. We next measured the level of procaspase 3, an early sign of apoptosis as well as expression of Mcl-1, an anti-apoptotic protein by western blot and similar results were obtained (Figure 4(b)). This clearly indicates that TGF-β induced PXR expression is crucial for resistance toward chemotherapy in HepG2 cells.
Figure 4.
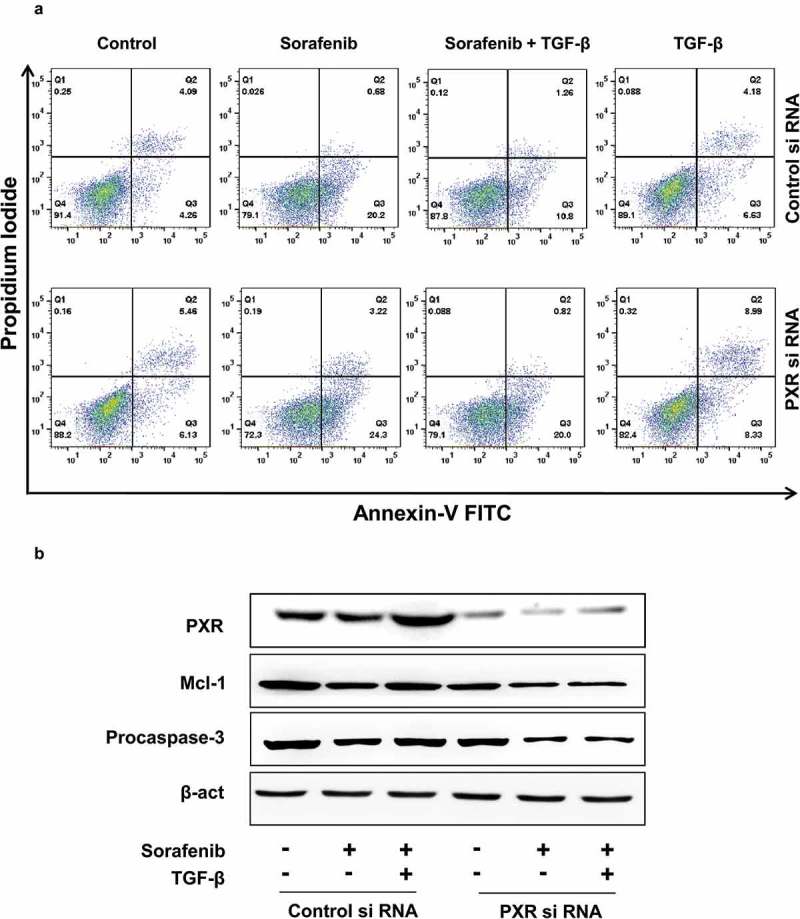
TGF-β inhibits chemotherapy-induced apoptosis. (a) Annexin-V and propidium iodide staining and (b) immunoblot analysis of procaspase-3 and Mcl-1 in control and PXR knock down HepG2 cells. Data are representative of three independent experiments performed in triplicate.
TGF-β induces endogenous PXR expression through non-canonical signaling cascade
Binding of TGF-β family ligands to its cognate receptor initiates a highly complex and cell type-dependent downstream signaling pathways, involving Smad-dependent and Smad-independent signaling cascades. In Smad-dependent pathway, activated TGF-β receptor phosphorylates downstream effector Smads (SMAD2/3) and forms a complex with SMAD4. This complex is then translocated to the nucleus where it binds to SMAD-binding elements (SBEs) which are specific DNA sequence motifs. Interaction of these pSMAD2/3-SMAD4 complexes with other transcriptional regulators leads to induction or suppression of various target genes. Besides the canonical Smad pathway, TGF-β can also propagate downstream signaling through non-canonical Smad-independent signaling cascades like c-Jun N-terminal kinase JNK, p38, extracellular‐signal‐regulated kinase ERK1/2 and AKT [37]. We were keen to identify the signaling mechanisms by which recombinant TGF-β induces PXR expression in HepG2 cell, for which HepG2 cells were pretreated with Smad and non-Smad signaling inhibitors and then sensitized with TGF-β and the expression of PXR was confirmed by qRT-PCR and western blotting (Figure 5(a,b)). The results showed that ERK inhibitor treatment abrogates the TGF-β induced PXR expression. These results clearly suggest that TGF-β induces xenobiotic nuclear receptor PXR through non-canonical Smad-independent ERK activation.
