

Abstract
Serine hydroxymethyltransferases (SHMTs) reversibly transform serine into glycine in a reaction accompanied with conversion of tetrahydrofolate (THF) into 5,10-methylene-THF (5,10-meTHF). In vivo, 5,10-meTHF is the main carrier of one-carbon (1C) units, which are utilized for nucleotide biosynthesis and other processes crucial for every living cell, but hyperactivated in overproliferating cells (e.g. cancer tissues). SHMTs are emerging as a promising target for development of new drugs because it appears possible to inhibit growth of cancer cells by cutting off the supply of 5,10-meTHF. Methotrexate (MTX) and pemetrexed (PTX) are two examples of antifolates that have cured many patients over the years but target different enzymes from the folate cycle (mainly dihydrofolate reductase and thymidylate synthase, respectively). Here we show crystal structures of MTX and PTX bound to plant SHMT isozymes from cytosol and mitochondria—human isozymes exist in the same subcellular compartments. We verify inhibition of the studied isozymes by a thorough kinetic analysis. We propose to further exploit antifolate scaffold in development of SHMT inhibitors because it seems likely that especially polyglutamylated PTX inhibits SHMTs in vivo. Structure-based optimization is expected to yield novel antifolates that could potentially be used as chemotherapeutics.
Subject terms: X-ray crystallography, Transferases
Introduction
Serine hydroxymethyltransferase (SHMT) reversibly interconverts serine and tetrahydrofolate (THF, Fig. 1) into glycine and 5,10-methylene-THF (5,10-meTHF)1,2. As a part of one-carbon (1C) metabolism, SHMTs fuel the folate cycle by providing 5,10-meTHF3. The folate cycle is responsible for biotransformation of folate species: THF, 5,10-meTHF, 5-methyl-THF, and 10-formyl-THF, with each of them playing distinct and crucial cellular roles. Intracellular membranes appear impermeable to 1C-loaded THF species—hence the need to generate 5,10-meTHF separately in subcellular compartments4. In mammals, SHMT isoforms operate in cytosol (e.g. human cytosolic SHMT (hcSHMT or SHMT1); UniProt ID: P34896) and mitochondria (human mitochondrial SHMT (hmSHMT or SHMT2); UniProt ID: P34897). Moreover, direction of the SHMT-catalyzed reaction is controlled independently in organelles via supply and demand of 1C units5. Therefore, depending on the metabolic status on cellular and subcellular levels, serine can be either catabolized or anabolized by SHMTs with simultaneous production or degradation of 5,10-meTHF5. Interestingly, plant genomes encode additional isozymes that localize in chloroplasts and nuclei6,7. It has been shown that in photorespiration the flux of serine-to-glycine interconversion is shifted towards serine synthesis that is thermodynamically less favored8,9. Increased activity of the glycine cleavage system (GCS) in the mitochondrial matrix results in high 5,10-meTHF/THF ratio10–12. This way, the SHMT-catalyzed reaction is one of the three pathways used by plants for serine biosynthesis13.
Figure 1.
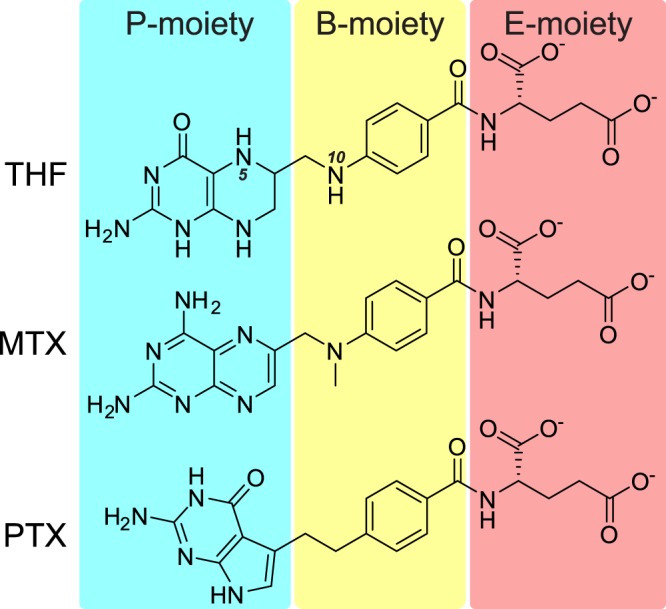
Structural formulas of tetrahydrofolate (THF), methotrexate (MTX), and pemetrexed (PTX). The compounds are divided into P-, B-, and E-moieties, as referenced throughout the text.
