

ABSTRACT
Microbial detoxification of plant toxins influences the use of plants as food sources by herbivores. Stephen's woodrats (Neotoma stephensi) specialize on juniper, which is defended by oxalate, phenolics and monoterpenes, while closely related N. albigula specialize on cactus, which only contains oxalate. Woodrats maintain two gut chambers harboring dense microbial communities: a foregut chamber proximal to the major site of toxin absorption, and a cecal chamber in their hindgut. We performed several experiments to investigate the location and nature of microbial detoxification in the woodrat gut. First, we measured toxin concentrations across gut chambers of N. stephensi. Compared to food material, oxalate concentrations were immediately lower in the foregut, while concentrations of terpenes remained high in the foregut, and were lowest in the cecal chamber. We conducted metagenomic sequencing of the foregut chambers of both woodrat species and cecal chambers of N. stephensi to compare microbial functions. We found that most genes associated with detoxification were more abundant in the cecal chambers of N. stephensi. However, some genes associated with degradation of oxalate and phenolic compounds were more abundant in the foregut chambers. Thus, microbial detoxification may take place in various chambers depending on the class of chemical compound.
Keywords: host-microbe interactions, oxalate, PICRUSt, terpenes, woodrat
Herbivorous mammals rely on gut microbes to degrade plant toxins for them; this process happens in several gut regions.
INTRODUCTION
Mammalian herbivores are often challenged by the consumption of toxic plant secondary compounds (PSCs) that negatively impact their physiology in various ways (Dearing, Foley and McLean 2005a). In response, mammalian herbivores have evolved myriad adaptations to cope with dietary toxins, such as altered behavior, efflux pumps in the gut and enhanced liver detoxification (Dearing, Foley and McLean 2005a). Recently, we demonstrated that the gut microbiota of herbivores also play an important role in allowing them to consume PSCs (Kohl et al. 2014b). However, it is still unclear where the process of microbial detoxification occurs in the gut.
It has been proposed that microbial detoxification should occur early in the gastrointestinal tract, to facilitate rapid degradation of PSCs and minimize the potential for their absorption in the digestive tract. In fact, it has been hypothesized that the rumen evolved first for detoxification and was later adopted for processes of fermentation (Hume and Warner 1980; Mackie 2014). Examples of animals in which foregut detoxification has been documented for particular PSCs include reindeer (Rangifer tarandus; Sundset et al. 2010) that have rumen communities that degrade usnic acid, a toxin produced by lichen, and hoatzins (Opisthocomus hoazin), that maintain communities of microbes in their foregut capable of metabolizing saponins (Garcia-Amado et al. 2007), and domesticated ruminants (Jones and Maegarrity 1986; Derakhshani, Corley and Al Jassim 2016). There is also evidence of microbial detoxification processes occurring in the hindguts of some animals. For example, Sage-grouse (Centrocercus urophasianus), specialize on toxic sagebrush (Artemisia spp.) and maintain a cecal microbiota enriched in detoxification genes (Kohl et al. 2016a). Additionally, the hindguts of koalas (Phascolarctos cinereus) harbor microbes capable of degrading phenolic PSCs (Osawa 1992), which likely aid the host in detoxification (Kohl, Stengel and Dearing 2016b). Understanding the location of microbial processes in the gut is likely to be important in the context of animal physiology. For example, the location of fermentation (foregut vs hindgut), seems to facilitate or constrain evolution of herbivore body size (Clauss et al. 2003) and dietary niche breadth (Alexander 1991). The location of microbial detoxification could have similar impacts on animal ecology and evolution.
In contrast to many above described examples, woodrats (genus Neotoma) are particularly interesting from a digestive standpoint as these herbivorous rodents possess both a foregut chamber and a large cecal chamber in their hindgut (for a diagram of the woodrat gut see Graphical Abstract or Kohl et al. 2011). The ‘foregut’ chamber is a semi-segmented portion of the fornix ventricularis or fundus of the stomach that maintains a pH of 4.4, while the gastric stomach chamber maintains a pH of 1.4 (Kohl et al. 2014a). The cecal chamber maintains a pH of 6.3 and is largely used for the selective retention of fluid and small particles of digesta for fermentation by gut microbes (Hume, Morgan and Kenagy 1993). Both the foregut and cecal chambers harbor dense communities of microbes and high concentrations of volatile fatty acids (Kohl et al. 2014a), which are the microbial products of fermentation. Following the hypothesis that the bovine rumen first evolved for detoxification (Hume and Warner 1980; Mackie 2014), we predict that microbial detoxification of PSCs might occur prior to the small intestine in the woodrat gut such that the animal experiences limited absorption and systemic exposure to toxic compounds.
To evaluate this hypothesis, we conducted a series of experiments focusing on a species of woodrat with a highly specialized diet. Stephen's woodrat (Neotoma stephensi) is a dietary specialist, with juniper (Juniperus monosperma) composing ∼90% of its diet (Vaughan 1982). Juniper defends itself from herbivory with various toxic compounds, including monoterpenes (Schwartz, Nagy and Regelin 1980), phenolics, such as condensed tannins (Holechek et al. 1990; Dearing, McLister and Sorensen 2005b), and oxalate (Justice 1985). These classes of defensive compounds vary in modes of action as feeding deterrents. Monoterpenes are small, lipophilic compounds that are absorbed rapidly and act as neurotoxins (Rattan 2010). Phenolics can bind to dietary protein and digestive enzymes, preventing digestion and absorption and thus limiting nutrient availability (Min et al. 2003). Oxalate crystals cause physical damage through the corrosion of the mouth and gut, and the formation of kidney stones (Miller and Dearing 2013).