Figure 5.
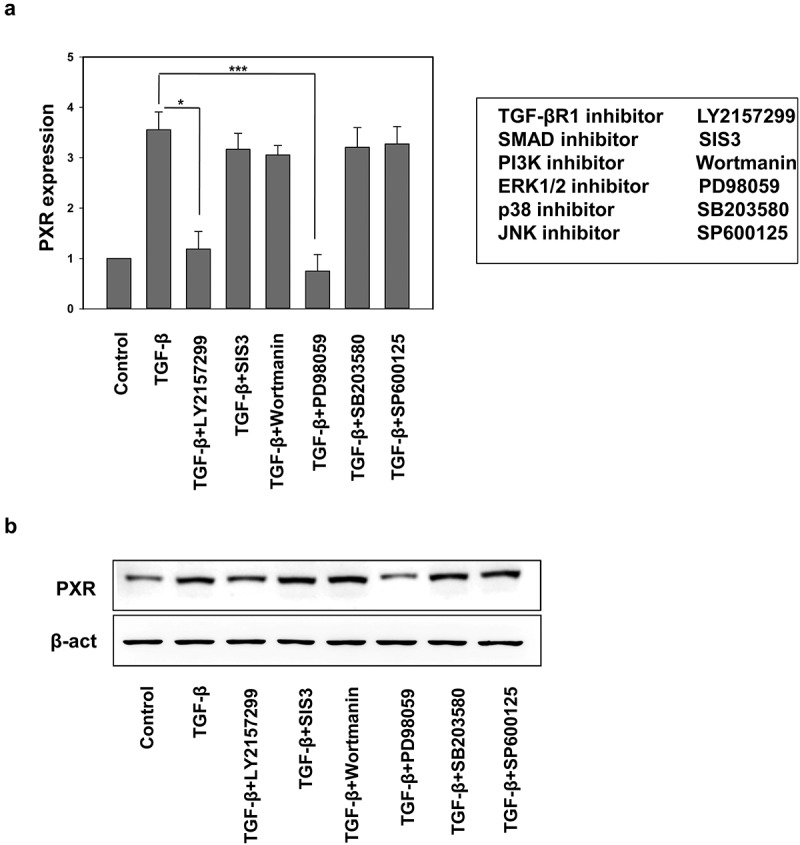
TGF-β induces endogenous PXR expression through non-canonical signaling cascade. (a) qRT-PCR and (b) immunoblot analysis of PXR in HepG2 cell treated with TGF-β alone or in combination with SMAD and non-SMAD pathway inhibitors. Asterisks represent significant differences (*p < 0.05, **p < 0.005, ***p < 0.0005). Data are representative of three independent experiments performed in triplicate.
ETS1 is essential for the TGF-β induced PXR expression
Next, we explored the transcriptional mechanism by which TGF-β mediated ERK activation leads to induction of PXR in liver cancer cells. Recent reports suggest that transcription factor E26 transformation specific sequence 1 (ETS1) plays a crucial role in PXR gene expression in liver cancer cells and it has also been reported that ETS1 is phosphorylated and activated by the ERK signaling pathway [38]. We examined whether ETS1 is involved in the TGF-β mediated induction of PXR via activation of ERK signaling in HepG2 cells. HepG2 cells were treated with vehicle and TGF-β with or without ERK inhibitor and evaluated for the activation of ETS1 by monitoring the levels of phospho-ETS1 (Figure 6(a)). We observed an increase in phospho-ETS1 in TGF-β treatment which was abrogated upon treatment with ERK inhibitor. This clearly suggests that TGF-β signaling activates ETS1 in HepG2 cells via a non-canonical ERK signaling. To further investigate the functional significance of this ETS1 activation in the PXR expression, we knock down ETS1 in HepG2 cells by using si-RNA (Figure 6(b)). We observed PXR induction upon TGF-β treatment in control but not in ETS1 knockdown cells (Figure 6(c)). It is previously shown that ETS1 binds to PXR promoter thereby playing a role in its transcriptional regulation [39,40].To further confirm the involvement of ETS1 in TGF-β mediated PXR induction, chromatin immunoprecipitation assay was performed in the presence and absence of TGF-β. Presence of TGF-β led to enhanced ETS1 binding to PXR promoter region as can be seen by fold enrichment (Figure 6(d)). This clearly suggests that ETS1 plays a regulatory role in the TGF-β mediated induction of PXR expression in HepG2 cells.