In general, 1C-related enzymes have been actively studied for many years, mainly because 1C units are utilized in crucial biosynthetic pathways, such as purine and thymidine biosynthesis, regeneration of S-adenosyl methionine, etc. These processes are vital to all living cells but in overproliferating cells (e.g. cancer tissues) operate at enhanced rates14,15. Therefore, by inhibiting SHMTs, it appears possible to limit the supply of 1C units, which are in highest demand by cancer cells. Such approach would impact not only the aforementioned biosynthetic pathways, but through them, would also influence epigenetic maintenance, amino acid homeostasis, and redox defense.
Methotrexate (MTX) (Fig. 1), a methylated derivative of aminopterin, was the first selective chemotherapeutic targeting 1C metabolism. Since its introduction in 195016, the use of MTX has gradually increased and now it is used to cure millions of cancer patients annually. MTX treatment, as a single agent or as a part of multidrug chemotherapy, is effective against leukemia, non-Hodgkin lymphomas—particularly with central nervous system involvement—and a number of other cancer types17. As a chemotherapeutic MTX is used in high doses over a short period of time and its efficacy depends on the duration of infusion and on intracellular drug concentration. Due to its very poor ability to cross the blood-brain barrier, MTX is administered at remarkably high doses to obtain therapeutic concentrations in the tumor tissue and in lymphoma-surrounding neural tissue18. Of course, such toxic levels of the drug also impact non-cancer cells. Therefore, MTX treatment is followed by folate supplementation to the patient until blood concentration of MTX becomes sufficiently low. Additionally, to limit systemic exposure, intra-arterial, intrathecal or intraventricular delivery is used to reach higher concentration of the drug in the central nervous system19. In low doses, MTX is generally well tolerated, and prolonged low-dose therapy is given to patients suffering from autoimmune diseases, such as lupus, psoriasis, sarcoidosis, and rheumatoid arthritis20.
Pemetrexed (PTX) is a relatively new drug, introduced in early 2000s21,22. From the chemical standpoint, PTX (unlike MTX) lacks the pteridine ring (Fig. 1). Non-small cell lung cancer and pleural mesothelioma are the only approved indications for PTX usage; however, trials are currently under way for testing it against other cancers (e.g. esophageal squamous cell carcinoma)23. Similarly to the MTX therapy, patients taking PTX are usually also supported with folate.
MTX and PTX inhibit folate-related enzymes. Specifically, MTX is most potent against dihydrofolate reductase (DHFR), whereas PTX targets mostly thymidylate synthase (TS) but also 5-aminoimidazole-4-carboxamide ribonucleotide transformylase (AICART), and glycinamide ribonucleotide formyltransferase (GARFT)24,25. However, there have been implications that MTX and PTX inhibit SHMTs as well26. This is not surprising, considering that MTX and PTX are chemically similar to THF, although they are not able to accept 1C units. However, because of chemical differences, it was rather obvious that binding of MTX, PTX, and THF by SHMTs should be different. Since SHMT enzymes are promising targets for drug design the need for such structures is quite apparent, as emphasized by an attempt to model the interactions by in silico docking26. Importantly, computational drug design methods, which minimize the cost of development and increase the chance of finding a good candidate, are most successful if a crystal structure is known27. After completion of experiments for this article, structures of hmSHMT in complexes with PTX and lometrexol showed first structural insights into SHMT interactions with antifolates28. However, it has been impossible to obtain structures of MTX bound to human SHMT isoforms or any structure showing a cytosolic isoform complexed with antifolate. Our recent work on plant SHMTs7 inspired us to use plant models, as we found that two Arabidopsis thaliana isoforms produce crystals diffracting to much higher resolution than the human isozymes. The two A. thaliana enzymes: mitochondrial AtSHMT2 (UniProt ID: Q94C74) and cytosolic AtSHMT4 (UniProt ID: O23254) are counterparts of hmSHMT and hcSHMT, respectively (of corresponding subcellular localizations). Amino acid sequence identities between corresponding plant and human isoforms are 60%, whereas hmSHMT and hcSHMT share 66% identity, and AtSHMT2 and AtSHMT4 share 56% identity (excluding mitochondrial target peptides).
