

Abstract
Several symptomatic treatments for Parkinson's disease (PD) are currently available. Still, the challenge today is to find a therapy that might reduce degeneration and slow down disease progression. The identification of pathogenic mutations in familial Parkinsonism (fPD) associated to the monogenic forms of PD provided pathophysiological insights and highlighted novel targets for therapeutic intervention. Mutations in the VPS35 gene have been associated with autosomal dominant fPD and a clinical phenotype indistinguishable from idiopathic PD. Although VPS35 mutations are relatively rare causes of PD, their study may help understanding specific cellular and molecular alterations that lead to accumulation α-synuclein in neurons of PD patients. Interacting with such mechanisms may provide innovative therapeutic approaches. We review here the evidence on the physiological role of VPS35 as a key intracellular trafficking protein controlling relevant neuronal functions. We further analyze VPS35 activity on α-synuclein degradation pathways that control the equilibrium between α-synuclein clearance and accumulation. Finally, we highlight the strategies for increasing VPS35 levels as a potential tool to treat PD.
Keywords: endosomal trafficking, retromer complex, therapeutic targets, Parkinson's disease, alpha-synucein, amyloid protein A (AA)
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder affecting 1% of people over 65 and 4.3% of those older than 85 (1).
The clinical hallmark of PD is bradykinesia, in combination with rest tremor and rigidity (1). This phase is preceded by a preclinical stage, when neurodegenerative processes have commenced, but there are no evident symptoms or signs, and by a prodromal phase, when symptoms and signs are present, but are yet insufficient to define disease (2). The motor phenomenology is attributed to the selective loss of dopaminergic neurons (DNs) in the substantia nigra pars compacta (SNpc) resulting in degeneration of the nigrostriatal tract and dopamine deficiency (3).
The neuropathological hallmark of PD is the occurrence of intracellular inclusions, Lewy bodies (LBs) and Lewy neurites, mainly consisting of aggregated α-synuclein (4). A remarkably convincing set of data supports the view that accumulation and aggregation of α-synuclein in neurons may represent a key pathogenic event (5–9). Several approaches aim to reduce the accumulation and propagation of α-synuclein toxic aggregates in the brain (10–12) (Figure 1). Ideally, an intervention aimed to reduce α-synuclein should be performed in the preclinical disease phase or—at least—in the prodromal phase, although it may be argued that the early clinical phase may also provide a suitable time window.
Figure 1.

Therapeutic strategies used to reduce α-synuclein toxic aggregates accumulation in neurons. To reduce α-synuclein accumulation in neurons, several therapeutic approaches have been evaluated and tested in murine PD models: (1) antiamyloidogenic agents, such as small molecules or biocomposts to block α-synuclein aggregation and fibrillogenesis; (2) immunotherapy using α-synuclein single-chain antibody able to attenuate neuronal degeneration in vivo; (3) peptide design to block the interaction between α-synuclein oligomer and membrane, able to improve deficits in murine PD models; (4) boosting α-synuclein degradation pathways. CMA, chaperone mediated autophagy; UPS, ubiquitin proteosome system; LAMP2A, lysosome-associated membrane protein 2A.
Monogenic PD subtypes represent a powerful tool for understanding the pathogenesis of PD and for developing treatment strategies. Mutations of the VPS35-retromer subunit have been recently shown to cause PD and to play a key role in neurodegenerative processes, as they control balance between degradation and recycling of fundamental proteins for neuronal survival (13). Moreover, this review outlines the genetic and biochemical evidence that pathogenic missense mutation and depletion of VPS35 affect dopamine (DA) neurons function at different levels. More recently, a direct link has been established between VPS35 and α-synuclein in a rodent PD model where VPS35 deficiency causes α-synuclein accumulation (14). This effect of VPS35 could unravel its key role in controlling macroautophagy, chaperone-mediated autophagy (CMA), and lysosomal function, which are the main degradation pathways of α-synuclein (15–17). A growing body of data converges to establish how VPS35 controls the transport and the localization of protein markers involved in α-synuclein degradation pathways, thus linking VPS35 to α-synuclein accumulation (18–21). An increase in VPS35 levels in PD mice rescues α-synuclein accumulation and induces neuroprotection (14), pointing to a possible role of VPS35 as a therapeutic target for PD. Moreover, VPS35 could represent a diseases-modifying target, and its neuroprotective role could be due to the control on neuronal signaling events (22), synaptic plasticity (23–25), trafficking of protein in dendritic spines (26), membrane dopamine transporter (DAT) recycling in DA neurons (27), and the regulation mitochondrial functions (28).
Pathogenic Role Of VPS35 In PD
Two independent studies with exome sequencing identified point mutations in the VPS35 gene causing an autosomal dominant form of PD (PARK17) (29, 30). The p.Asp620Asn (c.1858G>A) mutation has been proven to be pathogenic with a frequency ~1.3% in familial cases and 0.3% in sporadic PD cases (31, 32). In addition, non-pathogenic VPS35 variants (33) and variant of uncertain significance (34) have been described. The VPS35 gene maps on chromosome 16q11.2 encompasses 29.6 kb and 17 exons (35) (Figure 2).
Figure 2.

VPS35 gene, protein structure and interactions. Retromer cargo recognition complex is formed by vacuolar sorting protein 26 (VPS26; 38 kDa), vacuolar sorting protein 29 (VPS29; 20 kDa), vacuolar sorting protein 35 (VPS35; 92 kDa); the dimer of sinexin (SNXs). The N-terminal region (in blue) from the amino acid residues 1 to 172 [1–172] and C-terminal region (violet) from 307 to 796 (307–796) are important, respectively, for the interaction with VPS26 and VPS29 (36). The amino acid residues involved in the interaction with the SNXs are at N-terminal region (1–35, 37–54) and C-terminal region (307–796). A structural level VPS35 is a right-handed α-helix solenoid, and it is predicted to have 34 helices, 13 of which are in C-terminal. Different missense mutations have been identified in fPD. VPS35 variant of uncertain significance (VUS) are localized between exon 9 (P316S), exon 13 (R542W), and exon 14 (I560T, H599R, M607V), and the confirmed pathogenic mutation (D620N) is localized on the exon 15 (genes in light violet).
The clinical presentation of PARK17 PD patients bearing pathogenic VPS35 gene mutations is that of a typical clinically definite PD. The mean age at disease onset is 50.3 ± 7.3 years; cognitive and psychiatric features do not predominate, and levodopa response is sustained and not associated to severe dyskinesias (37).
