

ABSTRACT
The development of antibody therapeutics relies on animal models that accurately recapitulate disease biology. Syngeneic mouse models are increasingly used with new molecules to capture the biology of complex cancers and disease states, and to provide insight into the role of the immune system. The establishment of syngeneic mouse models requires the ability to generate surrogate mouse counterparts to antibodies designed for humans. In the field of bispecific antibodies, there remains a dearth of technologies available to generate native IgG-like mouse bispecific antibodies. Thus, we engineered a simple co-expression system for one-step purification of intact mouse IgG1 and IgG2a bispecific antibodies from any antibody pair. We demonstrated proof of concept with CD3/CD20 bispecific antibodies, which highlighted both the quality and efficacy of materials generated by this technology.
KEYWORDS: Mouse bispecific antibodies, electrostatic steering, inter-chain disulfide bond shifting, co-expression, T-cell engaging bispecific antibody, syngeneic mouse model
Introduction
Therapeutic antibodies are increasingly used to treat diseases and ailments, and considerable effort has been devoted to engineering next-generation antibodies with greater specificity and expanded functionality. In particular, bispecific antibodies capable of targeting multiple targets or epitopes simultaneously have risen in prominence, with over 85 in preclinical and clinical development.1 While the concept of bispecific antibodies is not new, their rise in clinical relevance is commensurate with the technology required to produce them in sufficient quantities.
One of the primary challenges of generating IgG-like bispecific antibodies is the heterogeneity associated with combining four unique peptide chains, two heavy (HC) and two light chains (LC). Bispecific antibodies can be produced by the somatic fusion of two hybridoma cell lines resulting in quadromas capable of secreting whole IgG molecules.2 The limitation of this approach is the expression of 10 possible combinations of HC and LC, only one of which is correct. Various strategies have been devised to reduce the number of potential HC and LC mis-pairings, thereby increasing the likelihood of forming the correct pair. These technologies include, but are not limited to, species-restricted pairing,3 knobs-into-holes,4 SEEDbodies,5 electrostatic steering,6,7 CrossMabs,8 the Azymetric platform,9 DuetMab,10 and antigen-binding fragment (Fab)-arm exchange,7,11 as has been extensively reviewed.1,12 However, all of these technologies focus on producing human antibodies, which limit their use in preclinical mouse models such as immune-compromised settings, and studies that require longer dosing durations.
Surrogate mouse bispecific antibodies are needed to accommodate repeated dosing schedules in immuno-oncology studies using syngeneic mouse models, which require intact immune systems. Surprisingly, in contrast to the variety of different technologies that have been developed over the last 20 years for producing whole IgG bispecific molecules, only one murine bispecific technology has been described.13 This technology extends the controlled Fab-arm exchange (cFAE) protocol, initially developed for human antibodies, to mice. In brief, parental antibodies which possess bispecific-enabling mutations are expressed separately, mixed in vitro under mild reducing conditions for the exchange, and subsequently purified. Mouse bispecific antibodies produced by cFAE have been used successfully to generate surrogate CD3/GP75 molecules for targeting mouse melanoma in syngeneic mouse models.13,14
We sought to engineer a mouse bispecific platform that would bypass the need for the downstream exchange, be simple and efficient to use, and allow the flexibility to produce any mouse antibody pair. To this end, we engineered the mouse HCs to favor heterodimeric HC-HC pairing, and added inter-chain disulfide bonds for cognate HC-LC pairing. The combination of these two approaches enabled us to produce both mouse IgG1 (mG1) and IgG2a (mG2a) bispecific antibodies by co-expression of two LCs and two HCs. We further validated this technology by generating CD3/CD20 mouse bispecific antibodies capable of effectively depleting B-cells in vitro and in vivo. This method is efficient, does not require downstream processing or extensive purification strategies, and yields well-behaved mouse bispecific antibodies that can be used in immuno-oncology and other relevant settings.
Results
Design and engineering of mouse IgG1 and IgG2a bispecific antibodies
The four unique polypeptide chains of bispecific antibodies must pair correctly to form a functional complex. In our mouse bispecific platform, we pursued a co-expression strategy, which would entail intracellular expression and assembly of all four chains, enabling traditional antibody purification with Protein A. This strategy requires simultaneous heterodimerization of two different HCs and pairing of cognate HC-LC.
To address the issue of heterodimeric HC-HC pairing, we designed electrostatic steering mutations into the CH3 domain to ensure specific HC pairing. In wild-type mIgG1-CH3, there are two pairs of salt bridges at the dimer interface: two negatively charged residues (E356 and D399) from one Fc (Fc-A) interact with two positively charged residues (K439 and K409) from the other Fc (Fc-B) (Figure 1a). By switching the charge of the residues, E356K/D399K in Fc-a’ and K439D/K409E in Fc-b’, the two salt bridges are not affected, so the bispecific Fc-a’b’ heterodimer will be maintained. However, the homodimerization of either Fc-a’a’ or Fc-b’b’ will be minimized due to the same charge repulsion: K356/K399 in Fc-a’ against its own K439/K409 and D439/E409 in Fc-b’ against its own E356/D399 (Figure 1b). This design promotes bispecific Fc formation while minimizing the homodimerization of two parental arms. We named this bispecific pair mG1KD.
Figure 1.
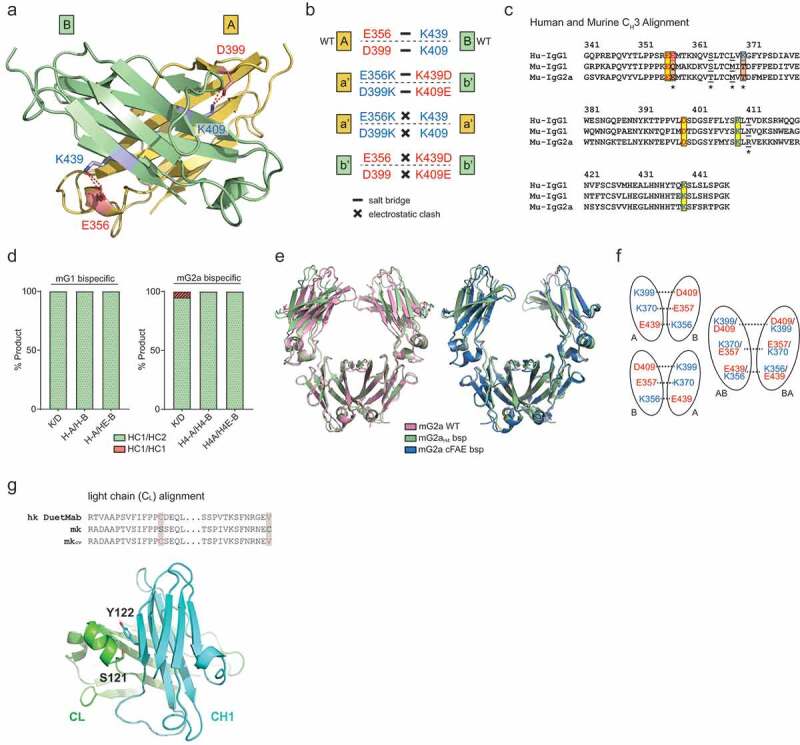
Design and engineering of bispecific mIgG1 and mIgG2a antibodies. (a) Location of two salt bridges (E356:K439 and D398:K309) in the CH3 domain of mG1 (PDB: 3HKF). (b) Switching residues to opposite charge still maintains salt bridges between heterodimer (a’- b’), but causes electrostatic clashes between homodimer (a’ – a’ and b’ – b’). (c) Sequence alignment of human (IgG1) and murine (IgG1 and IgG2a) CH3 domain. Salt bridge E357:K370 in human is missing in murine IgGs. Underlined residues were mutated to a human when the third salt bridge was included. (d) Testing bispecific heavy chain design in co-expression. Percentage of heterodimeric heavy chain pair were quantified via mass spectrometry. In mIgG1, K/D and designs with additional engineered salt-bridge produce complete heterodimer. In mIgG2a, K/D design produces 95% heterodimer with 5% HC1:HC1 homodimer. Designs with additional engineered salt bridge enhances bispecific HC formation to completion. (e) Superposition of bispecific mG2aH4 Fc (green) with mIgG2a wild-type Fc (pale red, PDB: 3zo0) and mIgG2a cFAE bispecific (blue, PDB: 5VAA). (f) Two Fc orientations exist in crystal packing, resulting in an AB and BA average structure at bispecific mutations. (g) Sequence alignment of human kappa light chain with DuetMab design and murine kappa light chain. S121 from CL and Y122 from CH1 (PDB: 1IGT) were selected for Cys substitutions to form inter-chain disulfide bonds.