In this work, we have performed functional in vitro studies using purified isozymes to investigate whether the behavior of cytosolic AtSHMT4 resembles that of hcSHMT, and if mitochondrial AtSHMT2 is similar to hmSHMT. Next, we have obtained high resolution crystal structures of AtSHMT2 complexed with MTX and PTX, as well as of AtSHMT4-MTX complex. Detailed analysis of the interacting residues and the environment in and near the binding cavities allowed us to indicate key structural elements responsible for antifolate binding. We have found features that are either similar or vary between MTX and PTX complexes. Comparisons of amino acid sequences and structures of the binding sites show that our results very likely apply to the human SHMTs as well. In our view, these observations should stimulate design of SHMT-targeted antifolates. Quoting Sir James Whyte Black: “the most fruitful basis for the discovery of new drug is to start with an old drug”29,30. We hope to utilize MTX and PTX as starting points for the development of novel chemotherapeutics targeted at SHMTs. Such an attempt will benefit from the fact that starting molecules are already approved drugs and have desired or acceptable ADME-Tox properties (absorption, distribution, metabolism, excretion, and toxicity). Therefore, it is expected that agents based on MTX and/or PTX but targeted at SHMT will become valuable.
Results
Dependence of AtSHMT2 and AtSHMT4 activity on pH is similar to that of their human counterparts of corresponding subcellular localizations
Measurements of the initial velocity of the SHMT forward reaction (serine + THF → glycine + 5,10-meTHF) were carried out with AtSHMT2 and AtSHMT4 at different pH values, keeping one substrate at fixed concentration while varying the concentration of the other substrate (Fig. 2). As with all other characterized SHMTs isolated from different sources31, a clear THF substrate inhibition is visible (Table 1). The behavior of A. thaliana SHMTs is similar to that of human isoforms. The THF substrate inhibition constant (Ki) with human and plant mitochondrial SHMTs (hmSHMT and AtSHMT2, respectively) stays more or less constant as pH is varied, whereas this parameter is greatly increased as pH increases with human and plant cytosolic isoforms (hcSHMT and AtSHMT4). Moreover, with both AtSHMT2 and AtSHMT4, KM for L-serine decreases as pH is increased (Table 1). This characteristic is in common with human SHMT isoforms31. Dependence of KM for THF is also similar between human and A. thaliana SHMT isoforms: this parameter stays constant with the mitochondrial isoforms and increases as pH is increased with the cytosolic isoforms. With both A. thaliana isoforms, kcat increases as pH is increased. In this respect, AtSHMT4 is different from the human cytosolic isoform that shows an opposite behavior32. This discrepancy in behavior might be due to the different experimental conditions used to characterize the enzymes, which include buffering compounds and salts that impact ionic strength. AtSHMT2 is also somewhat different from hmSHMT, for which kcat stays constant between pH 6.9 and 7.8 pH and increases when pH is increased to 8.431. Unfortunately, based on current data we cannot provide rational explanations of such differences between human and Arabidopsis SHMTs.
Figure 2.

Dependence of AtSHMT2 and AtSHMT4 activity on pH. The initial velocity of the SHMT forward reaction was measured at different pH values with AtSHMT2 and AtSHMT4 (0.2 μM) using a fixed concentration of one substrate while varying the concentration of the other substrate. The fixed L-serine concentration was saturating. THF fixed concentration was giving the maximum activity when varying L-serine. All experimental points are the average ± standard deviation of three independent measurements. Panel (A) AtSHMT2, 30 mM L-serine; Panel (B) AtSHMT2, 180 µM THF; Panel (C) AtSHMT4, 30 mM L-serine; Panel (D) AtSHMT4, 30 µM THF at pH 6.5 and 7.0, 50 µM THF at pH 8.0, 80 µM THF at pH 8.5 and 100 µM THF at pH 9.5. Reactions were carried out at 30 °C.