To evaluate the pathogenic the role of VPS35 deletion and the VPS35–D620N mutation, different transgenic mouse models have been developed. Knockout mice with the deletion of the endogenous VPS35 at embryonic stage resulted in early lethality under embryonic day 10 (38). VPS35 heterozygote mice (expressing 50% of protein respect to the wild type) survived to adulthood (38) and did not presented any PD-relevant deficit up to 6 months of age. However, the neuropathological evaluation at late stage (12 months) in VPS35 heterozygote mice revealed DA neuronal loss in SNpc and increase in α-syn levels (16). VPS35DAT−CRE mice demonstrated as the depletion of VPS35 protein in SNpc-DA neurons at 2–3 months of age already showed a reduction in TH+ DA neurons and TH+ fibers in the striatum (28). To evaluate the effect of VPS35–D620N mutation, a viral-mediated gene transfer model in adult rat has been developed, showing that the expression of the mutant in the nigrostriatal pathway was sufficient to induce DA neuronal loss and axonal pathology (39). VPS35–D620N knockin (KI) mice (40) at 3 months of age have shown normal motor functions and no loss of striatal DA neurons (41, 42). However, it has been reported that KI mice showed increased capacity to evoke dopamine release in dorsolateral striatum, consistent with elevated extracellular dopamine (42). The evaluation of the effect of VPS35–D620 KI mice on PD pathology with chronic aging provided evidence that the increase in the endogenous D620N mutant levels has a gain of function or partial-negative dominant mechanism (43). Thus, the endogenous expression of VPS35–D620N is sufficient to recapitulate some features of PD pathology inducing (i) the progressive degeneration of the nigrostriatal pathway, (ii) modest motor deficit with age consistence, and (iii) widespread axonal damage (43). In VPS35–D620N KI mice, α-synuclein accumulation and LB pathology were not detected, but tau-positive pathology, characterized by the abnormal accumulation of early “pretangle,” has been described (43). Moreover, VPS35–D620N KI mice or knockdown or knockout of VPS35 mice impacted LRRK2-mediated Rab protein phosphorylation (44) showing that VPS35 could play a major role in controlling LRRKK2 kinase activity and supporting the hypothesis that VPS35–D620N mutation resulted in a gain of function (44).
VPS35 Subunit Of the Retromer Complex
The VPS35 gene encodes for a subunit of the retromer complex that is composed of 796 amino acids with a molecular weight of 92 kDa (45). The retromer is an evolutionary conserved complex of eukaryotic cells that structurally comprises two subcomplexes: (i) a cargo recognition complex VPS26–VPS29–VPS35 heterotrimer mainly involved in the transport of proteins and (ii) a membrane-targeting dimer of sorting nexin, important for the adhesion of the complex to the endosomal membrane (SNX1, SNX2, SNX5, SNX6, and SNX32) (45) (Figure 2). Mammals have two paralogues of VPS26 subunit (VPS26A and VPS26B) that compete for a single binding site on VPS35 (46). Two variants of VPS26A have been identified in a patient with familial PD (46).
The retromer complex represents the master “conductor” of the sorting in the endosomal network; it is localized at the early endosome (EE) and mainly controls the retrograde trafficking of cargo proteins from EE to the trans-Golgi network (TGN) or to the plasma membrane (Figure 3). Moreover, the retromer complex mediates vesicular transport from mitochondria to peroxisomes (47). This complex represents the main sorting hub that receives, dissociates, and sorts cargoes of different origin: (i) plasma membrane (recycling of membrane receptors), (ii) biosynthetic pathways (retrieval of trafficking from Golgi), and (iii) lysosomal pathway (cargoes direct to lysosomes) (48) (Figure 3). Overall, these activities control the homeostasis of transmembrane proteins at plasma membrane and endolysosomal levels and regulate receptor abundance, signaling receptors, adhesion molecules, and hydrolase receptors.
Figure 3.
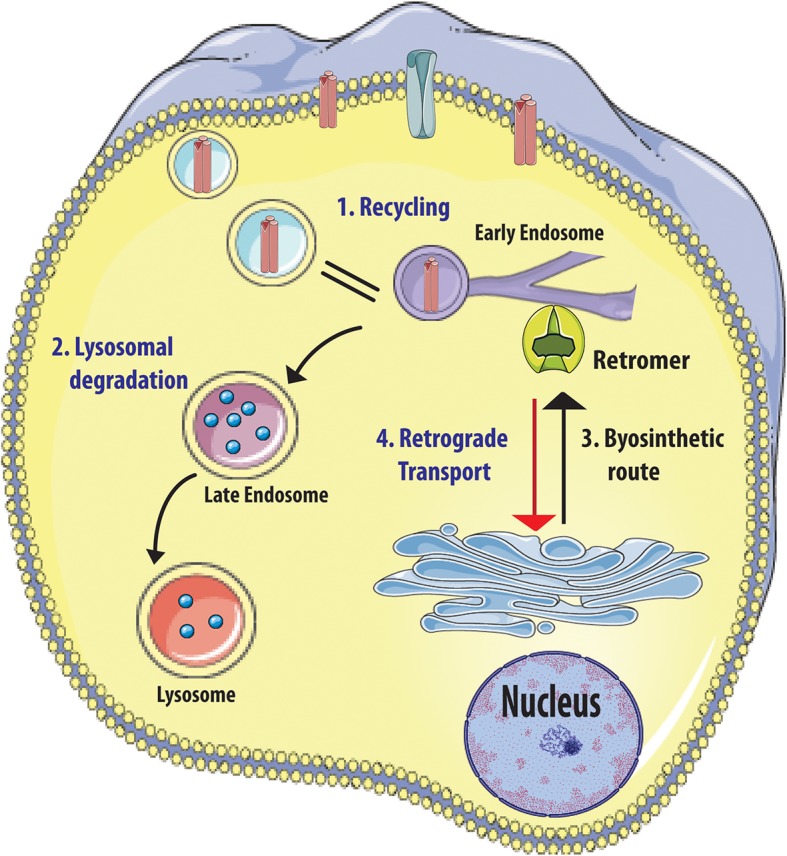
Retromer complex functions. Retromer complex (in green) controls the sorting of cargoes (1) originate from plasma membrane (receptors in red), (2) direct to lysosome for degradation, and (3) trafficking between trans-Golgi Network (TGN) (3, 4). Figure was adapted from Servier Medical Art images (http://smart.servier.com/).