Sequence and structural alignment between mIgG1, mIgG2a, and hIgG1 indicated the two salt bridges previously described are well conserved, suggesting that the same mG1KD strategy will likely be transferable to the mIgG2a. Thus, similar “K/D” mutations were designed for mIgG2a. Interestingly, while the backbone and core residues align very closely, hIgG1 has one additional salt bridge between the two CH3 domains (E357 and K370), which is missing in mIgG1 (Q357 and T370) and mIgG2a (T370) (Figure 1c). Therefore, we hypothesized that combining the third human salt bridge with the “K/D” design to mIgGs (Q357E/T370K in mIgG1 and T370K in mIgG2a) would further stabilize the heterodimeric bispecific Fc. Since this third salt bridge is taken from human IgG1, we reasoned that converting the nearby residues that pack around it to their human counterparts would create a more natural and stable structure. Thus, we replaced the following mouse residues with human: M368L and N411T in mIgG1 and T364S, M368L, and R411T in mIgG2a. These bispecific pairs are named mG1 H-A/H-B and mG2a H4-A/H4-B. We additionally replaced D409 in mG1 H-B and mG2a H4-B with E409 to potentially strengthen salt-bridge formation with K399. These bispecific pairs were named mG1 H-A/HE-B and mG2a H4-A/H4E-B.
To test our designs, we selected two of our in-house generated mouse antibodies and cloned their variable regions into the six bispecific HC pairs (three mIgG1 pairs and three mIgG2a pairs) as described above. Each bispecific HC DNA pair was co-transfected at the equal ratio with a common mouse kappa LC, as this test only focused on HC pairing efficiency. After Protein A purification, the level of bispecific HC heterodimer was examined via intact mass spectrometry. The “K/D” design worked best with mIgG1, which showed 100% heterodimeric formation of HC1-HC2 (Figure 1d; left panel). In mIgG2a, the “K/D” heterodimerization was robust (95%), but 5% of The HC1–HC1 homodimer byproduct was detected (Figure 1d; right panel). With the additional engineering described above, the heterodimerization of mIgG2a was enhanced to completion (Figure 1d; right panel), which confirmed our hypothesis that the addition of a salt bridge present in human antibodies favors the more stable HC1-HC2 heterodimer. In summary, our bispecific HC designs can promote specific and efficient HC pairing. mIgG1 with “K/D” mutations (mG1KD) and mIgG2a with H4-A/H4-B mutations (mG2aH4) were selected for generating subsequent bispecific antibodies.
The crystal structure of the heterodimeric mG2aH4 Fc was determined to further characterize the CH3–CH3 interface and to ensure that our designs did not cause any significant structural changes to the Fc dimer. The structure clearly revealed that the bispecific Fc structure closely resembled that of a wild-type Fc (PDB: 3ZO0),15 as well as a previously reported bispecific Fc generated by cFAE (PDB: 5VAA)13 (Figure 1e). Similar to previously determined crystal structures for human bispecific Fcs,7,9 the Fc dimer is situated within the crystal lattice such that the electron density is an average of the individual electron densities for a given mutant pair. To account for this averaging in the refinement, we assigned each mutation 50% occupancy during structure refinement (Figure 1f). The crystal structure suggests that our bispecific design did not alter overall Fc structure, a conclusion further supported by surface plasmon resonance (SPR), which showed that binding to mouse FcRn and mouse Fc gamma receptors (FcγR) is preserved (Table 1, Table 2, Fig S1 and Fig S2).
Table 1.
Characterization of FcRn binding by SPR at 25°C.
| Antibody | Analyte | ka (1/Ms) | kd (1/s) | KD (M) |
|---|---|---|---|---|
| CD3 mG1 D265A | FcRn | 6.6E+04 | 4.2E-02 | 6.4E-07 |
| CD3/CD20 mG1 D265A bsp | FcRn | 6.2E+04 | 4.2E-02 | 6.7E-07 |
| CD3 mG2a | FcRn | 1.0E+05 | 3.4E-02 | 3.2E-07 |
| CD3/CD20 mG2a bsp | FcRn | 9.8E+04 | 3.5E-02 | 3.5E-07 |
| CD3 mG2a D265A | FcRn | 1.1E+05 | 3.6E-02 | 3.1E-07 |
| CD3/CD20 mG2a D265A bsp | FcRn | 1.1E+05 | 3.6E-02 | 3.4E-07 |
Data from the representative experiment shown
Table 2.
Characterization of Fc gamma receptor binding by SPR at 25°C.
| FCGR1 |
FCGR2 |
FCGR3 |
FCGR4 |
|||||
|---|---|---|---|---|---|---|---|---|
| KD (nM) | N | KD (nM) | N | KD (nM) | N | KD (nM) | N | |
| CD3 mG1 D265A | nb | 8 | lb | 4 | lb | 4 | lb/nb | 4 |
| CD3/CD20 mG1 D265A bsp | nb | 12 | lb | 6 | lb | 6 | lb/nb | 6 |
| CD3 mG2a | 21 ± 3 | 12 | 1570 ± 438 | 6 | 625 ± 87 | 6 | 18 ± 4 | 5 |
| CD3/CD20 mG2a bsp | 33 ± 8 | 12 | 1993 ± 538 | 6 | 1030 ± 157 | 6 | 39 ± 22 | 6 |
| CD3 mG2a D265A | nb | 8 | lb | 4 | lb | 4 | lb/nb | 4 |
| CD3/CD20 mG2a D265A bsp | nb | 8 | lb | 4 | lb | 4 | lb/nb | 4 |
KD values ± standard deviation shown. N: number of measurement spots (ROIs) used; “lb”: low binding; “nb”: no binding.
To expand the functionality of mG1 and mG2a bispecifics, we also introduced D265A mutations in CH2, which has been shown to silence effector function by abrogating FcγR binding.16–18 We did not anticipate D265A would affect bispecific formation because the mutation occurs away from the CH3 heterodimerization interface. We confirmed the effectiveness of silencing with the D265A mutation in both mG1KD and mG2aH4 bispecific antibodies, as the SPR results showed that D265A was sufficient to render molecules FcγR inert (Table 2 and Fig S2).