Table 1.
Steady-state kinetic parameters of the SHMT forward reaction with AtSHMT2 and AtSHMT4.
| pH | KM | kcat (s−1) | kcat/KM | KiTHF (μM) | |||
|---|---|---|---|---|---|---|---|
| L-Ser (mM) | THF (μM) | L-Ser | THF | ||||
| AtSHMT2 | 6.5 | 11.65 ± 2.03 | 83.6 ± 8.1 | 5.3 ± 0.4 | 0.45 | 0.06 | 189.4 ± 12.8 |
| 7 | 8.05 ± 1.36 | 153.6 ± 10.9 | 8.1 ± 0.7 | 1.01 | 0.05 | 191.3 ± 9.5 | |
| 8 | 2.86 ± 0.30 | 143.9 ± 12.6 | 13.0 ± 0.7 | 4.53 | 0.09 | 186.4 ± 11.7 | |
| 8.5 | 1.78 ± 0.22 | 196.1 ± 14.6 | 20.6 ± 1.3 | 11.57 | 0.11 | 151.8 ± 8.7 | |
| 9.5 | 0.71 ± 0.09 | 192.4 ± 10.4 | 21.8 ± 2.3 | 30.73 | 0.11 | 127.4 ± 5.5 | |
| AtSHMT4 | 6.5 | 2.56 ± 0.26 | 7.87 ± 1.62 | 11.9 ± 1.3 | 4.63 | 1.51 | 48.13 ± 9.69 |
| 7 | 0.54 ± 0.05 | 14.97 ± 1.87 | 16.2 ± 1.1 | 29.97 | 1.08 | 57.49 ± 6.56 | |
| 8 | 0.30 ± 0.02 | 23.83 ± 3.99 | 17.4 ± 1.7 | 57.89 | 0.73 | 141.0 ± 23.23 | |
| 8.5 | 0.24 ± 0.02 | 33.75 ± 5.63 | 19.6 ± 1.9 | 81.53 | 0.58 | 163.3 ± 25.96 | |
| 9.5 | 0.20 ± 0.02 | 49.24 ± 6.62 | 18.2 ± 1.4 | 90.75 | 0.37 | 288.1 ± 39.18 | |
Methotrexate and pemetrexed inhibit mitochondrial AtSHMT2 and cytosolic AtSHMT4
Because of the specific activity assay used to characterize the inhibition by PTX and MTX, we needed a full characterization of the equilibrium binding between SHMT, glycine and 5-formyl-THF. This was carried out by measuring the absorbance change at around 500 nm observed when SHMT binds both glycine and the folate ligand forming a quinonoid intermediate. From a series of experiments, in which 5-formyl-THF was varied as glycine was kept fixed at different concentrations (Fig. S1), Kd and αKd were determined, which are respectively the dissociation constant of one ligand in the absence of the other and the dissociation constant of one ligand in the presence of a saturating concentration of the other ligand. We noticed that the formation of the quinonoid with AtSHMTs was much slower than with the human SHMT isoforms (data not shown).
MTX and PTX inhibition was analyzed in competitive binding assays, in which 5-formyl-THF and the antifolate compete for binding to SHMT. In this assay, the absorbance at 500 nm of the quinonoid is measured at variable 5-formyl-THF concentrations and glycine is kept at a fixed, saturating concentration (10 mM) while antifolate is fixed at different concentrations (Figs. 3, S2). In this system, the obtained Ki values refer to binding of the antifolates to the enzyme-glycine complex. The Ki values obtained for human and plant SHMTs are compared in Table 2.
Figure 3.