We have demonstrated that microbes aid in the detoxification of phenolics (Kohl et al. 2014b) and oxalate in other woodrat species (Miller et al. 2016b; Miller, Dale and Dearing 2017). For example, some populations of the white-throated woodrat (N. albigula) specialize on cactus (Opuntia spp.), which is rich in oxalate, but lacking in other defensive compounds. The foregut microbial communities of these N. albigula contain several taxa of oxalate-degrading microbes (Miller, Kohl and Dearing 2014). There has also been metagenomic evidence obtained from other systems suggesting that microbes aid in the detoxification of terpenes (mountain pine beetles: Adams et al. 2013; sage grouse: Kohl et al. 2016a), as well as experimental work demonstrating the detoxification function of the gut microbiota of pine weevils (Berasategui et al. 2017). However, the detoxification contribution of the microbiome has not been compared between the foregut and hindgut in any wild vertebrate herbivore.
We addressed several questions in the woodrat-juniper system. First, we measured concentrations of two classes of toxins (oxalate and monoterpenes) across regions of the gut. We predicted that if detoxification is occurring proximally in the gut, then toxins concentrations in the foregut would be significantly lower compared to food material. Next, we conducted a series of metagenomic analyses to inventory and compare the potential functions of microbial communities. We first compared the metagenomes of foregut communities between the juniper specialist N. stephensi and the cactus specialist N. albigula. We predicted that if the foregut microbiota of N. stephensi were specialized for detoxification of juniper, which contains oxalate, monoterpenes and phenolics, we would observe enrichment of detoxification genes associated with monoterpenes and phenolics compared to the foregut of N. albigula, which are only exposed to the oxalate in cactus (juniper and cactus both have high oxalate concentrations; Justice 1985). We also compared the metagenomes of foregut and cecal chambers within N. stephensi and predicted that the metagenomes of foregut chambers would be enriched in detoxification genes compared to the cecal communities.
MATERIALS AND METHODS
All experiments below were approved by the University of Utah Institutional Animal Care and Use Committee (IACUC) under protocols #12–12 010 and #16–0 2011.
Toxin levels throughout the gut
We conducted a feeding trial on N. stephensi to document changes in toxin concentrations across gut regions. Six individuals of N. stephensi were fed increasing concentrations of J. monosperma (3 days on 25%, 3 days on 50% and 4 days on 75%). On the last morning, animals were euthanized using isoflurane. Luminal contents of each gut region (foregut, stomach, small intestine, cecum, large intestine and feces) were collected. A small portion (∼0.25 g) of wet gut contents was placed in airtight headspace autosampler vials and frozen at −20°C until analysis. Concentrations of individual monoterpenes were determined using headspace gas chromatography with an Agilent 7694 headspace sampler coupled with an Agilent 6890N gas chromatograph (GC). One mL of headspace gas was injected into a J&W DB-5 capillary column (30 m × 250 µm × 0.25 µm). Operating conditions for the headspace sampler were: oven temperature at 100°C, loop temperature at 110°C, transfer line temperature at 120°C, a vial equilibrium time of 20 min, a pressurization time of 0.20 min, a loop fill time of 0.50 min, a loop equilibrium time of 0.20 min and an injection time of 0.50 min. Operating conditions for the GC were: splitless injector at 250°C, flame ionization detector at 300°C, oven temperature at 40°C for 2 min, then increasing 3°C/min to 60°C, then increasing 5°C/min to 120°C, then increasing 20°C/min to 300°C and held at 300°C for 7 min. The make-up gas was nitrogen and the carrier gas was helium. The inlet pressure was 80 kPa with a flow rate of 1.0 mL/min. Retention times and peak areas of individual monoterpenes were calculated using Agilent OpenLab version A.01.05. We verified the relative retention times of individual monoterpenes using standards ran under the same headspace and GC conditions as samples. We used standards to estimate the relative concentrations of individual monoterpenes in the intestinal content material by comparing AUC of peaks in diet or gut samples to AUC of a known volume of 10 mg/mL monoterpene standards (α-pinene, β-pinene, camphene, carene and terpinolene) that were dissolved in GC grade methylene chloride.
Remaining gut contents were placed in weighing boats and dried at 40°C for 48 h. The dried gut contents were then used to measure oxalate concentrations using a previously established protocol (Miller et al. 2016a). Roughly 0.4g of dried gut contents were ground and added to 5 mL of 6 N H2SO4 to solubilize the oxalate. After 15 min, 25 mL of distilled water was added, and the entire solution was filtered through grade 4 Whatman filter paper. The filtrate was brought up to a pH of 7 with NaOH, and 0.1 g of CaCl2 was added to precipitate the oxalate. The samples were centrifuged and decanted. After centrifugation, a volume of distilled water equal to that recovered after filtration was added, and the samples were titrated.