With heterodimeric HC-HC pairing addressed, we next focused on resolving cognate HC-LC pairing. In hIgG1 bispecific antibodies, cognate HC-LC pairing between human CH1 and CL interfaces can be achieved using the DuetMab technology.10 In this technology, the native inter-chain disulfide bond in one arm of the hIgG1 bispecific is shifted (HC F126C, LC S121C) such that covalent disulfide bond formation occurs preferentially when HCs are correctly paired to cognate LCs. Given that the DuetMab technology was not adapted for murine bispecifics, we investigated whether cognate HC-LC pairing in mIgG bispecific antibodies can be enhanced by disulfide bond engineering. To this end, we aligned human and mouse kappa (mκ) LC sequences, which revealed the conservation of S121, enabling the possibility of engineering a similar disulfide bond shift. To determine an amino acid in mouse CH1 that could pair with S121C, we utilized the crystal structure of a mIgG2a antibody (PDB: 1IGT)19 and identified Y122 (CH1) as a promising candidate for a mutation to cysteine given its proximity and orientation toward S121. Thus, the S121C (mκ-CL) – Y122C (mG2a-CH1) shifted disulfide bond was chosen for further characterization (Figure 1g). As in the human designs, the native inter-chain CH1-CL disulfide bond was eliminated by substituting C128 (mG2a-CH1) and C214 (mκ-CL) with valines. mG1 was engineered in a similar fashion by mutating Y122 (mG1-CH1) to cysteine and C209 (mG1-CH1) to valine (Figure 1g). In this work, we applied disulfide bond shifts to mG1K or mG2aH4B HC arms for pairing with LC (mκcv).
We next combined both HC-HC and HC-LC pairing technologies to generate a panel of bispecific mG1 and mG2a antibodies (Figure 2). Variable regions from in-house antibodies were cloned into mouse bispecific vectors and transfected at a 1:1:1:1 (HC1:LC1:HC2:LC2) ratio for protein expression. Bispecific antibodies were purified by one-step Protein A chromatography and subjected to intact mass spectrometry analysis. Most bispecific antibodies surveyed were purified as fully bispecific when all four chains were transfected at an equal DNA ratio (Figure 2a,b). In cases where 100% bispecific was not formed, HCs were always paired correctly, but one of two HC-LC pairings was not. For example, antibody 1/3 mG2aH4 (Figure 2b) purified was 89.3% bispecific. The 10.7% of incorrect product was identified to have correct HC pairing, but incorrect HC-LC pairing, since the variable 1 LC was found on both targeting arms. This suggested an imbalance in LC protein expression, and we found that varying the ratio of variable 1 to variable 3 LC DNA transfected restored 100% bispecific product (Fig S2). This optimization methodology was applied successfully to restore complete pairing to all samples with less than 100% initial bispecific formation (Figure 2a,b, asterisks).
Figure 2.
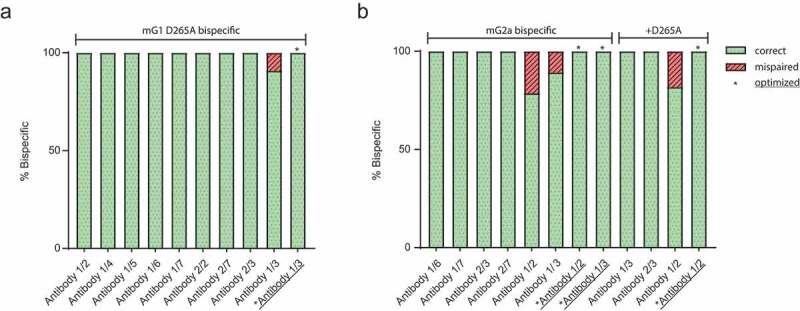
Purification and optimization of a panel of mG1 and mG2a bispecific antibodies. Intact mass spectrometry has conducted a panel of (a) mG1KD D265A, (b) mG2aH4, and mG2aH4 D265A bispecific antibodies transfected at equal DNA ratios for all four polypeptide chains. For constructs that exhibited some degree of mispairing DNA transfection ratios were optimized (asterisks) to restore complete bispecific formation.
Purification and characterization of CD3/CD20 mG1kd and mG2ah4 bispecific molecules
One area where bispecific antibodies could offer therapeutic benefit is through redirecting T-cells for targeted lysis of cancer cells.20 For example, CD3/CD20 and CD3/CD19 bispecific antibodies have been used to effectively treat human B-cell malignancies.21–23 Previous work has also demonstrated the ability of a human IgG2 bispecific to deplete mouse B-cells, when variable domains binding mouse CD3 and mouse CD20 were used with a human IgG backbone.7 Given the previous characterization of CD3/CD20 bispecific antibodies, we chose to use our mouse bispecific technology to generate a fully mouse CD3/CD20 surrogate molecule for the proof of concept.
To this end, we first selected variable regions for both CD3 and CD20 targeting arms of the bispecific. For targeting T-cells, we chose the variable region of the anti-mouse CD3ϵ antibody 145-2C11;24,25 to target B-cells, we used the variable region of the anti-mouse CD20 antibody, 18B12.26 Dose-dependent binding of 18B12 mG1 D265A to CD20 was verified by generating a Chinese hamster ovary (CHO) cell line expressing mouse CD20, and measuring binding by flow cytometry (Fig S4A & Fig S4B).
Next, we generated CD3/CD20 mG1KD D265A and CD3/CD20 mG2aH4 bispecific antibodies by co-expression. Secreted antibodies were affinity purified from supernatant in one step with Protein A, dialyzed into phosphate-buffered saline (PBS), and further characterized. To assess correct heavy-heavy chain pairing and to rule out the possibility that our purified product was a mixture of non-bispecific CD3 and CD20 antibodies, we analyzed CD3/CD20 mG1KD and CD3/CD20 mG2aH4 by intact mass spectrometry. This revealed that purified material yielded an intact mass corresponding to the presence of one each of all four unique polypeptide chains (Figure 3a,b). To simplify the analysis, samples for mass spectrometry were first treated with PNGase F for the removal of N-linked oligosaccharides. However, as is characteristic for mG2a antibodies, complete deglycosylation was not achieved for CD3/CD20 mG2aH4, which yielded a mass that corresponded to the bispecific with the addition of one or more G0F glycans (Figure 3b). Correct HC-LC pairing was further confirmed by intact mass spectrometry of CD3 and CD20 Fabs generated by papain proteolytic digestion (data not shown). Taken together these data demonstrate that bispecific mG1KD and mG2aH4 molecules purified by co-expression are homogenous and correctly paired.
Figure 3.