AtSHMT2 and AtSHMT4 inhibition by MTX and PTX. Panel (A) shows data for AtSHMT2 inhibition by MTX; (B) AtSHMT4 by MTX; C, AtSHMT2 by PTX; D AtSHMT4 by PTX. Secondary plots of slopes as functions of antifolate concentrations were obtained from fitting of inhibition data (Fig. S2). Intercept on the X-axis gives an estimate of the inhibition constant (Ki) related to antifolate binding to the enzyme-glycine complex. Since it is known that the mitochondrial matrix has an alkaline pH (pH 868) as compared to the cytosol (pH 7.368), inhibition measurements were performed in 20 mM KPi buffer at pH 8.0 for AtSHMT2 and pH 7.3 for AtSHMT4 at 30 °C. Measurements were carried out varying 5-formyl-THF concentration, while keeping glycine at 10 mM and antifolates at different fixed concentrations. Units of slopes are μM.
Table 2.
Methotrexate and pemetrexed Inhibition constant values obtained with human cytosolic (hcSHMT) and plant SHMT isoforms.
| SHMT isoform | KiMTX (µM) | KiPTX (µM) |
|---|---|---|
| hcSHMT | a201.0 ± 19 | b19.1 ± 3.1 |
| AtSHMT4 | 179.3 ± 9.3 | 95.1 ± 9.3 |
| AtSHMT2 | 270.2 ± 62.5 | 119.4 ± 13.2 |
Methotrexate binding mode
From structures obtained by soaking AtSHMT2 and AtSHMT4 crystals with MTX we learn that the antifolate can bind in three distinct ways. At this point it is also worth to mention that each pose of antifolate (MTX and PTX) was verified by calculating Polder maps33 and by analysis of the map shapes and correlation coefficients. For clarity, in this work we divide THF, MTX, and PTX molecules into three moieties: (i) pteridine or its mimic (P-moiety, Fig. 1); (ii) benzoyl (B-moiety); and (iii) glutamate (E-moiety).
Both AtSHMT2 and AtSHMT4 are tetramers based on size-exclusion chromatography. The subunits are marked A-D (counterclockwise in Fig. 4). Interfaces between subunits A/B and C/D are much more extensive than e.g. between A and D. In the crystal structure of AtSHMT2-MTX complex, we noted two binding modes of MTX, one corresponding to external PLP-Ser aldimine state (EA-type; chains A, B, C) and the other to PLP internal aldimine + free serine state (IA-type; chain D). At this moment, we can only assume that different reaction states result from different neighborhood of subunits in the crystal lattice and therefore different access to solvent channels—although cooperativity between subunits cannot be excluded. The EA-type of binding (Fig. 5A,B) will be described based on chain A due to the higher quality of electron density maps and the lower B-factors. In this case, direct hydrogen bonds dock exocyclic N4 of MTX to Oε of Glu104* (an asterisk indicates a residue from another, tightly bound subunit), and N2 of MTX to backbone O of Lys414. A network of water-mediated hydrogen bonds link the antifolate to Thr416, Ala423, and Glu104*. Altogether, five out of six heteroatoms of the P-moiety (pteridine-2,4-diamine) participate in hydrogen bonding. Tyr111* π-stacks with the pteridine ring. B-moiety is sandwiched by the side chains of Ile189 and Tyr182 from one side and Ala423 from the other side. Finally, E-moiety is mostly disordered but appears to point towards the residues 184–186, with Oε of the E-moiety interacting with Oγ of Thr186.
Figure 4.

Electrostatic potential and antifolate binding site. AtSHMT2-MTX complex (tetramer) is shown with marked subunits (A–D).
Figure 5.

MTX and PTX binding by SHMTs. Green mesh (in A,C,E,G,I) represents Polder electron density maps contoured at 4.5 σ calculated for 5 Å volume around the antifolates. Panels (B,D,F,H,J) show networks of interactions between SHMTs and antifolates. Internal (LLP) or external aldimines are yellow, free serine is in magenta, residues from another protein subunit than indicated in caption, involved in interactions with MXT or PTX, are marked by asterisks.