Sample collection for metagenomics
We compared the metagenomic sequencing results across species and gut regions to investigate differential abundance of potential microbial functions. We collected individuals of the cactus specialist N. albigula, from Castle Valley, UT, USA (38°30′ N, 109°18′ W). The microbial diversity and community structure across gut regions of N. albigula have been reported elsewhere (Miller, Kohl and Dearing 2014; Kohl et al. 2014a), but here we add metagenomics sequencing of the foregut chamber. Individuals of N. stephensi (different individual from those described in the experiment above) were collected from near Wupatki National Monument, AZ, USA (35°30′ N, 111°27′ W). Animals were immediately transported back to the University of Utah animal facility, provided with native diets: either cactus pads or juniper overnight (for N. albigula and N. stephensi, respectively). Woodrats are nocturnal animals, and upon visible inspection it was clear that plant material had been consumed overnight. The following morning animals were euthanized and dissected. The contents of the foregut chamber were collected from three individuals of each species, and the contents of the cecal chamber were collected from N. stephensi. We sequenced the metagenome of each sample (described below), and conducted several comparisons. First, we compared the foregut metagenomes of N. albigula and N. stephensi, with the prediction that the foregut of N. stephensi would be enriched in genes associated with detoxification of juniper, which contains more chemical classes of defensive compounds than cactus. Second, we compared the metagenomes of foregut contents and cecal contents in N. stephensi with the prediction that the abundance of detoxification genes would be higher in the foregut to facilitate detoxification prior to the major site of absorption in the small intestine.
Comparative metagenomics
We used metagenomics sequencing to inventory the functional potential of the woodrat gut microbiome. Inventorying the metagenomic content of a community is a commonly used technique in microbial ecology, though it should be recognized that this method only investigates the potential functions of a community (Franzosa et al. 2014). Total DNA was isolated from foregut and cecal contents using MoBio PowerFecal DNA extraction kit (MoBio, Carlsbad, California). Extracted DNA was sent to Argonne National Laboratory for sequencing. Genomic DNA was sheared using a Covaris Sonicator, to roughly 150 bp and metagenomic shotgun libraries were prepared using the Illumina TruSeq DNA sequencing preparation kits. Libraries were sequenced on the Illumina HiSeq2000 platform using a 2 × 100 bp run and V3 chemistry, which resulted in overlapping sequences.
Metagenomic sequences were uploaded to the MG-RAST website (Meyer et al. 2008). Sequences were screened against the genome of Arabidopsis thaliana to remove potential contamination from the plant-based diet. The reads were then filtered using dynamic trimming with a quality threshold of 15, such that any sequences with more than five low-quality bases were removed. Several foregut metagenomes contained high abundances of sequences (∼30%–50%) identified as plant DNA, likely resulting from the diet. Therefore, we partitioned our analysis by domain (bacteria, archaea and eukaryotes) by selecting sequences identified as bacteria using the non-redundant multi-source protein annotation database, M5nr (Wilke et al. 2012). Abundances of non-plant eukaryotic and archaeal sequences were too low for sufficient analysis. Sequences identified as bacterial in origin were then annotated to identify putative functions using the KEGG Orthology database (Kanehisa and Goto 2000; Kanehisa et al. 2016) with the following thresholds: (1) e-values <1e-5, (2) a minimum percentage identity to database sequences of 60% and (3) a minimum alignment length of 15 bases. We compared the abundances at Level 3 of the KEGG hierarchical classification, as well as specific genes associated with ‘Xenobiotic degradation and metabolism’, ‘Geraniol degradation’ and ‘Limonene and pinene degradation’. Abundances of functional categories were compared using JMP 12.0 (SAS Institute Inc.) using t-tests and the robust Response Screening function that performs the Benjamini-Hochberg false discovery rate (FDR) correction for multiple tests (Benjamini and Hochberg 1995).
We also conducted specific comparisons of the abundances of several detoxification genes: oxalyl-CoA decarboxylase, aryl alcohol dehydrogenase and several genes associated with terpene metabolism. The gene oxalyl-CoA decarboxylase (oxc) degrades the compound oxalate. The gene aryl alcohol dehydrogenase is upregulated in the woodrat foregut when individuals are fed diets containing phenolic-rich resin from creosote bush (Kohl et al. 2014b). Last, a number of genes associated with the ‘Limonene and pinene degradation’ are enriched or present in the gut microbiota of specialist insect and avian herbivores that feed on terpene-rich plants (Adams et al. 2013; Kohl et al. 2016a). We compared the abundances of these genes using JMP 12.0 (SAS Institute, Inc.) using pairwise t-tests (foregut N. stephensi vs foregut N. albigula and N. stephensi foregut vs cecum) with the Dunn's correction.
Metagenome prediction
We also conducted microbial inventories by sequencing the variable regions 1–3 of 16S rRNA gene following experimental and bioinformatic methods described in detail elsewhere (Phillips et al. 2017). Taxonomic comparison of the communities between foregut and cecal chambers has already been conducted with larger sample sizes (Kohl et al. 2014a). In the current study, we used 16S rRNA inventories to predict the metagenomic content of gut samples using genomic inference via PICRUSt (Langille et al. 2013). We compared the relative abundances of functional categories between the shotgun metagenomic and predicted metagenomics to assess if predictive profiling via PICRUSt is informative for woodrat gut samples. First, we averaged the relative abundances of functional categories within a sample type (foregut and cecum), and transformed the relative abundances using a log(x+1) transformation. We then conducted a linear regression between transformed PICRUSt abundances and metagenomic abundances. We also plotted the relative abundances of functional categories that exhibited the highest fold difference between the foregut and cecal chambers to qualitatively inspect whether trends were similar for PICRUSt and metagenomic data.
Accession numbers
Metagenomic sequences can be found in the NCBI Short Read Archive (SRA) database under BioProject PRJNA336354, and on MG-RAST under Metagenome Project IDs mgp10174 and mgp10296. 16S rRNA sequences from the foregut and cecal chambers of N. stephensi that were used in PICRUSt analyses can be found in the SRA database under BioProject PRJNA449358.