The Retromer Complex Controls Synaptic Function
It has been recently clarified that the retromer complex plays a key role in neurons, where it can, directly or indirectly, mediate essential neuronal functions through cargo selector accessory proteins (SNX27 and WASH complex). Thus, the retromer influences neuronal signaling events (such as downregulation of receptors activity) (22), synaptic plasticity (23–25), trafficking of protein in dendritic spines (26), and membrane DAT recycling in DA neurons (27). The VPS26-retromer subunit belonging to the β-arrestin family controls the downregulation and recycling to cell surface of β2-adrenergic receptor A (22). This is a G-protein-coupled receptor characterized by a cyclical pattern of activation/inactivation through endosomal internalization: the retromer balances the recycling and the degradation of this receptor (49). The retromer could also influence neuronal activity indirectly, controlling function and localization of accessory proteins, such as SNX27 and WASH complex. The SNX27 protein controls recycling of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, involved in long-term potentiation, a process linked to learning and memory (23, 25). The D620N VPS35 pathogenic mutation impairs interaction with the WASH complex (15) and may lead to mistrafficking protein cargoes, such as the GluA1 glutamate receptors subunit, the Glut-1 glucose transporter (26, 49, 50) and in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (51). DAT, which is strictly associated to the activity of DA neurons, undergoes constitutive internalization: its recycling is mediated by the retromer complex (27).
Therefore, retromer disruption via short hairpin RNA-mediated VPS35 knockdown results in DAT depletion from plasma membrane and decrease in DAT recycling (27). In summary, the retromer influences trafficking of signaling receptors and controls rapid signaling events involved in synaptic plasticity and dendritic spine formation that are specifically relevant to DA neuron function (27).
VPS35 Influences α-Synuclein Accumulation
Recent studies highlighted the crucial role of the retromer complex in controlling the accumulation of α-synuclein in PD mouse models and in an in vitro model of α-synuclein spreading (14). Reduction in VPS35 levels or the expression of mutated VPS35 protein in PD mouse hippocampus displayed defective α-synuclein clearance resulting in widespread accumulation of aggregates (14). These data were confirmed in mouse models showing that VPS35 deficiency or a pathogenic VPS35 mutation leads to accumulation and aggregation of α-synuclein in the SN, accompanied by degeneration of DA neurons, reduction in DA levels, impairment of locomotor behavior, and alteration of lysosomal morphology (16). On the contrary, an excess of wild-type VPS35 expression rescues the accumulation of α-synuclein aggregates and leads to the reduction in neuronal loss and astrogliosis in a PD mouse model overexpressing α-synuclein (14). Using an in vitro model of cortical neurons, it has been demonstrated that an increase in wild-type VPS35 levels may counteract the propagation of α-synuclein aggregates from neuron to neuron (14). In keeping with this line of evidence, VPS35 ablation in Drosophila results in the accumulation of α-synuclein in lysosomes (17). Furthermore, silencing of endogenous VPS35 in an α-synuclein transgenic fly does not only promote the accumulation of human wild-type α-synuclein but also causes eye degeneration and motor disability (17). These data suggest that endosomal dysfunction caused by VPS35 deficiency impairs the ability of neurons to cope with the accumulation of α-synuclein, thus facilitating the spread of PD pathology.
How Does VPS35 Control α-Synuclein Accumulation?
The hypothesis that VPS35 could promote α-synuclein clearance is related to the observation of a central role of this retromer protein in controlling interaction and transport of key proteins in α-synuclein degradation pathways.
In normal conditions, the quality control network dealing with α-synuclein accumulation is efficient in promoting its refolding or degradation. In general, to induce α-synuclein degradation, a cell can activate the ubiquitin protein system or alternatively—in the presence of intracytoplasmatic aggregates and LBs—can enhance the endosome-lysosomal degradation system, macroautophagy, or CMA (52) (Figure 4). Recent studies highlighted that VPS35 can act at different levels of the α-synuclein degradation pathways (Figure 5). Particularly, VPS35 acts indirectly on lysosomal activity by sorting receptors of lysosomal hydrolases, including Sortilin and cation independent 6 mannose phosphate receptor (CI-MPR) (18). VPS35 regulates macroautophagy controlling the localization of WASH complex and the trafficking of ATG9a protein (15). Finally, VPS35 manages CMA by mediating the retrieval and transport of LAMP-2A receptor to lysosomes (16) (Figure 5).
Figure 4.
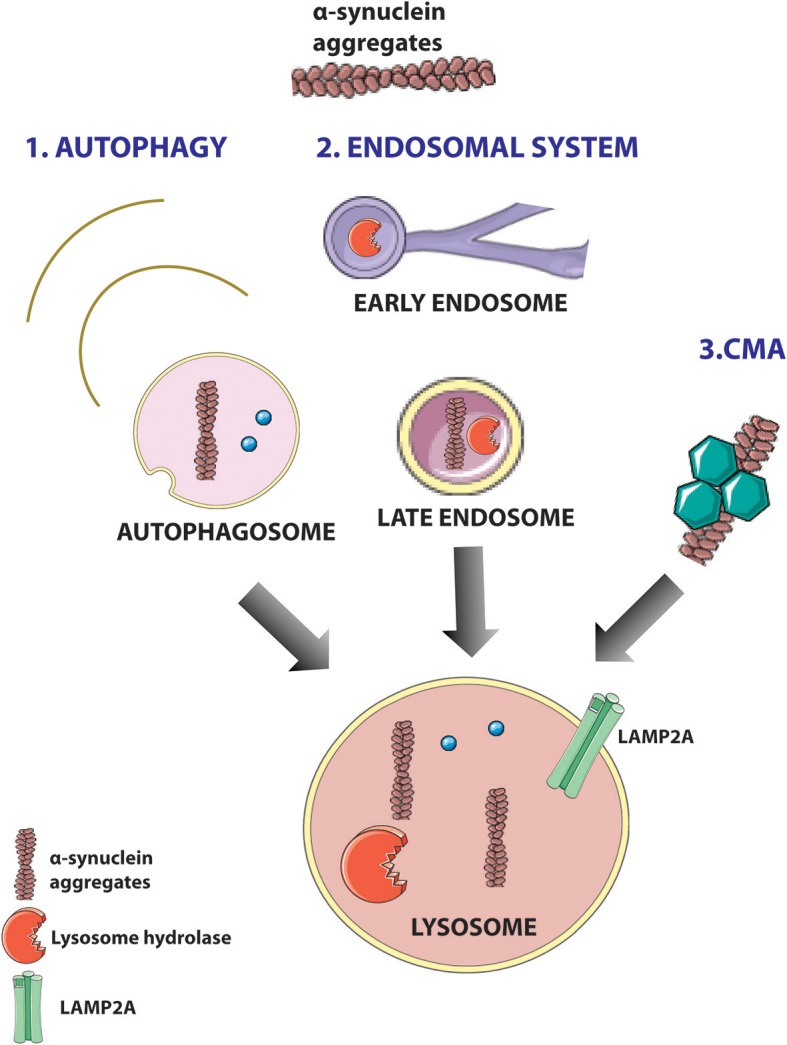
α-Synuclein degradation pathways in cells. Toxic aggregates of α-synuclein can be degraded in the cells through three pathways: macroautophagy (1), endosomal system (2), and chaperone-mediated autophagy (CMA) (3). (1) In the autophagy, key molecules can lead the autophagosome formation and the degradation of aggregates of α-synuclein; (2) lysosomal hydrolases generated in trans-Golgi Network (TGN) are sorted from specific hydrolase sorting receptors [cation-independent 6 mannose phosphate receptor (CI-MPR) and Sortilin], through the endosomal system to the lysosome, and mediate α-synuclein degradation; (3) chaperone-dependent selection of soluble α-synuclein aggregates that are targeted to lysosomes and directly translocated across the lysosomal membrane for degradation. Figure was adapted from Servier Medical Art images (http://smart.servier.com/).