The IA-type binding mode (Fig. 5C,D; chain D) is characterized by fewer H-bonds. In the P-moiety, exocyclic N2 H-bonds to carbonyl O of Tyr111* and to Nζ of Lys414. The pteridine ring is again stacked with the side chain of Tyr111*, however, it is rotated by 66° with respect to the ring in the EA-type binding. Surroundings of the B-moiety are similar to those observed in the EA-type, but the benzoyl ring is tilted by 23°. The benzoyl O atom interacts with Oγ of Thr184 via a water molecule. The two Oε atoms of the E-moiety are also involved in binding; Oε1 to the backbone amide of Asp185 and to (apparently protonated) Oδ of Asp185, whereas Oε2 interacts with Nη of Arg223 from the A-chain.
In AtSHMT4, (Fig. 5E,F, chain D) N1 of pteridine-2,4-diamine (with sp3 hybridization) interacts with Nζ of Lys373, whereas exocyclic N2 binds to carbonyl O of Arg67. The pteridine ring is stacked with the side chain of Tyr68*, equivalent to Tyr111* in AtSHMT2. The B-moiety is shielded by Ile147, Tyr139, and Ala382, corresponding to Ile189, Tyr182, and Ala423 of AtSHMT2, respectively. The E-moiety interacts with the backbone amide of Ala382 and Oγ of Ser142. Despite most of the interacting residues being conserved in AtSHMT2 and AtSHMT4, positioning of the P-moiety of MTX is different. In AtSHMT4, all four subunits within the crystal structure are in the external aldimine PLP-Ser form. Therefore, it is striking to see that the P-moiety in AtSHMT4 structure is posed in the same plane but rotated 180° with respect to the IA-type in AtSHMT2. Consistently, the stacking Tyr68* has a conformation similar to that of the corresponding Tyr111* of AtSHMT2 in the IA-type, and not to the EA-type as expected. This conformation of Tyr68* is even more striking when we consider that conformation of Tyr69* (AtSHMT4) is very different from that of Tyr112* in AtSHMT2 (IA-type).
Pemetrexed binding mode
Soaking crystals of AtSHMT2 with PTX and serine revealed two distinct forms, resembling situation observed for AtSHMT2-MTX complex. External PLP-Ser aldimine state (EA-type) is seen in chains B and C (PTX was not placed in chain A as suggested by poor Polder map coefficients33). Internal aldimine + free serine state (IA-type) is present in chain D.
In the EA-type of binding (Chain B, Fig. 5G,H), a direct H-bond binds exocyclic N2 of PTX to backbone O of Leu172. Water-mediated interactions involve each of the five heteroatoms in the P-moiety. Specifically, N1 is bonded to backbone N and Oγ of Thr416; N2 to Nδ of Asn415 and backbone O of Gly176; N3 to backbone O of Gly176; O4 to backbone N of Leu178; N7 to Oγ of Thr416 and carbonyl O of Ala423. Importantly, there is no π-stacking interaction involving PTX. Hydrophobic environment is ensured mostly by side chains of Leu172, Leu178, and Ile189 adjacent to PTX B-moiety. The linker between P- and B-moieties is shielded by the ring of Tyr111*. The E-moiety is partially disordered and located next to the loop fragment 184TDTK187, interacting with Oγ of Thr184 and Oγ of Thr186 via a water molecule. The carboxyl group (at Cα) forms a salt bridge with Nζ of Lys187.
In the IA-pose (Chain D, Fig. 5I,J), PTX forms fewer H-bonds. N1 directly H-bonds N of the free serine; N2 binds via water molecules to Nδ of Asn413, Oδ of Asn415, carbonyl O of Gly176 and Leu172; O4 is linked to Oγ of Thr416 via H2O; N7 directly docks to Oε of Glu104*. Like in the EA-pose, there is no π-stacking interaction of the ring moieties. The salt bridge to Lys187 is also conserved.