RESULTS
Concentrations of PSCs across the gut of N. stephensi
Water concentration varied over the length of the gut, with a significant reduction in the large intestine (Fig. 1). Oxalate concentrations in the entire gut were significantly less than in the food material (Fig 2; P < 0.0001). Oxalate concentrations also varied significantly across gut regions (P < 0.001), and were highest in the foregut and lowest in the stomach. Conversely, foregut concentrations of monoterpenes were not significantly lower than the food material (Fig. 1; P > 0.05 for all). Concentrations of monoterpenes did vary across gut regions, with the lowest concentrations always being in the cecum (Fig. 1; P < 0.0001 for all). In fact, camphene and β-pinene were not detectable in any cecal samples. For data on toxin concentrations relative to dry mass of gut contents, see Fig. S1 (Supporting Information).
Figure 1.
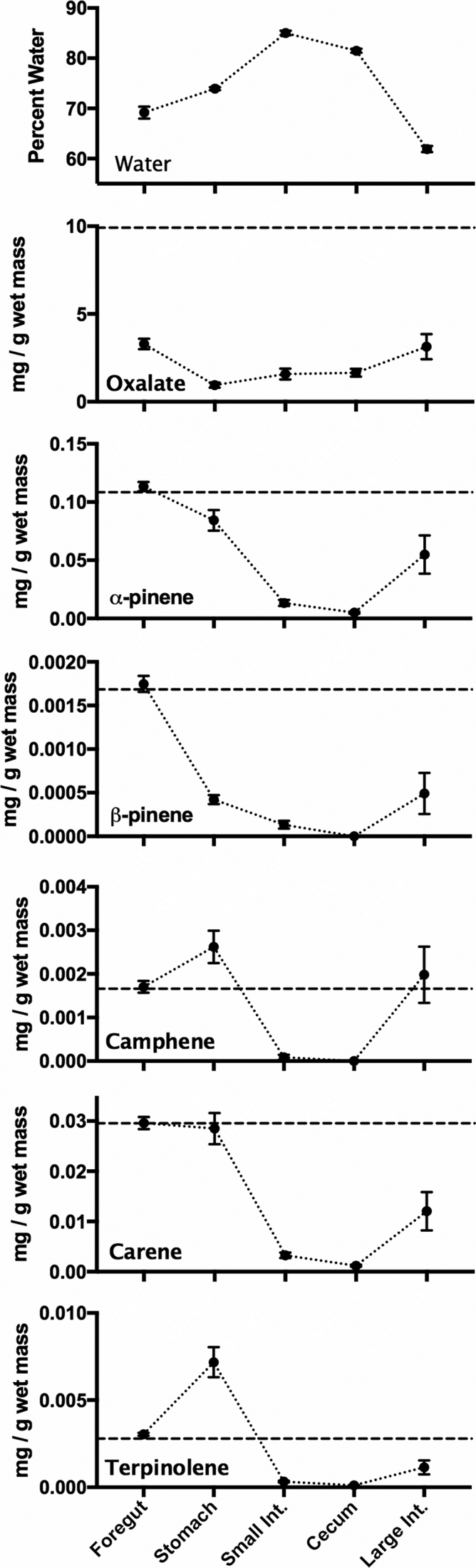
Concentrations of water and toxins across gut regions. The dotted line on toxin graphs represents concentrations as measured in the food material presented to woodrats. Points represent means ± 1 s.e.m. Data normalized to dry mass of food and gut contents can be found in Fig. S1 (Supporting Information).
Figure 2.

Functional categories (KEGG Level 3) that differed significantly between the foregut metagenomes of N. albigula and N. stephensi. Bars represent means ± 1 s.e.m. These differences are significant at a level of α = 0.05, after a Benjamini-Hochberg False Discovery Rate (FDR) correction for multiple tests.
Metagenomic sequencing
We obtained an average of 3.35 million reads per sample (± 517 000). We first compared the bacterial metagenomes of foregut chambers from N. stephensi (which specializes on juniper) and N. albigula (which specializes on cactus). There were no functions that were significantly enriched in the foregut bacterial metagenomes of N. stephensi. The foregut bacterial communities of N. albigula were enriched in functions related to the metabolism of several amino acids and vitamin B6 (Fig. 2). Further, when investigating detoxification genes, there were no detectable differences between the two species.
We next searched for differential functions between the foregut and cecal chambers of N. stephensi. The foregut bacterial metagenome was enriched in genes associated with the metabolism of nucleotides (purines), amino acids (glutathione, thiamine), vitamins (riboflavin, nicotinate and folate) and lipoic acid (Fig. 3A). The cecal bacterial metagenome was enriched for genes associated with the metabolism of starch and sucrose, as well as the biosynthesis of several essential amino acids (Fig. 3A). The cecum was also enriched in several functional categories related to detoxification (drug metabolism, chloroalkane and chloroalkene degradation; Fig. 3A).
Figure 3.

Differences in the metagenomic content of the foregut and cecal chambers of N. stephensi. Bars represent means ± 1 s.e.m. (A) Functional categories (KEGG Level 3) that differed significantly in abundance between the foregut and cecal metagenomes. (B) Detoxification genes that differed significantly in abundance between the foregut and cecal metagenomes. These differences are significant at a level of α = 0.05, after a Benjamini-Hochberg False Discovery Rate (FDR) correction for multiple tests.