Figure 5.
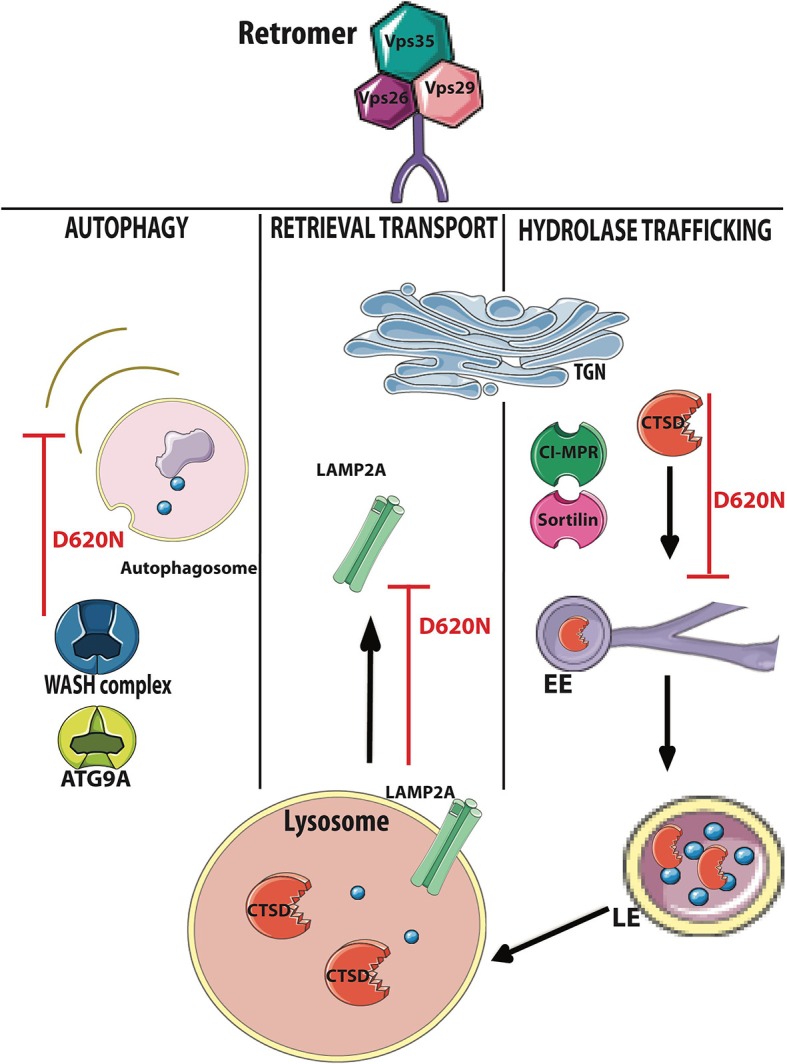
VPS35-retromer subunit controls the main α-synuclein degradation pathways. VPS35 has a relevant role in controlling the degradation of key molecules in the α-synuclein degradation pathways. In the autophagic process, vacuolar sorting protein 35 (VPS35) interacts and controls the trafficking of Wiskott–Aldrich syndrome protein (WASH) complex (blue) and autophagy-related protein 9A (ATG9A; blue) assuring the correct autophagosome formation; in chaperone-mediated autophagy (CMA), VPS35 controls the retrieval trafficking of lysosome-associated membrane protein 2A (LAMP2A) receptor (light green) to the trans-Golgi Network (TGN); VPS35 interacts with CI-MPR (dark green) and Sortilin (pink) leading indirectly to the trafficking of cathepsin D (CTSD, in red) through early endosomes (EE) and late endosomes (LE). VPS35–D620N mutation and VPS35 deficiency lead to dysfunctions in α-synuclein degradation. Figure was adapted from Servier Medical Art images (http://smart.servier.com/).
VPS35 Controls the Lysosomal Degradation Pathway
α-Synuclein enters through macroautophagy, CMA, and endosomal pathway into lysosome to be degraded by lysosomal hydrolases (53). VPS35 carries several cargoes involved in neurodegenerative disorders and more specifically in the clearance of α-synuclein (48). Two well-studied VPS35 cargoes are Sortilin (20, 21) and CI-MPR (18, 19), and VPS35–D620N mutant showed defects in sorting CI-MPR (54). These two cargoes operate as lysosomal hydrolase receptors, sorting acid hydrolases, such as cathepsins, from TGN to lysosomes. Aspartyl protease (cathepsin D) is the main lysosomal endopeptidase responsible for the degradation of α-synuclein (55). However, cysteine proteases (cathepsin B and L) have been shown to be effective in degraded α-synuclein (53). Direct interaction between α-synuclein and lysosomal proteases was shown in mice and human purified lysosomal extracts (53). CTSD-deficient mice showed accumulation of insoluble α-synuclein aggregates in the brain (55). Accordingly, CTSD-mutant brain from mice, sheep, and human showed selective synucleinopathy-like changes as well as α-synuclein accumulation and formation of ubiquitin-positive inclusions (55). Moreover, haploinsufficiency of CTSD resulted in the reduction in lysosomal functions and also acceleration of the propagation of LB pathology (56). Furthermore, recent studies in Drosophila have shown a direct link between VPS35 depletion and defects in CTSD trafficking and maturation (17) (Figure 5).