Discussion
Although having a similar scaffold, THF, MTX, and PTX are remarkably different from the chemical standpoint. Their P-moieties show highest variability (Fig. 1). MTX and PTX have dual-ring aromatic P-moieties, in contrast to THF. This is the most profound difference, making MTX and PTX unable to accept 1C units. The P-moieties of MTX and PTX vary significantly, too. PTX has a six-membered ring fused with a five-membered ring. As a consequence, it lacks what would be the N5 atom of MTX. It is therefore interesting to see that in the SHMT-MTX complexes, N5 never interacts with the protein. Removing an unnecessary H-bond donor seems like a step towards higher specificity and agrees with our kinetic data. Another feature that likely explains higher inhibitory efficiency of PTX against SHMTs is that it is able to bind deeper inside the cavity (Fig. 6). When we look at the P-moieties as if they were nucleobases, we see that MTX and PTX molecules (Fig. 1) have different “Watson-Crick” faces resulting in their different ability to be H-bond donors/acceptors. In this context, PTX is a closer mimic of THF. This explains why PTX can penetrate deeper into the cavity (Fig. 6) and be a more potent inhibitor of SHMTs (Table 2).
Figure 6.

Comparison of MTX, PTX, and THF binding modes. Positioning of THF (black, ball-and-stick) was acquired by superposing THF complex with murine SHMT (PDB ID: 1eji; chain A69). EA-poses are shown for AtSHMT2-MTX complex (chain A), and AtSHMT2-PTX complex (chain B).
The antifolate binding presented in this work shows that water molecules play a vital role in creating the extensive network of hydrogen bonds (Figs. 5 and 7). We propose to utilize positions of those water molecules (with respect to antifolates) as placeholders for specific functional groups which could be either H-bond acceptors or donors. This approach could likely enhance specificity and inhibitory properties of antifolates against SHMTs. It is also very important to note that resolving positions of water molecules inside the antifolate-binding cavity was possible due to high resolution diffraction data obtained particularly for AtSHMT2 structures. Lower resolution data from hmSHMT crystals28 (2.3 Å, Protein Data Bank, PDB ID: 6qvl) allowed to trace only a very limited number of water molecules.
Figure 7.

Detailed binding mode of PTX to AtSHMT2. PTX (orange bonds) interacts with the protein via an extensive network of hydrogen bonds (dashed, black lines; distances are given in Å), mostly mediated by water molecules (light blue circles). Hydrophobic interactions are indicated by “eyelashes”. Chain B of AtSHMT2-PTX complex is shown; EG, ethylene glycol molecule.
Considering the three moieties (P, B, and E) as pharmacophores, it is worth looking at the sequence and structure conservation between human and plant SHMTs with respect to regions of pharmacophore binding (Fig. 8). The sequence alignment shows a nearly complete conservation of residues that bind P and B moieties. Variability is present in the E-moiety binding region, however, the residue that ensures positive charge of the loop (Lys145 in AtSHMT4 and Lys187 in AtSHMT2) is universally conserved. The sequence-based conclusions are supported by structural superpositions (Fig. 8B,C). More precisely, for hcSHMT structure34 without a folate (PDB ID: 1bj4; RMSD = 0.75 Å with AtSHMT4-MTX) positions of most ligand-interacting residues are conserved with exceptions of loops containing Ala395 and Lys158 of hcSHMT that are in “open conformations” (Fig. 8B). It is likely, however, that upon folate binding the loops close and their conformations resemble those of AtSHMT4-MTX complex. Comparison of the mitochondrial isoforms, hmSHMT (PTX complex, PDB ID: 6qvl) with AtSHMT2-PTX (chain D, RMSD = 0.56 Å) shows even more similarities. Strikingly, for most residues involved in interaction with antifolates not only positions but also side chain conformations are the same (Fig. 8C). A deeper insight into PTX binding mode presented in AtSHMT2-PTX complex revealed that PTX pose in chain D is very similar to that presented in hmSHMT-PTX complex (PDB ID: 6qvl, Fig. 8C). P- and B-moieties of PTX are bound alike and small (~0.5 Å) discrepancies can likely be due to free serine that is bound to AtSHMT2 in addition to PTX. In fact, this free serine is involved in interaction between the protein and PTX. This means that the two complexes likely represent different PTX binding scenarios, (i) a pre-catalytic state at high serine concentration (AtSHMT2-PTX, chain D) or (ii) in serine-deficient state (hmSHMT-PTX complex).