We also specifically investigated genes associated with ‘Xenobiotic degradation and metabolism’, ‘Geraniol degradation’ and ‘Limonene and pinene degradation’. Here, the only gene enriched in the foregut was 4-oxalocrotonate tautomerase, which was 24 × more abundant in the foregut compared to the cecum (Fig. 3B). There were five other detoxification genes that were significantly more abundant in the cecum compared to the foregut chamber (Fig. 3B). Three of these genes (carboxylesterase 2, hydroxyatrazine ethylaminohydrolase and 2-hydroxycyclohexanecarboxyl-CoA dehydrogenase) were 2–3 × more abundant in the cecal chambers compared to the foregut chambers. Two genes, anthraniloyl-CoA monooxygenase and dehalogenases, were not detected in the foregut chambers, but were present in the cecal chambers of the juniper-feeding N. stephensi (Fig. 3B).
Last, we focused on a subset of detoxification enzymes highlighted from previous studies. The gene oxalyl-CoA decarboxylase, which is anticipated to function in oxalate degradation, did not exhibit differential abundance between the foregut chambers of N. stephensi and N. albigula. However, the abundance of this gene was 4.5 × higher in the foregut of N. stephensi compared to the cecum (Fig. 4). Similarly, aryl-alcohol dehydrogenase, which is more abundant in the woodrat foregut when animals are fed phenolic-rich creosote resin (Kohl et al. 2014b), did not exhibit significant differences between the foregut chambers of N. stephensi or N. albigula, but was 41 × higher in the N. stephensi foregut compared to the cecum (Fig. 4). Last, we investigated several genes in the ‘Limonene and pinene degradation’ pathway that are enriched in other herbivores that feed on terpene-rich plants (Adams et al. 2013; Kohl et al. 2016a; Berasategui et al. 2017). Enoyl-CoA hydratase (E.C. 4.2.1.17) was more abundant in the foregut of N. stephensi compared to N. albigula, with no difference between the foregut and cecal chambers of N. stephensi (Fig. 4). Additionally, aldehyde dehydrogenase (E.C. 1.2.1.3) exhibited no difference between the foregut chambers of N. stephensi and N. albigula, but was 2.5 × more abundant in the cecal chambers of N. stephensi (Fig. 4).
Figure 4.

Relative abundances of specific detoxification enzyme genes associated with a priori hypotheses from other systems (see Methods). Bars represent means ± 1 s.e.m. Relative abundances were compared using t-tests (foregut N. stephensi vs foregut N. albigula and N. stephensi foregut vs cecum) with the Dunn's correction.
We found a significant positive relationship between the relative abundances of functional categories as estimated by PICRUSt and determined by metagenomic sequencing (Fig. 5; foregut: R2 = 0.49, slope = 0.922 ± 0.074, P < 0.0001; cecum: R2 = 0.51, slope = 0.978 ± 0.072, P < 0.0001). Some functional categories, such as those representing ‘Peptidases’ and ‘Amino acid Related Enzymes’, were not well predicted by PICRUSt, in that they were present on the higher end of the x-axis (predicted to be present by PICRUSt but not detected by metagenomic sequencing). However, categories that exhibited the greatest fold-differences between the two chambers were also predicted to differ by PICRUSt (Fig. 5; with the exception of Starch and Sucrose Metabolism).
Figure 5.

(A) A comparison of predictive profiling by PICRUSt and metagenomic data. (B) Bar graphs depict the functional categories that exhibited the highest fold-differences between the foregut and cecal metagenomes. Bars represent means ± 1 s.e.m.
DISCUSSION
Here, we investigated the nature and location of microbial detoxification in the guts of Stephen's woodrat, a juniper specialist. The location of detoxification within the gut seems to vary depending on the chemical class of defensive compounds. We discuss our findings below, as well as the potential ecological and evolutionary consequences of foregut vs hindgut detoxification by the microbiome.
We first compared concentrations of PSCs along the length of the gastrointestinal tract. We predicted that if toxins were metabolized early in the gut, their concentrations in the foregut chamber would be lower than those measured in food material, similar to what has been observed for usnic acid in the reindeer rumen (Sundset et al. 2010). This pattern was observed in the case of oxalate, where foregut concentrations were roughly a third of those measured in the diet. Since oxalate is detected only at very low levels in the urine of woodrats (Justice 1985) and mammals do not produce the necessary oxalate-degrading enzymes (Miller and Dearing 2013), it seems that oxalate is detoxified efficiently by microbes in the foregut. Conversely, terpene concentrations in the foregut were the same as those measured in food material, suggesting that the foregut is not a significant site for detoxification. Instead, concentrations of terpenes were lowest in the cecum, where some terpene compounds were not even detected. These results are similar to those observed in sage-grouse, where terpene concentrations are also lowest in the cecal chambers (Kohl et al. 2015). Neotoma stephensi has physiological adaptations that reduce terpene absorption in the gut by 40% when compared to N. albigula (Sorensen, Turnbull and Dearing 2004), perhaps allowing terpene metabolism to be delayed until the food reaches the cecum. In the case of terpenes, concentrations increased in the large intestine compared to the cecum. We hypothesize that the selective absorption of water or the excretion of toxin compounds into the large intestine result in higher concentrations of toxins in this region. Further experiments, perhaps using radiolabeled compounds, are needed to understand the routes of absorption and detoxification of these compounds in different regions of the woodrat digestive system.