VPS35 Controls Macroautophagy
Macroautophagy has a profound importance in the clearance of intracytoplasmatic aggregate-prone proteins as well as α-synuclein in PD and huntingtin in Huntington's disease (57). The treatment with rapamycin, which stimulates autophagy, enhanced the clearance of aggregate-prone proteins (57). Dysregulation of the autophagy pathway has been observed in the brain of PD patients and in PD mouse models (58), and the overexpression of α-synuclein caused mislocalization of autophagic proteins and impaired macroautophagy (59).
VPS35 controls the endosomal recruitment of WASH complex, a scaffolding protein localized at the EE (15). WASH complex regulates (i) the autophagic process of the autophagosome development (15), (ii) the formation of actin patch on endosomes to promote protein sorting (60), and (iii) endosome-to-cell surface recycling (61). Recently, it has been demonstrated that retromer interacts directly with FAM21–WASH complex controlling its correct localization (15). VPS35–D620N mutant destabilized this interaction, impairing WASH complex recruitment to endosomes (15, 62). The mislocalization of WASH complex induces defects in autophagosome formation, perturbing protein sorting, and specifically altering ATG9A trafficking (15) (Figure 5). ATG9A is a multipass transmembrane protein that acts early in the autophagic pathway and controls LC3-positive compartment interaction with autophagosome (15). Moreover, parkin PD mutations could impair the ubiquitination of VPS35, inducing the regulation of retromer-dependent sorting (63). Thus, parkin deficiency leads to the reduction in retromer-associated WASH complex cargo in mouse brains (63).
VPS35 Controls CMA
The clearance of misfolded proteins and aggregates by CMA is essential for the normal cellular functions and its efficiency decline with age. Defects in CMA have been detected in PD patient (64). Therefore, the protein levels of CMA markers (LAMP2A and hsc70) were significantly reduced in SNpc and amygdala of PD patients (64). CMA is controlling the degradation of cytosolic proteins (as well as α-synuclein and LRRK2) carrying the recognition motif (KFERQ) (41). CMA presents multiple steps: (i) the recognition of KFERQ motif by HSPA8/HSC70 which target the substrate to lysosomes, (ii) the binding of the substrate to LAMP2A receptor, (iii) formation of substrate–translocation complex with membrane-bound LAMP2A, (iv) substrate degradation by lysosomal enzymes, and (v) disassembly of translocation complex and degradation of multimeric LAMP2A to be recycled (41). Recent studies have been shown that LAMP2A receptor trafficking from endosome to Golgi is altered in mice with reduced VPS35 levels or VPS35–D620N mutations (Figure 5). These mice showed alterations in lysosomal morphology with an increase in LAMP1A, reduction in LAMP2A vesicles, and an a relative increase in α-synuclein accumulation (16) (Figure 5).
Discussion And Outlook
Pathogenic mutations in different PD-related genes with autosomal dominant or autosomal recessive Mendelian inheritance (LRRK2, VPS35, DNAJC13, SYNJ1, TMEM230, RAB39B) are involved in endosomal trafficking, indicating that DA neurons are specifically susceptible to endosomal dysfunctions (36). Neuropathological and neuroanatomical studies have demonstrated that the impairment of cellular trafficking may preferentially affect SNc DA neurons (36, 65). VPS35 mutations identified in PD lead to retromer dysfunctions (29, 30). Recent studies highlighted the interplay between VPS35 and LRRK2, two endosomal proteins associate to late-onset PD, showing a common pathway for the sorting of proteins through TGN and endolysosomal system (66–68). The loss of VPS35 or LRRK2 in Drosophila has been reported to affect synaptic recycling (68). Furthermore, the overexpression of VPS35 or VPS26 retromer subunits ameliorated the pathogenic mutant LRRK2 eye phenotype, protecting from locomotor deficit (67). VPS35–D620N KI mice or knockdown of VPS35 mice impacted LRRK2-mediated Rab protein phosphorylation (44) supporting the hypothesis that VPS35–D620N mutation resulted in a gain of function (44).
Different studies have shown that VPS35 has a key role in controlling α-synuclein accumulation in different PD models (14, 16, 17) even if the development of VPS35–D620N KI mice lack the presence of α-synuclein accumulation (43). Here, we review evidence that the increase in VPS35 levels in neurons represents a potential therapeutic target for the treatment of PD. Boosting VPS35 functions could help DA neurons to degrade α-synuclein toxic aggregates and prevent neuronal loss (Figure 5).
Despite the understanding on VPS35 association to PD pathology, very little is known about the molecular mechanisms involved in the control of α-synuclein accumulation and in a possible neuroprotective role of VPS35 from α-synuclein-induced toxicity. The retromer complex is mainly involved in neurodegenerative disorders through the control of synaptic functions, trafficking, and localization of cargo proteins that are essential for neuronal homeostasis and α-synuclein accumulation vs. clearance. Therefore, VPS35 mutations could cause PD by acting on the α-synuclein degradation pathways (15–17, 69). A more detailed knowledge on the molecular mechanisms by which VPS35 promotes α-synuclein clearance could serve to clarify the role of VPS35-retromer protein in PD and to identify alternative and efficient new potential therapeutic targets downstream to VPS35 action. Besides, validation of new VPS35-based targets could be tested in PD mouse models before being developed in the clinical setting. VPS35 has a wide spectrum of actions within neurons, many of which are potentially relevant for the pathophysiology of PD, particularly synaptic functions (22–24, 26, 48) and control of DAT recycling (27) (Figure 6). Moreover, VPS35 controls mitochondrial function: VPS35 deficiency disrupts the mitophagic process, promoting mitochondrial fragmentation and DA neuron loss (28).
Figure 6.

VPS35 deficiency and dopamine (DA) neuronal loss. VPS35-retromer subunit controls different mechanisms in the cells as well as synaptic and mitochondrial functions other than dopamine transporter (DAT) recycling and α-synuclein degradation. Altogether, the data give an overview about the possible molecular mechanisms controlled by vacuolar protein sorting 35 (VPS35), which could lead to DA neuron degeneration in Parkinson's disease (PD). Figure was adapted from Servier Medical Art images (http://smart.servier.com/).