Figure 6.
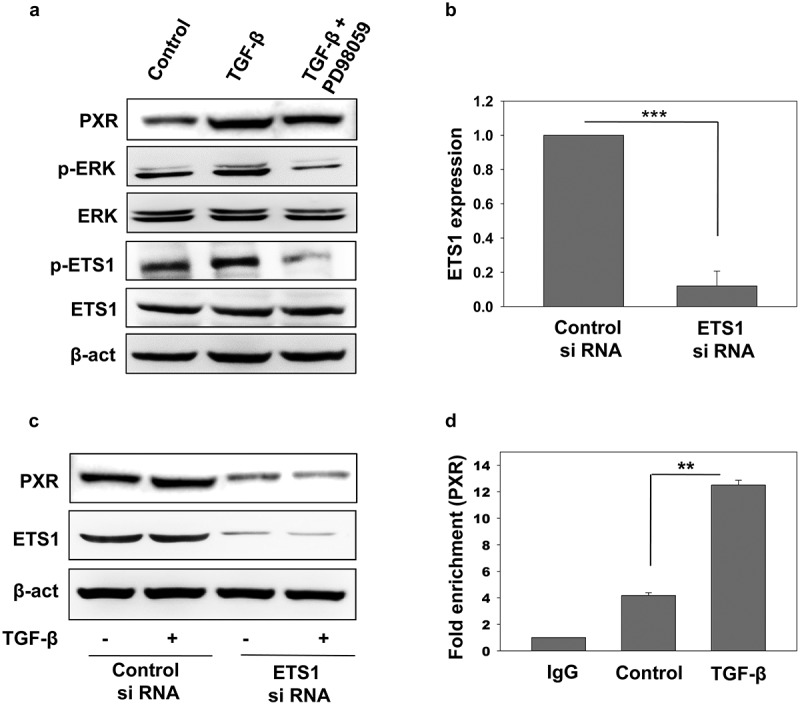
Role of ETS1 in the TGF-β induced PXR expression. (a) Immunoblot analysis of phospho-ETS1 and ETS1 in HepG2 cells sensitized with recombinant TGF-β alone or in combination with PD98059. (b) qRT-PCR of ETS1 in control and ETS1 knockdown HepG2 cells. (c) Immunoblot analysis of PXR expression in control and ETS1 knockdown HepG2 cells in the presence or absence of recombinant TGF-β. (d) Chip analysis of ETS1 binding at PXR promoter in the presence and absence of recombinant TGF-β. Asterisks represent significant differences (*p < 0.05, **p < 0.005, ***p < 0.0005). Data are representative of three independent experiments performed in triplicate.
Effect of TGF-β on PXR expression in Huh7 and PLC/PRF/5 cells
TGF-β mediated PXR induction was observed in TGF-β induced apoptosis resistant cell line HepG2. Next, we were intrigued to look for the expression of PXR in other cell lines which are sensitive to TGF-β induced cell death. We performed immunoblot analysis in two other liver cancer cell lines Huh7 and PLC/PRF/5. Intriguingly, the increase in the expression of PXR upon TGF-β treatment was not observed in these two cell lines. As this effect could be due to altered signaling, we looked for the activation of ERK pathway and observed that ERK signaling pathway was inhibited on TGF-β treatment as can be seen by lower levels of phosphorylated ERK. There was also decrease in the levels of p-ETS1 (Figure 7(a)). PXR up-regulation by TGF-β is only observed in cells that are refractory to its proapoptotic effect, such as HepG2 cells, but not in cells that are sensitive to its cytotoxic effect, such as Huh7 and PLC/PRF/5 cells. However, hyperactivation of survival signaling pathway (ERK pathway) by phorbol esters in TGF-β sensitive cell lines (Huh-7 and PLC/PRF/5) induced PXR expression (Figure 7(b,c)). This concludes that TGF-β-ERK-ETS1 axis is important for inducing the expression of PXR in liver cancer cell lines.
Figure 7.