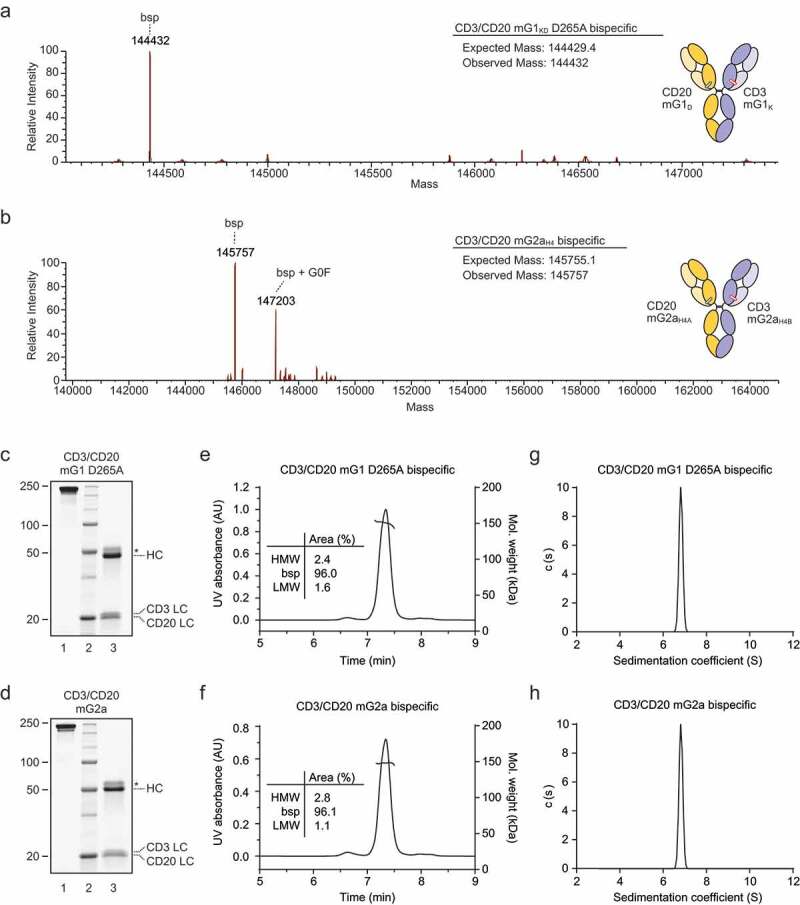
Co-expression and purification of CD3/CD20 bispecific antibodies. Intact mass spectrometry of (a) CD3/CD20 mG1KD D265A and (b) CD3/CD20 mG2aH4 treated with PNGase overnight. CD3/CD20 mG2aH4 was not completely deglycosylated, hence the presence of one or more G0F glycans in addition to the mass of the correct bispecific. (c) CD3/CD20 mG1KD D265A and (d) CD3/CD20 mG2aH4 were analyzed by SDS-PAGE in both non-reducing (lanes C-1 and D-1) and 50 mM dithiothreitol reducing (lanes C-3 and D-3) conditions. Asterisk (*) denotes glycosylated products, which run at a greater molecular weight. CD3/CD20 mG1KD D265A and CD3/CD20 mG2aH4 were also analyzed by SEC-MALS (e and f, respectively). High molecular weight (HMW) and low molecular weight (LMW) species were calculated as a percentage of the total area under the curve. Sedimentation coefficients for CD3/CD20 mG1KD D265A and CD3/CD20 mG2aH4 as measured by analytical ultracentrifugation (g and h, respectively).
Biophysical characterization of CD3/CD20 mG1kd and mG2ah4 bispecific molecules
CD3/CD20 mG1KD and CD3/CD20 mG2aH4 bispecific molecules were also characterized by denatured sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under both reducing and non-reducing conditions. In non-reduced samples we did not observe any non-covalently linked LCs, showing that HC-LC disulfide bonds are formed on both sides (Figure 3c,d – lanes 1). When reduced, both bispecific molecules yielded two distinct LCs corresponding to CD3 mκcv and CD20 mκ, as expected for a bispecific molecule (Figure 3c,d – lanes 3).
To assess aggregation and purity, we first analyzed each molecule by size exclusion chromatography multi-angle light scattering (SEC-MALS). CD3/CD20 mG1KD and CD3/CD20 mG2aH4 eluted predominantly as one peak with molecular weights that corresponded to correctly assembled bispecific antibodies (Figure 3e,f). We next analyzed each molecule by sedimentation velocity on an analytical ultracentrifuge (AUC). Both molecules sediment as a single peak at 6.82S with a sedimentation coefficient characteristic of antibodies (Figure 3g,h). Together these data show that both molecules purified are stably composed of two correctly paired heavy and light chains.
We next assessed the thermal stability of bispecific mG1KD and mG2aH4 molecules by differential scanning calorimetry (DSC). Antibodies typically have two to three transitions that correspond to the temperature-induced unfolding of Fab, CH2, and CH3 domains. In the case of bispecific antibodies, the theoretical number of transition states increases due to a greater number of unique domains. DSC profiles for CD3/CD20 mG1KD D265A and mG2aH4 both revealed three transition states that were mixtures of their parental counterparts (Table 3 and Fig S5). These data indicate that bispecific molecules exhibit antibody-like melting temperatures, with all melting transitions greater than 60°C.
Table 3.
Characterization of CD3/CD20 bispecific antibodies by differential scanning calorimetry.
| Molecule | T Onset (°C) | Tm1 (°C) | Tm2 (°C) | Tm3 (°C) |
|---|---|---|---|---|
| CD3/CD20 mG1 D265A bsp | 55.3 | 63.6 | 67.6 | 79.6 |
| CD3 mG1 | 59.7 | 66.5 | 68.6 | 78.9 |
| CD20 mG1 | 63.8 | 72.9 | 76.1 | - |
| CD3/CD20 mG2a bsp | 54.8 | 67.7 | 71.6 | 75.6 |
| CD3 mG2a | 58.1 | 65.7 | 67.7 | 76.0 |
| CD20 mG2a | 60.4 | 71.5 | 75.2 | - |
T-cell-engaging bispecific depletes B-cells in vitro
We next demonstrated that CD3/CD20 mG1KD D265A and CD3/CD20 mG2aH4 bispecific molecules could engage target-expressing cells. CD3/CD20 molecules were confirmed to bind both CHO-CD20 cells (Figure 4a,b) and primary CD3 + T-cells isolated from mouse spleens (Figure 4c,d) by flow cytometry. To test for B-cell depletion in vitro, we adapted a previously demonstrated cytotoxic assay in which primary mouse T-cells were incubated with the B-cell lymphoma line, A20.7 Both CD3/CD20 mG1KD and CD3/CD20 mG2aH4 depleted A20 cells in a dose-dependent manner while control molecules, CD3/Neg and Neg/CD20, did not (Figure 4e,f).
Figure 4.
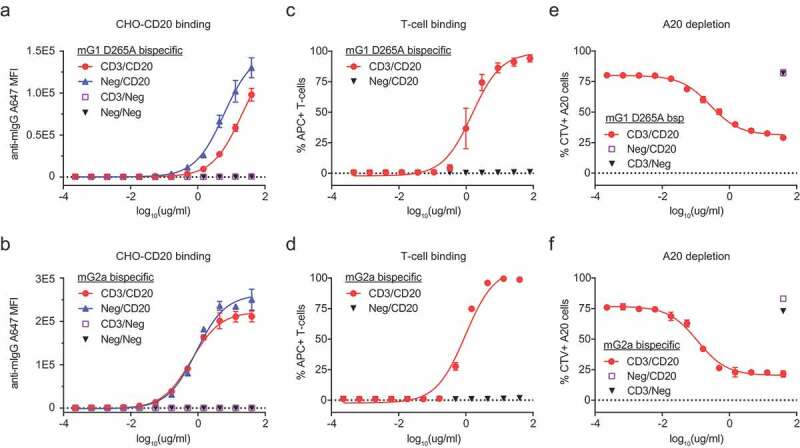
CD3/CD20 bispecific antibodies engage T-cells and target CD20+ cells in vitro. To assess CD20 binding, CD3/CD20, Neg/CD20, CD3/Neg, or Neg/Neg bispecific antibodies in both (a) mG1KD D265A and (b) mG2aH4 backbones were incubated with CHO-CD20 cells and assessed by flow cytometry (mean ± SD, n = 3). To assess CD3 binding, CD3/CD20 or Neg/CD20 bispecific antibodies in both (c) mG1KD D265A and (d) mG2aH4 backbones were incubated with primary T-cells and assessed by flow cytometry (mean ± SD, n = 3). T-cell-mediated depletion of A20 cells were conducted at a 1:5 T:E ratio for 72 h with CD3/CD20, Neg/CD20, or CD3/Neg in both (e) mG1KD D265A (EC50 = 1.85 nM) and (f) mG2aH4 (EC50 = 0.77 nM) backbones. The depletion of CellTrace Violet (CTV) labeled A20 cells were measured by flow cytometry (mean ± SD, n = 3).