However, the role of VPS35 could also be played in a more general level on the control of accumulation/degradation of misfolded proteins, hence representing a potential target for different neurodegenerative disorders, such as PD and AD. Indeed, dysregulation of retromer transport is also strongly implicated in AD (70–73). Alterations of retromer activity in AD mouse models leads to pronounced neurodegeneration (deficit in post-synaptic glutamatergic neurotransmission) and deficiency in development of hippocampal-dependent memory (impairment of long-term potentiation) (36, 65) compatible with the role of retromer complex in controlling synaptic functions (22–24, 26). Moreover, different studies addressing VPS35 loss of function reported an elevation of endogenous Aβ peptide levels, in keeping with a VPS35 role in controlling amyloid protein precursor (APP) sorting and processing (28, 70–73).
New research may seek to develop a VPS35-based therapy. A possible therapeutic strategy to boost VPS35 functions could be a drug promoting the stabilization of retromer complex. Earlier studies identified chemical molecules capable of stabilizing the retromer complex by binding the complex in a bind site between VPS35–VPS29 (74). The stabilization of the retromer complex resulted in the reduction in complex degradation and in the increase in VPS35 and retromer subunit levels in cell lines (74). Starting from this observation, it could be possible to identify a lead compound able to combine the effect on the target with the possibility to cross the blood–brain barrier, to be tested in a clinical trial on PD patients. On the other hand, a second disease-modifying approach to boost VPS35 functions could be the delivery of VPS35 gene in the brain by adeno-associated-2-viral vectors (AAV2). The gene therapy approach will allow the introduction of VPS35 gene in the cells that result in the upregulation of VPS35 expression. Recently, it has been demonstrated that AAV2 treatment represents promising human therapeutic applications, with increasing evidence for safety and efficacy (75). First, AAV2 is the most effective in delivery genes in vivo for the presence of the primary adenovirus receptor in human cells that allow easy infection with a high yield of transgene expression. Moreover, currently, there are many clinical trials using AAV2 applications, and doses and routes of administration are already established. Finally, the elucidation of the molecular mechanism used by VPS35 to control neuronal functions, neuroprotection, and α-synuclein clearance could be addressed and may provide a new way to identify new therapeutic strategies in PD and other neurodegenerative disorders where neurotoxic aggregates build up. The other challenges will be to overcome in the translational therapeutic pipeline, including biomarker development, clinical trial strategies, and understanding the potential utility of VPS35 as therapeutic target in different PD backgrounds (sporadic and genetic PD cases).
A clinically related question is when a potentially disease-modifying therapy should be prescribed. Patients who are in the early symptomatic phase are thought to have a significant proportion of viable DA neurons. The diverse functions of VPS35 and its ability to counteract α-synuclein accumulation would justify testing VPS35-based approach from the prodromal until the early symptomatic phase of PD.
Author Contributions
SE and AA conceived the study, wrote, and revised the manuscript. SE generated the figures.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary
Abbreviations
- AD
Alzheimer's disease
- APP
amyloid protein precursor
- ATG9A
autophagy-related protein 9A
- CI-MPR
cation independent 6 mannose phosphate receptor
- CMA
chaperone-mediated autophagy
- CTSD
cathepsin D
- DN
dopaminergic neurons
- DAT
dopamine transporter
- EE
Early endosome
- LAMP2A
Lysosome-associated membrane protein 2A
- PD
Parkinson's disease
- SN
substantia nigra
- TGN
trans-Golgi Network
- WASH
Wiskott–Aldrich syndrome protein
- VPS35
vacuolar sorting protein 35.
Footnotes
Funding. This work was supported by Fondazione Umberto Veronesi (grant to SE) and by Fondazione Grigioni (grant to AA).
References
- 1.Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. (2015) 30:1591–601. 10.1002/mds.26424 [DOI] [PubMed] [Google Scholar]
- 2.Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, et al. MDS research criteria for prodromal Parkinson's disease. Mov Disord. (2015) 30:1600–11. 10.1002/mds.26431 [DOI] [PubMed] [Google Scholar]
- 3.Braak H, Braak E. Pathoanatomy of Parkinson's disease. J Neurol. (2000) 247:3–10. 10.1007/PL00007758 [DOI] [PubMed] [Google Scholar]
- 4.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. (1997) 388:839–40. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- 5.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein : from structure and toxicity to therapeutic target. Nat Rev Neurosci. (2013) 14:38–48. 10.1038/nrn3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. (1998) 18:106–8. 10.1038/ng0298-106 [DOI] [PubMed] [Google Scholar]
- 7.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. α-Synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- 8.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. α-Synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. (2004) 364:1167–9. 10.1016/S0140-6736(04)17103-1 [DOI] [PubMed] [Google Scholar]
- 9.Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, et al. Causal relation between α-synuclein gene duplication and familial Parkinson's disease. Lancet. (2004) 364:1169–71. 10.1016/S0140-6736(04)17104-3 [DOI] [PubMed] [Google Scholar]
- 10.Wrasidlo W, Tsigelny IF, Price DL, Dutta G, Rockenstein E, Schwarz TC, et al. A de novo compound targeting α-synuclein improves deficits in models of Parkinson's disease. Brain. (2016) 139:3217–36. 10.1093/brain/aww238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spencer B, Emadi S, Desplats P, Eleuteri S, Michael S, Kosberg K, et al. ESCRT-mediated uptake and degradation of brain-targeted α-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol Ther. (2014) 22:1753–67. 10.1038/mt.2014.129 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Di Giovanni S, Eleuteri S, Paleologou KE, Yin G, Zweckstetter M, Carrupt PA, et al. Entacapone and tolcapone, two catechol O-methyltransferase inhibitors, block fibril formation of α-synuclein and β-amyloid and protect against amyloid-induced toxicity. J Biol Chem. (2010) 285:14941–54. 10.1074/jbc.M109.080390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brodin L, Shupliakov O. Retromer in synaptic function and pathology. Front Synaptic Neurosci. (2018) 10:37. 10.3389/fnsyn.2018.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhungel N, Eleuteri S, Li LB, Kramer NJ, Chartron JW, Spencer B, et al. Parkinson's disease genes VPS35 and EIF4G1 interact genetically and converge on α-synuclein. Neuron. (2015) 85:76–87. 10.1016/j.neuron.2014.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, et al. Mutation in VPS35 associated with Parkinson's disease impairs WASH complex association and inhibits autophagy. Nat Commun. (2014) 5:1–16. 10.1038/ncomms4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, et al. VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of Parkinson's disease. J Neurosci. (2015) 35:10613–28. 10.1523/JNEUROSCI.0042-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miura E, Hasegawa T, Konno M, Suzuki M, Sugeno N, Fujikake N, et al. VPS35 dysfunction impairs lysosomal degradation of α-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson's disease. Neurobiol Dis. (2014) 71:1–13. 10.1016/j.nbd.2014.07.014 [DOI] [PubMed] [Google Scholar]