Interestingly, we did not observe many differences related to detoxification between the foregut microbial metagenomes of N. stephensi and N. albigula, even though these two host species feed on different diets with contrasting chemical profiles. It could be that the foregut microbiome offers broad detoxification capacities, while cecal microbial communities are more specialized. That being said, we did observe a higher abundance of enoyl-CoA hydratase in the foregut of N. stephensi compared to N. albigula. This gene plays a role in the ability for microbes to degrade terpenes (Iurescia et al. 1999), and is enriched in the gut metagenomes of sage-grouse (Kohl et al. 2016a) and mountain pine beetles (Adams et al. 2013). Thus, there may be some functional enrichment for detoxification in the foregut of N. stephensi.
Within N. stephensi, the foregut and cecal microbial communities appear to provide different detoxification functions. In the foregut, we observed higher abundance of the gene 4-oxalocrotonate tautomerase, which is involved in the degradation of aromatic hydrocarbons by bacteria (Harayama and Timmis 1989). The foregut also had higher abundances of the gene oxalyl-CoA decarboxylase, which degrades oxalate, and aryl-alcohol dehydrogenase, which degrades aromatic hydrocarbons. The abundance of aryl-alcohol dehydrogenase increases in the foregut of another woodrat species (N. lepida) when animals are fed diets containing phenolic PSCs (Kohl et al. 2014b) and thus may be an important enzyme that is provided by the foregut microbial community.
We also obtained evidence for enrichment of detoxification enzymes in the cecal chamber. Higher-level gene categories, such as ‘Drug Metabolism’ and ‘Chloroalkane and Chloroalkene Degradation’ were more abundant in the cecal chambers than the foregut. Specifically, the cecum was enriched for genes such as hydroxyatrazine ethylaminohydrolase, which deaminates or dechlorinates triazine compounds (Boundy-Mills et al. 1997; Seffernick et al. 2002), and anthraniloyl-CoA monooxygenase, which plays a role in the catabolism of aromatic compounds (Carmona et al. 2009). Additionally, the aldehyde dehydrogenase gene, a component of the ‘Limonene and Pinene Degradation’ pathway, was significantly more abundant in the cecum.
Finally, we tested whether predictive profiling of metagenomics based on 16S rRNA sequences is appropriate for woodrat gut samples. We found a significant relationship between the relative abundances of functional categories as estimated by PICRUSt and determined by metagenomic sequencing. Additionally, results for PICRUSt and metagenomics were consistent for some of the categories that exhibited the largest differences between the two chambers. However, PICRUSt did not predict all of the categories accurately. Many functional categories (e.g. ‘Peptidases’ and ‘Amino acid Related Enzymes’) were predicted to be present by PICRUSt analysis, but were not detected by metagenomic sequencing. Interestingly, the gut microbial communities of herbivores tend to have lower abundances of genes encoding protease relative to carnivores, perhaps allowing more of these important nutrients to be digested and absorbed by the host (Muegge et al. 2011). Thus, the use of predictive metagenomic profiling may not always be feasible for non-model systems, especially given that most microbial genomes used for predictive profiling are from the human gut. Indeed, PICRUSt works significantly better for human samples than for samples from non-human mammalian guts (Langille et al. 2013). The sequencing of additional genomes from non-human mammals would help to improve the accuracy of programs like PICRUSt in non-model systems. It seems that results from predictive profiling in non-model systems should perhaps be verified with other methods, such as quantitative PCR.
Overall, we hypothesized that microbial detoxification should occur proximally in the gastrointestinal tract, so as to facilitate degradation of PSCs before they would be absorbed later in the gut. However, it seems that detoxification processes may be spatially structured along the length of the gut. The evidence for foregut detoxification is strongest for oxalate, where abundances of the detoxification enzyme (oxalyl-CoA decarboxylase) were high, and concentrations of oxalate were already much lower than in food material, and abundances of potentially oxalate-degrading bacteria are highest (Miller, Kohl and Dearing 2014). Terpene degradation may occur in the cecal chamber, where abundances of terpene-degradation genes were higher, and concentrations of terpenes were at their lowest. It is somewhat puzzling why woodrats would maintain this spatial arrangement of microbial communities, given that terpenes are absorbed very rapidly due to their small size and high lipophilicity (Sorensen, Turnbull and Dearing 2004). However, N. stephensi exhibits lower terpene absorption than other woodrat species (Sorensen, Turnbull and Dearing 2004), resulting in relatively higher terpene exposure and resultant detoxification by microbial communities in the hindgut. Laboratory rats are able to excrete ∼25% of absorbed monoterpenes back into the gut lumen through the biliary duct (Igimi et al. 1973), an excretion route that may be enhanced in a specialist herbivore such as N. stephensi. This excretion route may facilitate further microbial degradation of terpenes in the hindgut. While we did not measure concentrations of phenolics through the gut, various genes associated with degrading aromatic hydrocarbons were enriched in both the foregut and cecum. Phenolics can bind to dietary protein and digestive enzymes, preventing digestion and absorption and thus limiting nutrient availability (Min et al. 2003). Therefore, foregut detoxification of phenolics would be beneficial for avoiding the anti-nutritive effects of PSCs. Again, labeled chemicals to trace sites of absorption, excretion and metabolism would enhance our understanding of the location of these processes.