We next investigated whether CD3/CD20 mG1KD D265A and CD3/CD20 mG2aH4 could activate primary mouse T-cells in the presence of primary B-cells. To this end, we incubated total splenocytes with bispecific molecules for 1 day and showed activation of T-cells with both CD3/CD20 mG1KD D265A and CD3/CD20 mG2aH4 by CD25 expression (Figure 5a). Bispecific CD3 antibodies activate T-cells directly by inducing the formation of a cytolytic synapse between conjugated T-cells and target cells. Thus, to capture conjugated B- and T-cells, we isolated mouse splenocytes and treated with CD3/CD20 mG1KD. These data revealed a dose-dependent increase in conjugated B- and T-cells (Figure 5b-d) and showed that 19.5% of total events were B/T-cell conjugates (Figure 5b). There may be more conjugates than we observed because some proportion of conjugates may have dissociated during flow cytometry analysis.
Figure 5.
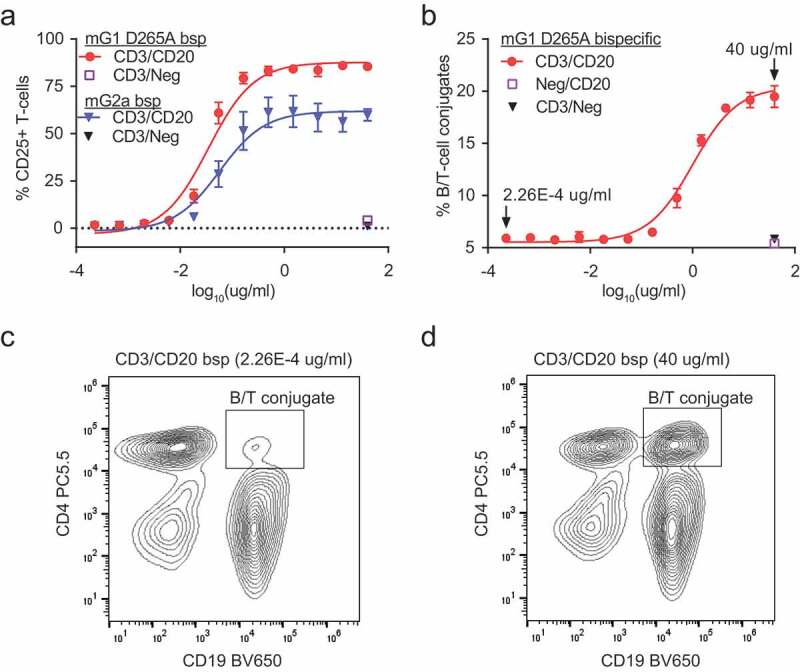
CD3/CD20 bispecific antibodies activate T-cells through direct conjugation of T-cells and B-cells. (a) Total splenocytes were incubated with CD3/CD20 and CD3/Neg in both mG1KD D265A and mG2aH4 backbones for 24 h. T-cell activation was assessed by CD25 staining followed by flow cytometry (mean ± SD, n = 3). (b) B/T-cell conjugates were captured by flow cytometry following incubation of total splenocytes with CD3/CD20, Neg/CD20, or CD3/Neg in the mG1KD D265A backbone for 1.5 h. The total percentage of conjugates were calculated from Σ(CD19+CD4+, CD19+CD8+, CD19+CD4+CD8+)/total events (mean ± SD, n = 3). Representative flow cytometry plots at (c) 2.26E-4 ug/ml and (d) 40 ug/ml CD3/CD20 antibody depicts the concentration-dependent increase of conjugated CD4+CD19+ B-cells and T-cells (top right quadrant).
T-cell-engaging bispecific depletes B-cells in vivo
We next evaluated the ability of CD3/CD20 mouse bispecific to deplete B-cells in vivo. Mice were injected with 1 mg/kg CD3/CD20 and CD3/Neg on inert mG1KD D265A and inert mG2aH4 D265A backbones. Thus, any depletion observed with these molecules would be primarily driven through T-cell-mediated cytotoxicity. Seven days after treatment, circulating B-cells were quantified by flow cytometry, which demonstrated that B-cells were depleted from the circulation of animals treated with CD3/CD20 mG1KD D265A (0.48% of leukocytes) and CD3/CD20 mG2aH4 D265A (0.09% of leukocytes) but not control CD3/Neg mG1KD D265A (43.8% of leukocytes) and CD3/Neg mG2aH4 D265A (44.6% of leukocytes) animals (Figure 6a).
Figure 6.
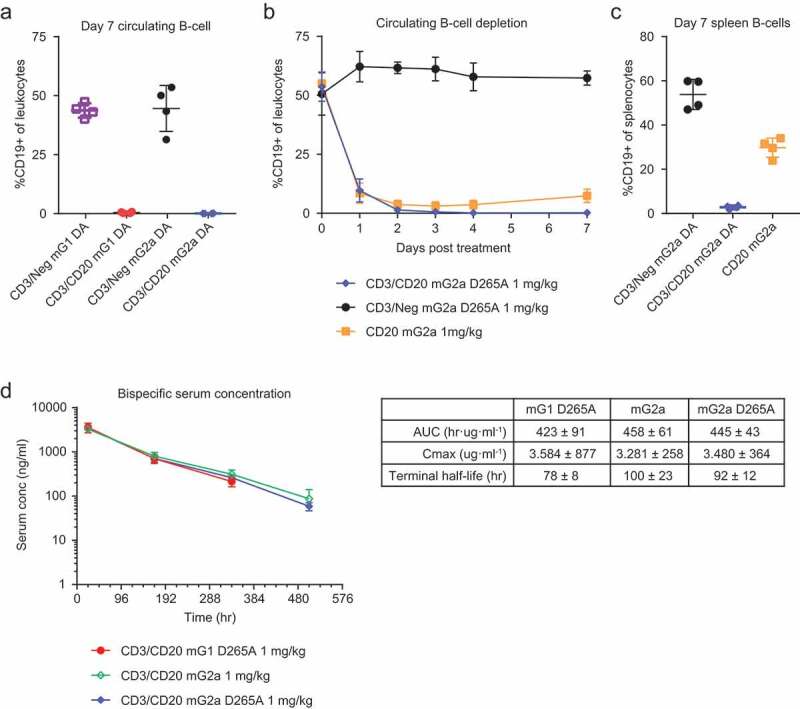
CD3/CD20 bispecific antibodies deplete B-cells in vivo. Wild-type C57BL/6 mice were treated with a single 1 mg/kg dose of CD3/CD20 or CD3/Neg in both mG1KD D265A and mG2aH4 D265A. (a) After 7 days, CD19 + B-cells were measured by flow cytometry (mean ± SD, n = 4). (b) In a second study, wild-type C57BL/6 were treated with a single 1 mg/kg dose of CD3/CD20 mG2aH4 D265A, CD3/Neg mG2aH4 D265A, or CD20 mG2a. Circulating B-cells were measured by flow cytometry at 0, 1, 2, 3, 4, and 7 days (mean ± SD, n = 4). (c) Spleens were harvested at day 7 and B-cells measured by flow cytometry (mean ± SD, n = 4). (d) Pharmacokinetic (PK) data were collected from wild-type C57BL/6 treated with 1 mg/kg CD3/CD20 or CD3/Neg in both mG1KD D265A, mG2aH4, and mG2aH4 D265A backbones. Serum concentrations were measured at 0, 1, 7, 14, and 21 days.