- 18.Arighi CN, Hartnell LM, Aguilar RC, Haft CR, Bonifacino JS. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J Cell Biol. (2004) 165:123–33. 10.1083/jcb.200312055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harbour ME, Breusegem SY, Antrobus R, Freeman C, Reid E, Seaman MN. The cargo-selective retromer complex is a recruiting hub for protein complexes that regulate endosomal tubule dynamics. J Cell Sci. (2010) 123:3703–17. 10.1242/jcs.071472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen MS, Madsen P, Christensen EI, Nykjaer A, Gliemann J, Kasper D, et al. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. (2001) 20:2180–90. 10.1093/emboj/20.9.2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim E, Lee Y, Lee HJ, Kim JS, Song BS, Huh JW, et al. Implication of mouse Vps26b-Vps29-Vps35 retromer complex in sortilin trafficking. Biochem Biophys Res Commun. (2010) 403:167–71. 10.1016/j.bbrc.2010.10.121 [DOI] [PubMed] [Google Scholar]
- 22.Temkin P, Lauffer B, Jäger S, Cimermancic P, Krogan NJ, von Zastrow M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat Cell Biol. (2011) 13:715–23. 10.1038/ncb2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Zhao Y, Zhang X, Badie H, Zhou Y, Mu Y, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat Med. (2013) 19:473–80. 10.1038/nm.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loo LS, Tang N, Al-Haddawi M, Stewart Dawe G, Hong W. A role for sorting nexin 27 in AMPA receptor trafficking. Nat. Commun. 5, (2014). 10.1038/ncomms4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussain NK, Diering GH, Sole J, Anggono V, Huganir RL. Sorting Nexin 27 regulates basal and activity-dependent trafficking of AMPARs. Proc Natl Acad Sci USA. (2014) 111:11840–5. 10.1073/pnas.1412415111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian Y, Tang FL, Sun X, Wen L, Mei L, Tang BS, et al. VPS35-deficiency results in an impaired AMPA receptor trafficking and decreased dendritic spine maturation. Mol Brain. (2015) 8:70. 10.1186/s13041-015-0156-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu S, Fagan RR, Uttamapinant C, Lifshitz LM, Fogarty KE, Ting AY, et al. The dopamine transporter recycles via a retromer-dependent postendocytic mechanism: tracking studies using a novel fluorophore-coupling approach. J Neurosci. (2017) 37:9438–52. 10.1523/JNEUROSCI.3885-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L, et al. VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. (2015) 12:1631–43. 10.1016/j.celrep.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in parkinson disease. Am J Hum Genet. (2011) 89:162–7. 10.1016/j.ajhg.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset parkinson disease. Am J Hum Genet. (2011) 89:168–75. 10.1016/j.ajhg.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guella I, Soldà G, Cilia R, Pezzoli G, Asselta R, Duga S, et al. The Asp620asn mutation in VPS35 is not a common cause of familial Parkinson's disease. Mov Disord. (2012) 27:800–1. 10.1002/mds.24927 [DOI] [PubMed] [Google Scholar]
- 32.Kumar KR, Weissbach A, Heldmann M, Kasten M, Tunc S, Sue CM, et al. Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch Neurol. (2012) 69:1360–4. 10.1001/archneurol.2011.3367 [DOI] [PubMed] [Google Scholar]
- 33.Sharma M, Ioannidis JP, Aasly JO, Annesi G, Brice A, Bertram L, et al. A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet. (2012) 49:721–6. 10.1136/jmedgenet-2012-101155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verstraeten A, Wauters E, Crosiers D, Meeus B, Corsmit E, Elinck E, et al. Contribution of VPS35 genetic variability to LBD in the Flanders-Belgian population. Neurobiol Aging. (2012) 33:1844.e11–3. 10.1016/j.neurobiolaging.2012.01.006 [DOI] [PubMed] [Google Scholar]
- 35.Deng H, Gao K, Jankovic J. The VPS35 gene and Parkinson's disease. Mov Disord. (2013) 28:569–75. 10.1002/mds.25430 [DOI] [PubMed] [Google Scholar]
- 36.Hunn BHM, Cragg SJ, Bolam JP, Spillantini MG, Wade-Martins R. Impaired intracellular trafficking defines early Parkinson's disease. Trends Neurosci. (2015) 38:178–88. 10.1016/j.tins.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheerin UM, Charlesworth G, Bras J, Guerreiro R, Bhatia K, Foltynie T, et al. Screening for VPS35 mutations in Parkinson's disease. Neurobiol Aging. (2012) 33:838.e1–5. 10.1016/j.neurobiolaging.2011.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wen L, Tang FL, Hong Y, Luo SW, Wang CL, He W, et al. VPS35 haploinsufficiency increases Alzheimer's disease neuropathology. J Cell Biol. (2011) 195:765–79. 10.1083/jcb.201105109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsika E, Glauser L, Moser R, Fiser A, Daniel G, Sheerin UM, et al. Parkinson's disease-linked mutations in VPS35 induce dopaminergic neurodegeneration. Hum Mol Genet. (2014) 23:4621–38. 10.1093/hmg/ddu178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishizu N, Yui D, Hebisawa A, Aizawa H, Cui W, Fujita Y, et al. Impaired striatal dopamine release in homozygous Vps35 D620N knock-in mice. Hum Mol Genet. (2016) 25:4507–17. 10.1093/hmg/ddw279 [DOI] [PubMed] [Google Scholar]