The location of detoxification within the gut could have important ecological and evolutionary implications for plants and herbivores. For example, when comparing several species of rodents, only those with foregut chambers were able to metabolize significant amounts of oxalate (Shirley and Schmidt-Nielsen 1967). Thus, evolution of this chamber may be important for the ability to consume oxalate-rich plants. Consumption of plant toxins might also drive changes in gut anatomy to enhance detoxification by facilitating a longer duration of residency in various gut chambers. For example, woodrats feeding on phenolic-rich oak exhibit an increase in cecal size (Skopec et al. 2008), which could be driven by increased fiber content of oak or to facilitate the detoxification of PSCs. Interestingly, supplementing diets with isolated phenolic compounds causes an increase in cecal size in lab rats (Zduńczyk, Juśkiewicz and Estrella 2006). Alterations of the size or function of gut chambers in response to toxins could have implications for dietary niche breadth or energetics. In herbivorous birds, there is evidence suggesting that microbial detoxification occurs in both foregut (Garcia-Amado et al. 2007) and hindgut chambers (Kohl et al. 2016a). The guts of birds are under strong selective pressure to be small so as to facilitate flight (Price et al. 2015), and thus sites of microbial detoxification may be especially important in these species. Studies on additional and phylogenetically diverse herbivores and classes of PSCs may help us to better understand the importance of microbial detoxification in the gut, and how the spatial arrangement of these processes contribute to animal morphology, performance, ecology and evolution.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Madelina James and KayLene Yamada for helping with feeding trials and Chelsea Merriman for analysis of monoterpenes.
FUNDING
DEB 1146194 This work was supported by the National Science Foundation [DEB 1210094 to M.D.D. and K.D.K.; DEB 1342615 to M.D.D.; to JSF; and DBI 1400456 to K.D.K].
Conflicts of interest. None declared.
REFERENCES
- Adams AS, Aylward FO, Adams SM et al. Mountain pine beetles colonizing historical and naive trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl Environ Microbiol. 2013;79:3468–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander RM. Optimization of gut structure and diet for higher vertebrate herbivores. Phil Trans R Soc. 1991;333:249–55. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Soc Stat Soc B. 1995;57:289–300. [Google Scholar]
- Berasategui A, Salem H, Paetz C et al. Gut microbiota of the pine weevil degrades conifer diterpenes and increases insect fitness. Mol Ecol. 2017;26:4099–110. [DOI] [PubMed] [Google Scholar]
- Boundy-Mills K, de Souza ML, Mandelbaum RM et al. The atzB gene of Pseudomonas sp. strain ADP encodes the second enzyme of a novel atrazine degradation pathway. Appl Environ Microbiol. 1997;63:916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona M, Zamarro MT, Blázquez B et al. Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol Mol Biol Rev. 2009;73:71–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss M, Frey R, Kiefer B et al. The maximum attainable body size of herbivorous mammals: morphophysiological constraints on foregut, and adaptations of hindgut fermenters. Oecologia. 2003;136:14–27. [DOI] [PubMed] [Google Scholar]
- Dearing MD, Foley WJ, McLean S. The influence of plant secondary metabolites on the nutritional ecology of herbivorous terrestrial vertebrates. Ann Rev Ecol Evol Syst. 2005a;36:169–85. [Google Scholar]
- Dearing MD, McLister JD, Sorensen JS. Woodrat (Neotoma) herbivores maintain nitrogen balance on a low-nitrogen, high-phenolic forage, Juniperus monosperma J Comp Physiol B. 2005b;175:349–55. [DOI] [PubMed] [Google Scholar]
- Derakhshani H, Corley SW, Al Jassim R. Isolation and characterization of mimosine, 3, 4 DHP and 2, 3 DHP degrading bacteria from a commercial rumen inoculum. J Basic Microbiol. 2016;56:580–5. [DOI] [PubMed] [Google Scholar]
- Franzosa EA, Morgan XC, Segata N et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci. 2014;111:E2329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Amado MA, Michelangeli F, Gueneau P et al. Bacterial detoxification of saponins in the crop of the avian foregut fermenter Opisthocomus hoazin. J Anim Feed Sci. 2007;16:82–5. [Google Scholar]
- Harayama S, Timmis KN. Catabolism of aromatic hydrocarbons by Pseudomonas. In: Hopwood DA, Chater KF (eds). Genetics of Bacterial Diversity. London: Academic Press; 1989, 152–75. [Google Scholar]
- Holechek JL, Munshikpu L, Saiwana L et al. Influences of six shrub diets varying in phenol content on intake and nitrogen retention by goats. Trop Grassl. 1990;24:93–8. [Google Scholar]
- Hume ID, Morgan KR, Kenagy GJ. Digesta retention and digestive performance in Sciurid and Microtine rodents: effects of hindgut morphology and body size. Physiol Zool. 1993;66:396–411. [Google Scholar]
- Hume ID, Warner ACI. Evolution of microbial digetion in mammals. In: Ruckebusch Y, Thivend P (eds). Digestive Physiology and Metabolism in Ruminants. Lancaster: MTD Press; 1980, 665–84. [Google Scholar]
- Igimi H, Nishimura M, Kodama R et al. Studies on the metabolism of d-limonene (p-mentha-1,8-diene): I. the absorption, distribution and excretion of d-limonene in rats. Xenobiotica. 1973;4:77–84. [DOI] [PubMed] [Google Scholar]