To further investigate the kinetics of depletion over the first 7 days, we conducted a second in vivo study in which mice were injected with 1 mg/kg CD3/CD20 and CD3/Neg bispecific molecules with mG2aH4 D265A backbones (Figure 6b). Additionally, mice were injected with 1 mg/kg CD20 mG2a antibody, which possesses active effector function and should deplete B-cells through antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity.
One day post-injection, we observed a depletion of B-cells from circulation with CD3/CD20 mG2aH4 D265A and CD20 mG2a molecules (9.63% and 8.51% of leukocytes), but not with CD3/Neg mG2aH4 D265A (62.2% of leukocytes) (Figure 6b). On day four following CD3/CD20 mG2aH4 D265A treatment, mice exhibited depletion of nearly all circulating B-cells (0.18% of leukocytes), which persisted through day seven (0.25% of leukocytes and 2.8% of splenocytes) (Figure 6b,c). Non-bispecific CD20 mG2a antibody demonstrated maximal depletion of B-cells by day three (3.03% of leukocytes); however, these cells began to recover by day seven (7.39% of leukocytes and 29.78% of splenocytes) (Figure 6b,c). CD3/Neg mG2aKD D265A-treated mice maintained B-cell numbers throughout the course of treatment (57.35% of leukocytes and 53.85% of splenocytes) (Figure 6b,c).
To determine if bispecific-forming mutations affected pharmacokinetic properties, we injected mice with a single 1 mg/kg dose of CD3/CD20 mG1 D265A, CD3/CD20 mG2a, or CD3/CD20 mG2a D265A and measured total plasma concentrations over 21 days (Figure 6d). The results indicate that the bispecific mutations did not affect pharmacokinetic properties, as the molecules behaved similar to native mouse IgGs. Furthermore, an anti-drug response or immunogenicity was unlikely, as evidenced by the linear clearance of the bispecific molecules through day 21.
Discussion
Successful research and development of new antibody therapeutics requires robust animal models that can recapitulate the biology of interest. Syngeneic mouse models are a vital component of preclinical studies and can offer a more natural tumor environment than cross-species xenograft models. Additionally, syngeneic mice possess intact immune systems that can be monitored following treatment with immune-oncology agents, where multiple pathways and cell-types are often involved in tumor clearance, and enable the possibility of longer and repeated dosing regimens. However, the use of syngeneic models necessitates the development of surrogate molecules that accurately represent their human counterparts. The design and engineering of native IgG-like bispecific molecules, which possess favorable pharmacokinetic and immunogenic properties, has largely focused on human IgG. Currently, native IgG mouse bispecific surrogates must be generated through extensive purification strategies or through cFAE.13
In this work, we designed and engineered an alternate technology for efficiently producing native IgG1 and IgG2a mouse bispecific antibodies. By combining principles of electrostatic steering and disulfide bond engineering, we engineered a system capable of generating intact mouse bispecific antibodies with any antibody pair by co-expression and one-step purification. In our hands, CH3 mutations that enable cFAE in mouse IgG molecules were insufficient for forming fully heterodimeric HCs by co-expression and require an exchange for proper pairing (data not shown). In contrast, our co-expression system simplifies downstream processing and allows the development of high-throughput screening assays and analysis.
One area of future improvement is the mispairing in some antibody pairs, which led to incorrect HC-LC arrangements when all four chains were co-transfected at equal DNA ratios. We surmised imbalanced expression rather than engineering of HC-LC pairs to be the primary driver of mispairing, since it was observed with both engineered and non-engineered arms. This imbalance may have occurred due to the use of variable regions originally derived from hamster antibodies, which may have affected expression levels. It is likely that these issues would be mitigated with the use of murine variable regions. Nonetheless, even in cases of imbalanced expression and mispairing, through a simple one-time optimization step involving titrating the transfected DNA, we were able to reproducibly restore 100% correct assembly.
We also extensively characterized the biophysical properties and biological activity of mG1 and mG2a bispecific antibodies in a CD3/CD20 system. The ability to silence effector function with a D265A mutation in CH2 enabled a more focused mechanism of action through T-cell cytotoxicity, and can be effectively utilized for other T-cell engaging bispecifics. CD3/CD20 bispecific antibodies were capable of depleting B-cells both in vitro and in vivo, and served as a proof of concept for this new technology. Taken together, we present a simple method for the one-step production of native mG1 and mG2a bispecific antibodies. This work fills a gap in available technologies for generating native mouse bispecific antibodies, and will serve to advance the preclinical development of promising therapeutics.
Materials and methods
Expression & purification of bispecific antibodies
Bispecific antibodies were generated by the simultaneous transfection of four expression plasmids each encoding different polypeptide chains into Expi293 cells (Thermo Fisher Scientific). Expi293 cells were selected for their high yield and ease of transient transfection. HCs and LCs were transfected with a DNA ratio (by weight) of 1:1:1:1 for HC1:HC2:LC1:LC2, respectively. However, due to unequal CD3 and CD20 LC expression, an optimized 1:1:1.2:0.8 ratio was used where LC1 = CD20 LC and LC2 = CD3 LC. Following 6 days of transient expression, the supernatant was harvested and purified on an AKTA Avant (GE Healthcare). Mouse IgG1 and IgG2a supernatants were loaded on either MabSelect Sure LX (GE Healthcare) or MabSelect Sure (GE Healthcare) columns, respectively, washed with PBS, eluted in 100 mM pH 3.6 sodium citrate buffer, and dialyzed into PBS.
Mass spectrometry analysis
Samples for intact liquid chromatography-mass spectrometry (LC-MS) were prepared and analyzed as follows: mouse bispecific samples were deglycosylated with PNGase F (Prozyme or New England Biolabs) in 1X PBS overnight at 37°C. An Exactive™ Plus EMR mass spectrometer coupled with Ultimate 3000 RS UPLC system (Thermo Fisher Scientific) was used for all LC-MS analysis. The separation was performed on a Waters BEH300 C4 2.1 mm × 50 mm column with a gradient of acetonitrile in 0.1% formic acid from 20% to 90% in 4 min at 80°C. Mass spectra were collected at an m/z range of 500–4200, and deconvoluted using BioPharma Finder 1.0 with manual ReSpect™ algorithm in output mass range of 20000–160000 Da.
SEC-MALS
The molecular weights of bispecific antibodies were determined by using multi-angle light scattering. Protein samples (1 mg/mL, 20 μL) were injected onto an Acquity UPLC Protein BEH SEC Column 200 size-exclusion column attached to a Waters Acquity UPLC H-Class system at a flow rate of 0.3 mL/min in 1x PBS, 0.1% Na azide. The eluted peaks were analyzed using a Wyatt DAWN HELEOS-II multi-angle light scattering instrument and a Wyatt Optilab T-rEX differential refractometer. Data were reevaluated in Astra 7.3.0 software using the Zimm model.