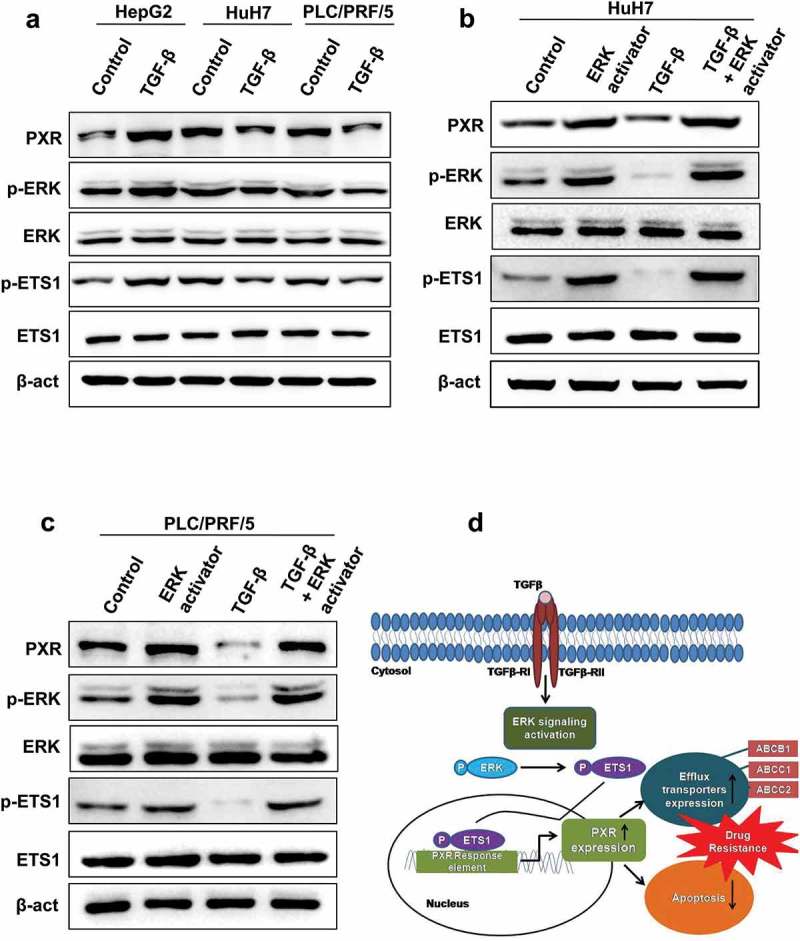
Effect of TGF-β on PXR expression in Huh7 and PLC/PRF/5. (a) Immunoblot analysis of PXR, phospho-ERK and phospho-ETS1 in HepG2, Huh7 and PLC/PRF/5 cells. (b and c) Hyperactivation of ERK pathway by phorbol esters induced PXR expression in Huh7 (b) and PLC/PRF/5 (c) cells. (d) Schematic representing TGF-β induced PXR expression lead to chemoresistance by upregulating the expression of drug efflux transporters such as ABCB1, ABCC1 and ABCC2 and inhibiting drug-induced apoptosis. TGF-β increase expression of PXR through non-canonical SMAD-independent ERK pathway which leads to phosphorylation of downstream gene ETS1. Binding of ETS1 on the specific promoter region of PXR resulted in elevated expression levels of PXR.
Discussion
HCC is the fifth leading cancer by incidence worldwide and constitutes a major global health problem. A clear perspective of the molecular mechanisms leading to HCC is imperative in order to design novel therapeutic strategies to either avert or alleviate the development of this cancer. Recent studies have revealed that TGF-β plays a key regulatory role in chronic liver disease and attenuation of TGF-β signaling leads to various cooperative down-stream effects, resulting in improved clinical outcome in HCC [18,19,41]. In addition to these, TGF-β signaling has been lately shown to play a critical role in resistance toward chemotherapy in many cancers [17,21]. In this study, we demonstrate the role of TGF-β in chemoresistance in HCC cell lines.