- Iurescia S, Marconi AM, Tofani D et al. Identification and sequencing of β-myrcene catabolism genes from Pseudomonas sp. strain M1. Appl Environ Microbiol. 1999;65:2871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RJ, Maegarrity RG. Successful transfer of DHP‐degrading bacteria from Hawaiian goats to Australian ruminants to overcome the toxicity of Leucaena. Aust Vet J. 1986;63:259–62. [DOI] [PubMed] [Google Scholar]
- Justice KE. Oxalate digestibility in Neotoma albigula and Neotoma mexicana. Oecologia. 1985;67:231–4. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Kawashima M et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl KD, Connelly JW, Dearing MD et al. Microbial detoxification in the gut of a specialist avian herbivore, the Greater Sage-Grouse. FEMS Microbiol Lett. 2016a;363:fnw144. [DOI] [PubMed] [Google Scholar]
- Kohl KD, Miller AW, Marvin JE et al. Herbivorous rodents (Neotoma spp.) harbour abundant and active foregut microbiota. Environ Microbiol. 2014a;16:2869–78. [DOI] [PubMed] [Google Scholar]
- Kohl KD, Pitman E, Robb BC et al. Monoterpenes as inhibitors of digestive enzymes and counter-adaptations in a specialist avian herbivore. J Comp Physiol B. 2015;185:425–34. [DOI] [PubMed] [Google Scholar]
- Kohl KD, Stengel A, Dearing MD. Inoculation of tannin-degrading bacteria into novel hosts increases performance on tannin-rich diets. Environ Microbiol. 2016b;18:1720–9. [DOI] [PubMed] [Google Scholar]
- Kohl KD, Weiss RB, Cox J et al. Gut microbes of mammalian herbivores facilitate intake of plant toxins. Ecol Lett. 2014b;17:1238–46. [DOI] [PubMed] [Google Scholar]
- Kohl KD, Weiss RB, Dale C et al. Diversity and novelty of the gut microbial community of an herbivorous rodent (Neotoma bryanti). Symbiosis. 2011;54:47. [Google Scholar]
- Langille MGI, Zaneveld J, Caporaso JG et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie RI. Mutualistic fermentative digestion in the gastrointestinal tract: diversity and evolution. Integr Comp Biol. 2014;42:319–26. [DOI] [PubMed] [Google Scholar]
- Meyer F, Paarmann D, D'Souza M et al. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AW, Dale C, Dearing MD. The induction of oxalate metabolism in vivo is more effective with functional microbial communities than with functional microbial species. mSystems. 2017;2:e00088–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AW, Dearing MD. The metabolic and ecological interactions of oxalate-degrading bacteria in the mammalian gut. Pathogens. 2013;2:636–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AW, Kohl KD, Dearing MD. The gastrointestinal tract of the white-throated woodrat (Neotoma albigula) harbors distinct consortia of oxalate-degrading bacteria. Appl Environ Microbiol. 2014;80:1595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AW, Oakeson KF, Dale C et al. Effect of dietary oxalate on the gut microbiota of the mammalian herbivore Neotoma albigula. Appl Environ Microbiol. 2016a;82:2669–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AW, Oakeson KF, Dale C et al. Microbial community transplant results in increased and long-term oxalate degradation. Microb Ecol. 2016b;72:470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min BR, Barry TN, Attwood GT et al. The effect of condensed tannins on the nutrition and health of ruminants fed fresh temperate forages: a review. Anim Feed Sci Technol. 2003;106:3–19. [Google Scholar]
- Muegge BD, Kuczynski J, Knights D et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa R. Tannin-protein complex-degrading enterobacteria isolated from the alimentary tracts of koalas and a selective medium for their enumeration. Appl Environ Microbiol. 1992;58:1754–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CD, Hanson J, Wilkinson JE et al. Microbiome structural and functional interactions across host dietary niche space. Integr Comp Biol. 2017;57:743–55. [DOI] [PubMed] [Google Scholar]
- Price ER, Brun A, Caviedes-Vidal E et al. Digestive adaptations of aerial lifestyles. Physiol. 2015;30:69–78. [DOI] [PubMed] [Google Scholar]
- Rattan RS. Mechanism of action of insecticidal secondary metabolites of plant origin. Crop Prot. 2010;29:913–20. [Google Scholar]
- Schwartz CC, Nagy JG, Regelin WL. Juniper oil yield, terpenoid concentration, and antimicrobial effects on deer. J Wildl Manage. 1980;44:107–13. [Google Scholar]
- Seffernick JL, Shapir N, Schoeb M et al. Enzymatic degradation of chlorodiamino-s-triazine. Appl Environ Microbiol. 2002;68:4672–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley EK, Schmidt-Nielsen K. Oxalate metabolism in the pack rat, sand rat, hamster, and white rat. J Nutr. 1967;91:496–502. [DOI] [PubMed] [Google Scholar]
- Skopec MM, Haley S, Torregrossa A-M et al. An oak (Quercus agrifolia) specialist (Neotoma macrotis) and a sympatric generalist (Neotoma lepida) show similar intakes and digestibilities of oak. Physiol Biochem Zool. 2008;81:426–33. [DOI] [PubMed] [Google Scholar]
- Sorensen JS, Turnbull CA, Dearing MD. A specialist herbivore (Neotoma stephensi) absorbs fewer plant toxins than does a generalist herbivore (Neotoma albigula). Physiol Biochem Zool. 2004;77:139–48. [DOI] [PubMed] [Google Scholar]
- Sundset MA, Barboza PS, Green TK et al. Microbial degradation of usnic acid in the reindeer rumen. Naturwissenschaften. 2010;97:273–8. [DOI] [PubMed] [Google Scholar]
- Vaughan TA. Stephen's woodrat, a dietary specialist. J Mammal. 1982;63:53–62. [Google Scholar]
- Wilke A, Harrison T, Wilkening J et al. The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinformatics. 2012;13:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zduńczyk Z, Juśkiewicz J, Estrella I. Cecal parameters of rats fed diets containing grapefruit polyphenols and inulin as single supplements or in a combination. Nutrition. 2006;22:898–904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.