Analytical ultracentrifugation
AUC was conducted using a ProteomeLab XL-I ultracentrifuge (Beckman Coulter) with an An-50 Ti rotor. Samples were loaded into two-channel sedimentation velocity cells in PBS. All runs were conducted at 20°C with a rotor speed of 30,000 rpm. Sedimentation was monitored at an absorbance of 280 nm. Data analysis was conducted with Sedfit27 using a non-model-based continuous distribution corrected for time-invariant and radial-invariant noise. Sedimentation coefficients were standardized to 20°C in water.
Differential scanning calorimetry
DSC samples were dialyzed overnight in pH7.4 PBS and diluted to 1.0 mg/ml prior to measurement on a MicroCal PEAQ-DSC instrument (Malvern Panalytical). Thermograms were collected from 20°C to 100°C at a scan-rate of 60°C/h. Scans were performed with no feedback and a 10-min pre-scan thermostat time. Data collected were analyzed and fit with MicroCal PEAQ-DSC software.
FcRn binding measurements
Mouse FcRn binding was characterized on a Proteon XPR 36 instrument (BioRad) at 25°C. All antibodies were biotinylated for 1 h using a 1.5-fold excess of EZ-Link NHS-LC-LC-Biotin (Thermo Scientific; catalog number 21343). Biotinylated antibodies were captured on an NLC chip (BioRad). Mouse FcRn was purified in-house and buffer-exchanged into running buffer (20 mM MES pH 6.0, 150 mM NaCl and 0.05% Tween-20) prior to analysis. Mouse FcRn was injected in two independent five-fold, five-membered concentration series with a top concentration of 1 µM or higher. The independent dilution series had different top concentrations and were analyzed globally. All data were double-referenced and fitted to a 1:1 Langmuir binding model with mass transport using the instrument software. A representative experiment is shown in Fig S1 and Table 1.
FcγR binding measurements
Mouse FcγR binding was characterized on a Carterra LSA instrument at 25°C. The running buffer was composed of 10 mM HEPES pH 7.4, 150 mM NaCl and 0.05% Tween-20. Biotinylated antibodies were captured on multiple spots (region of interest; ROIs) of a LSA Streptavidin (SAD) chip (XanTec bioanalytics GmbH) each. Different mouse FcγR produced inhouse were injected in a five-fold dilution series each with top concentrations ranging from 500 nM to 17 µM depending on expected affinity and availability. All data were double-referenced and fitted to a steady-state binding model using the instrument software.
Structure determination
Bispecific mG2aH4 Fc was purified by the co-transfection of CH2-CH3 domains of mG2aH4-A and mG2aH4-B into Expi293 cells (Thermo Fisher Scientific) at a 1:1 DNA ratio. Following 6 days of transient expression, the supernatant was harvested and purified on an AKTA Avant (GE Healthcare). Supernatant was run on a MabSelect Sure (GE Healthcare) column, washed with PBS, eluted in 100 mM pH 3.6 sodium citrate buffer. Elution was then applied to a HiLoad 26/600 Superdex 75 column (GE Healthcare) equilibrated in a buffer containing 50 mM NaCl and 25 mM Tris pH 7.5.
Fc crystals were grown in sitting drops at 22°C using a solution containing 21% PEG10000 and 0.1 M bicine pH 7.9. Crystals were harvested at 1 week and cryoprotected with glycerol before flash freezing in liquid nitrogen. Diffraction data were collected at the Advanced Light Source beamline IMCA. The crystals belonged to the space group P1 with unit cell dimensions a = 64.27 Å, b = 64.40 Å, c = 88.98 Å and diffracted to a resolution of 1.87 Å. Data were processed with XDS.28 The structure of the mIgG2a bispecific Fc complex was solved by molecular replacement using mIgG2a Fc structure (PDB: 3ZO0) with alanines substituted for bispecific mutations using PHASER.29 Alternate cycles of the model building using the program Coot30 and model refinement using Phenix 1.15.2–3472 of coordinates and B-factors with simulated annealing, NCS torsional restraints, and secondary structure restraints. Later rounds of the model building replaced alanines with both bispecific mutations at each position as alternative residue conformations set to 50% occupancy. Coordinates for the structure are deposited in PDB (PDB ID 6UQC).
Cell line generation
For detecting CD20 binding, we generated a CHO cell line expressing mouse CD20 N-terminally fused to mNeonGreen (mNG).31 The construct was cloned into a UCOE (EMD Millipore) expression vector and introduced into CHO-S cells (Gibco) by electroporation with an Amaxa Nucelofector II (Lonza). Cells were allowed to recover for 72 h before selection with 600 μg/ml hygromycin B (Roche). Cell lines were maintained in CD-CHO media (Gibco) supplemented with 600 μg/ml hygromycin B (Roche).
Cell binding experiment
For anti-CD20 binding, 1E5 CHO-CD20 cells were aliquoted into wells and incubated with a titration of the antibody of interest for 1 h at 4°C in a buffer containing 10% fetal bovine serum (FBS) in PBS. Cells were then washed 3x, incubated with anti-mIgG Alexa 647 secondary (Jackson ImmunoResearch; #715-606-151) for 1 h at 4°C, washed 3x, and re-suspended in 100 μl 10% FBS/PBS. Cells were then analyzed on a Cytoflex Flow Cytometer (Beckman Coulter Life Sciences). For anti-CD3 binding, total splenocytes were harvested from fresh mouse spleens by homogenization in a gentleMACS Dissociator (Miltenyi Biotec), treated with red blood cell lysis buffer (BioLegend), and washed 2x with 10% FBS in PBS. Cells were then incubated with 1:100 Mouse BD FC block (BD Biosciences; #553141) in PBS before the addition of CD3/CD20 or Neg/CD20 antibodies directly labeled with Alexa 647 (Invitrogen; #A20347). After 1 h of incubation at 4°C, cells were washed 3x with 10% FBS/PBS and analyzed by flow cytometry. For B- and T-cell conjugation experiments, splenocytes were treated as described for anti-CD3 binding, stained with fluorescein isothiocyanate (FITC)-CD8 (Biolegend; #100804), PerCP/Cy5.5-CD4 (Biolegend; #100434), and BV650-CD19 (BD Biosciences; #563235).
B-cell depletion assay
Total splenocytes were harvested from fresh mouse spleens by homogenization in a gentleMACS Dissociator (Miltenyi Biotec). T-cells were then isolated by magnetic negative selection with an EasySep Mouse T-cell isolation kit (Stemcell Technologies). For the B-cell depletion assay, A20 cells were labeled with CellTrace Violet (Thermo Fisher Scientific) and incubated with purified T-cells at a target-to-effector ratio of 1:5. After 72-h cells were then stained with FITC-CD8 (Biolegend; #100804), PerCP/Cy5.5-CD4 (Biolegend; #100434), BV650-CD19 (BD Biosciences; #563235). Cells were then analyzed on a Cytoflex Flow Cytometer (Beckman Coulter Life Sciences).