- 41.Ho PW, Leung CT, Liu H, Pang SY, Lam CS, Xian J, et al. Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy. (2019) 14:1–24. 10.1080/15548627.2019.1603545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cataldi S, Follett J, Fox JD, Tatarnikov I, Kadgien C, Gustavsson EK, et al. Altered dopamine release and monoamine transporters in Vps35 p.D620N knock-in mice. NPJ Parkinsons Dis. (2018) 4:27. 10.1038/s41531-018-0063-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X, Kordich JK, Williams ET, Levine N, Cole-Strauss A, Marshall L, et al. Parkinson's disease-linked D620N VPS35 knockin mice manifest tau neuropathology and dopaminergic neurodegeneration. Proc Natl Acad Sci USA. (2019) 116:5765–74. 10.1073/pnas.1814909116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mir R, Tonelli F, Lis P, Macartney T, Polinski NK, Martinez TN, et al. The Parkinson's disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J. (2018) 475:1861–83. 10.1042/BCJ20180248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seaman MNJ. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. (2004) 165:111–22. 10.1083/jcb.200312034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shannon B, Soto-Ortolaza A, Rayaprolu S, Cannon HD, Labbé C, Benitez BA, et al. Genetic variation of the retromer subunits VPS26A/B-VPS29 in Parkinson's disease. Neurobiol Aging. (2014) 35:1958.e1–2. 10.1016/j.neurobiolaging.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol. (2010) 20:1310–5. 10.1016/j.cub.2010.05.066 [DOI] [PubMed] [Google Scholar]
- 48.Wang S, Bellen HJ. The retromer complex in development and disease. Development. (2015) 142:2392–6. 10.1242/dev.123737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGarvey JC, Xiao K, Bowman SL, Mamonova T, Zhang Q, Bisello A, et al. Actin-sorting nexin 27 (SNX27)-retromer complex mediates rapid parathyroid hormone receptor recycling. J Biol Chem. (2016) 291:10986–1002. 10.1074/jbc.M115.697045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seaman MNJ. Retromer and its role in regulating signaling at endosomes. Prog Mol Subcell Biol. (2018) 57:137–49. 10.1007/978-3-319-96704-2_5 [DOI] [PubMed] [Google Scholar]
- 51.Munsie LN, Milnerwood AJ, Seibler P, Beccano-Kelly DA, Tatarnikov I, Khinda J, et al. Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson's disease VPS35 mutation p.D620N. Hum Mol Genet. (2015) 24:1691–703. 10.1093/hmg/ddu582 [DOI] [PubMed] [Google Scholar]
- 52.Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. (2015) 47:e147. 10.1038/emm.2014.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGlinchey RP, Lee JC. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci USA. (2015) 112:9322–7. 10.1073/pnas.1500937112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, et al. The Vps35 D620N mutation linked to Parkinson's disease disrupts the cargo sorting function of retromer. Traffic. (2014) 15:230–44. 10.1111/tra.12136 [DOI] [PubMed] [Google Scholar]
- 55.Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. (2009) 2:5. 10.1186/1756-6606-2-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bae EJ, Yang NY, Lee C, Kim S, Lee HJ, Lee SJ. Haploinsufficiency of cathepsin D leads to lysosomal dysfunction and promotes cell-to-cell transmission of α-synuclein aggregates. Cell Death Dis. (2015) 6:e1901. 10.1038/cddis.2015.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ravikumar B. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. (2002) 11:1107–17. 10.1093/hmg/11.9.1107 [DOI] [PubMed] [Google Scholar]
- 58.Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:a009357. 10.1101/cshperspect.a009357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. α-Synuclein impairs macroautophagy: Implications for Parkinson's disease. J Cell Biol. (2010) 190:1023–37. 10.1083/jcb.201003122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gomez TS, Billadeau DD. A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev Cell. (2009) 17:699–711. 10.1016/j.devcel.2009.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Piotrowski JT, Gomez TS, Schoon RA, Mangalam AK, Billadeau DD. WASH knockout T cells demonstrate defective receptor trafficking, proliferation, and effector function. Mol Cell Biol. (2013) 33:958–73. 10.1128/MCB.01288-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGough IJ, Steinberg F, Jia D, Barbuti PA, McMillan KJ, Heesom KJ, et al. Erratum: retromer binding to FAM21 and the WASH complex is perturbed by the parkinson disease-linked VPS35(D620N) mutation. Curr Biol. (2014) 24:1678 10.1016/j.cub.2014.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williams ET, Glauser L, Tsika E, Jiang H, Islam S, Moore DJ. Parkin mediates the ubiquitination of VPS35 and modulates retromer-dependent endosomal sorting. Hum Mol Genet. (2018) 27:3189–205. 10.1093/hmg/ddy224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. (2010) 67:1464–72. 10.1001/archneurol.2010.198 [DOI] [PubMed] [Google Scholar]
- 65.Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. (2012) 27:1478–83. 10.1002/mds.25135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. (2013) 77:425–39. 10.1016/j.neuron.2012.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Linhart R, Wong SA, Cao J, Tran M, Huynh A, Ardrey C, et al. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson's disease mutant of Leucine-rich repeat kinase 2 (LRRK2). Mol Neurodegener. (2014) 9:1–10. 10.1186/1750-1326-9-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Inoshita T, Arano T, Hosaka Y, Meng H, Umezaki Y, Kosugi S, et al. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum Mol Genet. (2017) 26:2933–48. 10.1093/hmg/ddx179 [DOI] [PubMed] [Google Scholar]
- 69.Zavodszky E, Seaman MNJ, Rubinsztein DC. VPS35 Parkinson mutation impairs autophagy via WASH. Cell Cycle. (2014) 13:2155–6. 10.4161/cc.29734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muhammad A, Flores I, Zhang H, Yu R, Staniszewski A, Planel E, et al. Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Aβ accumulation. Proc Natl Acad Sci USA. (2008) 105:7327–32. 10.1073/pnas.0802545105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sullivan CP, Jay AG, Stack EC, Pakaluk M, Wadlinger E, Fine RE, et al. Retromer disruption promotes amyloidogenic APP processing. Neurobiol Dis. (2011) 43:338–45. 10.1016/j.nbd.2011.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fjorback AW, Seaman M, Gustafsen C, Mehmedbasic A, Gokool S, Wu C, et al. Retromer binds the FANSHY sorting motif in sorLA to regulate amyloid precursor protein sorting and processing. J Neurosci. (2012) 32:1467–80. 10.1523/JNEUROSCI.2272-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang CL, Tang FL, Peng Y, Shen CY, Mei L, Xiong WC. VPS35 regulates developing mouse hippocampal neuronal morphogenesis by promoting retrograde trafficking of BACE1. Biol Open. (2012) 1:1248–57. 10.1242/bio.20122451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, et al. Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol. (2014) 10:443–9. 10.1038/nchembio.1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niethammer M, Tang CC, LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, et al. Long-term follow-up of a randomized AAV2-GAD gene therapy trial for Parkinson's disease. JCI Insight. (2017) 2:e90133. 10.1172/jci.insight.90133 [DOI] [PMC free article] [PubMed] [Google Scholar]