We observed that the anti-tumor effect of sorafenib and doxorubicin was greatly impaired in HepG2 cells sensitized with recombinant TGF-β; however, this rescue in cell death was not observed in presence of TGF-β receptor inhibitor LY2157299 (Figure 1(a,b)). We identified the molecular mechanisms involved in the TGF-β mediated drug non-responsiveness in HepG2 cells and found that TGF-β signaling enhances the endogenous expression of xenobiotic nuclear receptor PXR (Figure 2(b,c)). PXR is an adopted orphan nuclear receptor activated by a diverse array of xenobiotic and pharmaceutical agents and plays a major role in their metabolism [34,42]. It has been implicated in the modulation of various infectious diseases [43] as well as different types of cancers. Notably, PXR expression is found to be up-regulated in different cancer patients and representative cell lines and its activation leads to compromised efficacy of many anticancer drugs but the signaling mechanism that governs PXR expression in these cells is largely unknown [7,44–46]. To the best of our knowledge, we have shown for the first time that tumor microenvironment cytokine TGF-β induces PXR expression in HepG2 cells by activating non-canonical ERK signaling pathway, which was validated by using pharmacological inhibitor of ERK (Figure 5(a,b)). This activated ERK phosphorylates and activates ETS1 transcription factor (Figure 6(a)), which was shown to be a critical regulator of endogenous PXR expression in hepatic cells. Loss of function of ETS1 abrogates the TGF-β induced PXR expression in HepG2 cells (Figure 6(c)). It is known that TGF-β is unable to induce cell death in HepG2 cells as the pro-survival ERK pathway is constitutively active in these cells, whereas other cell lines such as Huh7 and PLC/PRF/5 respond well to TGF-β induced cell death [47]. So, we looked for the TGF-β induced PXR expression in these cell lines. Interestingly, we were not able to see the PXR up-regulation by TGF-β in cells that are sensitive to its cytotoxic effect, such as Huh7 and PLC/PRF/5 cells (Figure 7(a)) but there was an increased expression of PXR in these cells when ERK pathway was activated by phorbol esters (Figure 7(b,c)). So, activating the pro-survival pathway (ERK) also led to a significant induction of PXR in these cell lines. This conclusively proves that TGF-β induces PXR expression in liver cancer cells via ERK-ETS1 axis.
We also demonstrated the plausible mechanisms by which TGF-β induced PXR expression protects the cells from the cytotoxic effect of anticancer drugs. We found that PXR modulates TGF-β mediated chemoresistance in HepG2 cells by promoting drug efflux and anti-apoptotic mechanisms (Figures 3 and 4). TGF-β treatment marginally enhances the drug efflux potential of HepG2 cells, but TGF-β treatment along with PXR activation by SR12813 leads to a significant increase in the drug efflux potential of HepG2 cells. TGF-β treatment also inhibits the apoptosis induced by anticancer drugs in HepG2 cells and this anti-apoptotic effect was partially abrogated through PXR knock down. Taken together, our data demonstrate that TGF-β induced chemoresistance in HCC cell line is primarily modulated by xenobiotic nuclear receptor PXR (Figure 7(d)).
In light of the above considerations, the suppression of TGF-β pathway could form one of the mechanisms that could be employed to overcome drug resistance. However, as TGF-β has a dual role in tumorigenesis, it is crucial to find new drugs that will selectively eliminate the tumor-promoting activities of TGF-β. The better approach could be PXR antagonism, as activated PXR leads to increased expression of drug-metabolizing enzymes, drug efflux transporters as well as reduced apoptosis which together lead to lowered drug efficacy and increased drug resistance. Therefore, antagonizing hPXR could be of therapeutic value. Efforts need to focus on developing nontoxic but potent PXR specific antagonists which may lead to increased bioavailability of drugs and their enhanced effectiveness.
Funding Statement
This work was supported by the Council of Scientific and Industrial Research (CSIR) 12th Plan Network project Bugs to Drugs, [BSC0211] to PG. This work is also supported by Department of Biotechnology, Ministry of Science and Technology, National Bioscience Award project [GAP-0162] to PG. We thank IMTECH, a constituent laboratory of the CSIR, for the facilities and financial support. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgments
We thank IMTECH, a constituent laboratory of the CSIR, for facilities and financial support.