Mouse study
In a first mouse study, wild-type C57BL/6 were dosed (n = 4) with 1 mg/kg CD3/CD20 mG1 D265A, CD3/CD20 mG2a D265A, CD3/Neg mG1 D265A, or CD3/Neg mG2a D265A. On day 7 mice were bled and circulating B-cells were characterized as follows. Ten microliter of mouse blood was cleared of red blood cell with ACK lysing buffer (Thermo Fisher Scientific). Cells were then pre-treated with 1:100 Mouse BD FC block (BD Biosciences; #553141) in PBS before staining with FITC-CD45 (Biolegend; #103108), PerCP/Cy5.5-CD4 (Biolegend; #100434), Alexa647-CD8 (Biolegend; #100724), and BV650-CD19 (BD Biosciences; #563235). Cells were then analyzed by flow cytometry on a Cytoflex (Beckman Coulter Life Sciences). In a second study, wild-type C57BL/6 were dosed (n = 4) with 1 mg/kg CD3/CD20 mG2a D265A, CD3/Neg mG2a D265A, or CD20 mG2a. Circulating B- and T-cells were measured before injection (day 0) and on days 1, 2, 3, 4, and 7. Additionally, spleens were harvested for analysis on day 7. Characterization of cells was conducted by flow cytometry as previously described.
Acknowledgments
The authors thank Ishita Barman, Julian Low, Barbara Aguilar and Natalie Bezman for reagents, technical advice and helpful discussions. The authors thank Steven Sheriff, Jodi Muckelbauer, and staffs from beamline IMCA at Argonne National Laboratory for assistance in data collection.
Disclosure of interest
All authors are current or former employees of Bristol-Myers Squibb.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Labrijn AF, Janmaat ML, Reichert JM, Parren P.. Bispecific antibodies: A mechanistic review of the pipeline. Nat Rev Drug Discov. 2019;18(8):585–12. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 2.Milstein C, Cuello AC. Hybrid hybridomas and their use in immunohistochemistry. Nature. 1983;305(5934):537–40. doi: 10.1038/305537a0. [DOI] [PubMed] [Google Scholar]
- 3.Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol. 1995;155:219–25. [PubMed] [Google Scholar]
- 4.Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16(7):677–81. doi: 10.1038/nbt0798-677. [DOI] [PubMed] [Google Scholar]
- 5.Davis JH, Aperlo C, Li Y, Kurosawa E, Lan Y, Lo KM, Huston JS. Seedbodies: fusion proteins based on strand-exchange engineered domain (seed) CH3 heterodimers in an Fc analogue platform for asymmetric binders or immunofusions and bispecific antibodies. Protein Eng Des Sel. 2010;23(4):195–202. doi: 10.1093/protein/gzp094. [DOI] [PubMed] [Google Scholar]
- 6.Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285(25):19637–46. doi: 10.1074/jbc.M110.117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strop P, Ho WH, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, Rickert M, Appah CT, Pascua E, Radcliffe T, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol. 2012;420(3):204–19. doi: 10.1016/j.jmb.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 8.Schaefer W, Regula JT, Bahner M, Schanzer J, Croasdale R, Durr H, Gassner C, Georges G, Kettenberger H, Imhof-Jung S, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A. 2011;108(27):11187–92. doi: 10.1073/pnas.1019002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Von Kreudenstein TS, Escobar-Carbrera E, Lario PI, D’Angelo I, Brault K, Kelly J, Durocher Y, Baardsnes J, Woods RJ, Xie MH, et al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: quality by molecular design. MAbs. 2013;5(5):646–54. doi: 10.4161/mabs.25632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs. 2015;7(2):377–89. doi: 10.1080/19420862.2015.1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Labrijn AF, Meesters JI, de Goeij BE, van den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A. 2013;110(13):5145–50. doi: 10.1073/pnas.1220145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labrijn AF, Meesters JI, Bunce M, Armstrong AA, Somani S, Nesspor TC, Chiu ML, Altintas I, Verploegen S, Schuurman J, et al. Efficient generation of bispecific murine antibodies for pre-clinical investigations in syngeneic rodent models. Sci Rep. 2017;7(1):2476. doi: 10.1038/s41598-017-02823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benonisson H, Altintas I, Sluijter M, Verploegen S, Labrijn AF, Schuurhuis DH, Houtkamp MA, Verbeek JS, Schuurman J, van Hall T. CD3-bispecific antibody therapy turns solid tumors into inflammatory sites but does not install protective memory. Mol Cancer Ther. 2019;18(2):312–22. doi: 10.1158/1535-7163.MCT-18-0679. [DOI] [PubMed] [Google Scholar]
- 15.Keeble AH, Khan Z, Forster A, James LC. TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc Natl Acad Sci U S A. 2008;105(16):6045–50. doi: 10.1073/pnas.0800159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nature Medicine. 2000;6(4):443–46. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 17.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. Fcgammariv: A novel FcR with distinct IgG subclass specificity. Immunity. 2005;23(1):41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 18.Baudino L, Shinohara Y, Nimmerjahn F, Furukawa J, Nakata M, Martinez-Soria E, Petry F, Ravetch JV, Nishimura S, Izui S. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol. 2008;181(9):6664–69. doi: 10.4049/jimmunol.181.9.6664. [DOI] [PubMed] [Google Scholar]
- 19.Harris LJ, Larson SB, Hasel KW, McPherson A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry. 1997;36:1581–97. [DOI] [PubMed] [Google Scholar]
- 20.Krishnamurthy A, Jimeno A. Bispecific antibodies for cancer therapy: A review. Pharmacol Ther. 2018;185:122–34. doi: 10.1016/j.pharmthera.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Loffler A, Kufer P, Lutterbuse R, Zettl F, Daniel PT, Schwenkenbecher JM, Riethmuller G, Dorken B, Bargou RC. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95(6):2098–103. doi: 10.1182/blood.V95.6.2098. [DOI] [PubMed] [Google Scholar]
- 22.Smith EJ, Olson K, Haber LJ, Varghese B, Duramad P, Tustian AD, Oyejide A, Kirshner JR, Canova L, Menon J, et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci Rep. 2015;5:17943:1–12. doi: 10.1038/srep17943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun LL, Ellerman D, Mathieu M, Hristopoulos M, Chen X, Li Y, Yan X, Clark R, Reyes A, Stefanich E, et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of b cell malignancies. Sci Transl Med. 2015;7(287):287ra270. doi: 10.1126/scitranslmed.aaa4802. [DOI] [PubMed] [Google Scholar]
- 24.Leo O, Foo M, Sachs DH, Samelson LE, Bluestone JA. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci U S A. 1987;84(5):1374–78. doi: 10.1073/pnas.84.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alegre ML, Tso JY, Sattar HA, Smith J, Desalle F, Cole M, Bluestone JA. An anti-murine CD3 monoclonal antibody with a low affinity for Fc gamma receptors suppresses transplantation responses while minimizing acute toxicity and immunogenicity. J Immunol. 1995;155:1544–55. [PubMed] [Google Scholar]
- 26.(Dunn RJSD, CA, US), Mertsching, Elisabeth (San Diego, CA, US), Peach, Robert (San Diego, CA, US), Kehry, Marilyn R. (San Diego, CA, US) . Anti-mouse CD20 antibodies and uses thereof. In: Anti-mouse CD20 antibodies and uses thereof. United States: Biogen Idec Inc; 2007. (Cambridge, MA, US). [Google Scholar]
- 27.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78(3):1606–19. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W. XDS Acta Crystallogr D Biol Crystallogr. 2010;66(Pt2):125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. Phenix: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, et al. A bright monomeric green fluorescent protein derived from branchiostoma lanceolatum. Nat Methods. 2013;10(5):407–09. doi: 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.