Author contribution
EB, NA and PG designed the experiments, EB, NA, SK, DT, SG and RK performed the experiments, EB, NA and PG analyzed the data, EB, NA and PG wrote the manuscript, PG supervised the project.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- [3].Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47(Suppl):S2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cervello M, McCubrey JA, Cusimano A, et al. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012;3:236–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chappell WH, Steelman LS, Long JM, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011;2:135–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Feng F, Jiang Q, Cao S, et al. Pregnane X receptor mediates sorafenib resistance in advanced hepatocellular carcinoma. Biochim Biophys Acta Gen Subj. 2018;1862:1017–1030. [DOI] [PubMed] [Google Scholar]
- [8].Yang SF, Chang CW, Wei RJ, et al. Involvement of DNA damage response pathways in hepatocellular carcinoma. Biomed Res Int. 2014;2014:153867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chun E, Lee KY. Bcl-2 and Bcl-xL are important for the induction of paclitaxel resistance in human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2004;315:771–779. [DOI] [PubMed] [Google Scholar]
- [10].Jodo S, Kobayashi S, Nakajima Y, et al. Elevated serum levels of soluble Fas/APO-1 (CD95) in patients with hepatocellular carcinoma. Clin Exp Immunol. 1998;112:166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Urtasun R, Latasa MU, Demartis MI, et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology. 2011;54:2149–2158. [DOI] [PubMed] [Google Scholar]
- [12].Nies AT, Konig J, Pfannschmidt M, et al. Expression of the multidrug resistance proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int J Cancer. 2001;94:492–499. [DOI] [PubMed] [Google Scholar]
- [13].Takano M, Otani Y, Tanda M, et al. Paclitaxel-resistance conferred by altered expression of efflux and influx transporters for paclitaxel in the human hepatoma cell line, HepG2. Drug Metab Pharmacokinet. 2009;24:418–427. [DOI] [PubMed] [Google Scholar]
- [14].Gupta DK, Singh N, Sahu DK. TGF-beta mediated crosstalk between malignant hepatocyte and tumor microenvironment in hepatocellular carcinoma. Cancer Growth Metastasis. 2014;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pickup MW, Owens P, Moses HL. TGF-beta, bone morphogenetic protein, and activin. Signaling and the tumor microenvironment. Cold Spring Harb Perspect Biol. 2017;9:a022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. [DOI] [PubMed] [Google Scholar]
- [17].Zhang Y, Zhang Y, Geng L, et al. Transforming growth factor beta mediates drug resistance by regulating the expression of pyruvate dehydrogenase kinase 4 in colorectal cancer. J Biol Chem. 2016;291:17405–17416. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [18].Ungerleider N, Han C, Zhang J, et al. TGFbeta signaling confers sorafenib resistance via induction of multiple RTKs in hepatocellular carcinoma cells. Mol Carcinog. 2017;56:1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Takahashi K, Yan IK, Kogure T, et al. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Bio. 2014;4:458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brown JA, Yonekubo Y, Hanson N, et al. TGF-beta-induced quiescence mediates chemoresistance of tumor-propagating cells in squamous cell carcinoma. Cell Stem Cell. 2017;21:650–64 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Brunen D, Willems SM, Kellner U, et al. TGF-beta: an emerging player in drug resistance. Cell Cycle. 2013;12:2960–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Saini A, Mahajan S, Gupta P. Nuclear receptor expression atlas in BMDCs: NR4A2 restricts immunogenicity of BMDCs and impedes EAE. Eur J Immunol. 2016;46:1842–1853. [DOI] [PubMed] [Google Scholar]
- [23].Nanduri R, Mahajan S, Bhagyaraj E, et al. The active form of vitamin D transcriptionally represses smad7 signaling and activates extracellular signal-regulated kinase (ERK) to inhibit the differentiation of a inflammatory T helper cell subset and suppress experimental autoimmune encephalomyelitis. J Biol Chem. 2015;290:12222–12236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Leopold Wager CM, Arnett E, Schlesinger LS. Macrophage nuclear receptors: emerging key players in infectious diseases. PLoS Pathog. 2019;15:e1007585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mahajan S, Saini A, Kalra R, et al. Frienemies of infection: a chronic case of host nuclear receptors acting as cohorts or combatants of infection. Crit Rev Microbiol. 2016;42:526–534. [DOI] [PubMed] [Google Scholar]
- [26].Moore LB, Parks DJ, Jones SA, et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–15127. [DOI] [PubMed] [Google Scholar]
- [27].Mani S, Huang H, Sundarababu S, et al. Activation of the steroid and xenobiotic receptor (human pregnane X receptor) by nontaxane microtubule-stabilizing agents. Clin Cancer Res. 2005;11:6359–6369. [DOI] [PubMed] [Google Scholar]
- [28].Nallani SC, Goodwin B, Maglich JM, et al. Induction of cytochrome P450 3A by paclitaxel in mice: pivotal role of the nuclear xenobiotic receptor, pregnane X receptor. Drug Metab Dispos. 2003;31:681–684. [DOI] [PubMed] [Google Scholar]
- [29].Bhagyaraj E, Tiwari D, Ahuja N, et al. A human xenobiotic nuclear receptor contributes to nonresponsiveness of Mycobacterium tuberculosis to the antituberculosis drug rifampicin. J Biol Chem. 2018;293:3747–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huang H, Chen J, Ding CM, et al. LncRNA NR2F1-AS1 regulates hepatocellular carcinoma oxaliplatin resistance by targeting ABCC1 via miR-363. J Cell Mol Med. 2018;22:3238–3245. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [31].Namisaki T, Schaeffeler E, Fukui H, et al. Differential expression of drug uptake and efflux transporters in Japanese patients with hepatocellular carcinoma. Drug Metab Dispos. 2014;42:2033–2040. [DOI] [PubMed] [Google Scholar]
- [32].Giannelli G, Mikulits W, Dooley Set al. The rationale for targeting TGF-beta in chronic liver diseases. Eur J Clin Invest. 2016;46:349–361. [DOI] [PubMed] [Google Scholar]
- [33].Kast HR, Goodwin B, Tarr PT, et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002;277:2908–2915. [DOI] [PubMed] [Google Scholar]
- [34].Synold TW, Dussault I, Forman BM. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7:584–590. [DOI] [PubMed] [Google Scholar]
- [35].Zhou J, Liu M, Zhai Y, et al. The antiapoptotic role of pregnane X receptor in human colon cancer cells. Mol Endocrinol. 2008;22:868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Masuyama H, Nakatsukasa H, Takamoto N, et al. Down-regulation of pregnane X receptor contributes to cell growth inhibition and apoptosis by anticancer agents in endometrial cancer cells. Mol Pharmacol. 2007;72:1045–1053. [DOI] [PubMed] [Google Scholar]
- [37].Derynck R, Zhang YE. Smad-dependent and smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. [DOI] [PubMed] [Google Scholar]
- [38].Plotnik JP, Budka JA, Ferris MW, et al. ETS1 is a genome-wide effector of RAS/ERK signaling in epithelial cells. Nucleic Acids Res. 2014;42:11928–11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kumari S, Saradhi M, Rana M, et al. Pregnane and xenobiotic receptor gene expression in liver cells is modulated by Ets-1 in synchrony with transcription factors Pax5, LEF-1 and c-Jun. Exp Cell Res. 2015;330:398–411. [DOI] [PubMed] [Google Scholar]
- [40].Shao Z, Li Y, Dai W, et al. ETS-1 induces sorafenib-resistance in hepatocellular carcinoma cells via regulating transcription factor activity of PXR. Pharmacol Res. 2018;135:188–200. [DOI] [PubMed] [Google Scholar]
- [41].Kang D, Han Z, Oh GH, et al. Down-regulation of TGF-beta expression sensitizes the resistance of hepatocellular carcinoma cells to sorafenib. Yonsei Med J. 2017;58:899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lehmann JM, McKee DD, Watson MA, et al. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bhagyaraj E, Nanduri R, Saini A, et al. Human xenobiotic nuclear receptor PXR augments mycobacterium tuberculosis survival. J Immunol. 2016;197:244–255. [DOI] [PubMed] [Google Scholar]
- [44].Chen Y, Tang Y, Wang MT, et al. Human pregnane X receptor and resistance to chemotherapy in prostate cancer. Cancer Res. 2007;67:10361–10367. [DOI] [PubMed] [Google Scholar]
- [45].Masuyama H, Hiramatsu Y, Kodama J, et al. Expression and potential roles of pregnane X receptor in endometrial cancer. J Clin Endocrinol Metab. 2003;88:4446–4454. [DOI] [PubMed] [Google Scholar]
- [46].Mensah-Osman EJ, Thomas DG, Tabb MM, et al. Expression levels and activation of a PXR variant are directly related to drug resistance in osteosarcoma cell lines. Cancer. 2007;109:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Caja L, Sancho P, Bertran E, et al. Overactivation of the MEK/ERK pathway in liver tumor cells confers resistance to TGF-{beta}-induced cell death through impairing up-regulation of the NADPH oxidase NOX4. Cancer Res. 2009;69:7595–7602. [DOI] [PubMed] [Google Scholar]