Figure 8.

Comparison of hcSHMT, hmSHMT, AtSHMT2, and AtSHMT4. In Panel (A), bars above the sequences indicate residue conservation. Residues interacting with P-, B-, and E-moieties are marked with blue squares, yellow circles, and red triangles, respectively, and labeled with a corresponding letter. Panels (B,C) show pairwise structural comparisons of cytosolic AtSHMT4 (gray and teal subunits; blue MTX) with hcSHMT (coral, green), and of mitochondrial AtSHMT2 (gray, teal; orange PTX) with hmSHMT (coral, green; navy PTX). Residues and their numbering are given for plant/human isoforms. Antifolates are shown in ball-and-stick representation.
After their uptake inside the cell by the reduced folate carrier35,36, MTX and PTX undergo polyglutamylation by ATP-dependent folylpoly-γ-glutamate synthetase (FPGS), analogously to the folate species37–40. Polyglutamylation renders folates (or antifolates) polyanions that are not recognized by cellular efflux systems, and therefore accumulate in cellular compartments41. More importantly, polyglutamylated folates and antifolates present increased binding affinity to the folate-dependent enzymes. It has been shown that pentaglutamylated PTX is roughly 100-fold more potent than its monoglutamyl form; for instance, Ki against TS decreases from 109 nM to 1.3 nM, whereas Ki against GARFT decreases from 9.3 μM to 65 nM42. Notably, the Ki value of PTX against hcSHMT of 19.1 μM is very close to Ki against GARFT26. Intracellular concentration of PTX (or its polyglutamylated forms) reach 10–30 μM43. This suggests that polyglutamylated PTX may already effectively inhibit hcSHMT in vivo.
The vital role of polyglutamylation is consistent with the fact that loss of FPGS activity in patients, usually due to recurrent exposure, causes resistance to antifolates such as MTX and PTX38,44. Higher inhibitory activity of polyglutamylated antifolates against TS and GARFT allows us to hypothesize that the in vivo activity against SHMTs is also significantly higher. This hypothesis is supported by the fact that in the case of AtSHMT3, KM for THF decreases ~340-fold (218 μM for THF, and 0.64 μM for tetraglutamylated THF)6. Although comparing Ki and KM values can be misleading, higher affinity for polyglutamylated antifolates appears clear. It is also consistent with location of the positively-charged surface area around the entrance to the antifolate binding cavity that is able to attract negatively-charged polyglutamyl tail of THF or antifolates (Fig. 4). Unfortunately, polyglutamated forms of MTX or PTX are currently not commercially available, thus we cannot verify whether they indeed inhibit SHMTs much stronger than the non-glutamylated forms. So far, research on anticancer drugs has yielded candidate scaffolds for future drugs45. Pyrazolopyran-based inhibitors are effective against hcSHMT46. However, search for new antitumor agents by no means has been the only attempt to target SHMTs. For instance, an extensive drug design campaign is aimed at SHMTs from a malarial parasite Plasmodium falciparum SHMTs47–50. Inhibitors based also on pyrazolopyran showed a remarkable potency against PfSHMT48. One of the inhibitors was able to reduce parasitemia in vivo in a mouse model by 73%49. Herbicidal and pharmaceutical use of pyrazolopyrans is a subject of a patent (WO2013182472A1). So far, low metabolic stability limits the use of many potent SHMT inhibitors48. In this context, exploiting commonly used drugs (with some inhibitory activity against SHMTs) as starting scaffolds appears to be a very auspicious concept.
Conclusions and Future Outlook
In light of recent discoveries, design of SHMT inhibitors seems to be promising in finding novel chemotherapeutics. However, we are challenged by (i) high dynamics of SHMTs, related to the reaction state, (ii) apparently multiple binding modes of antifolates, and (iii) the need for candidate drugs to be polyglutamylated by FPGS. Therefore, we are very optimistic to note that already available antifolate chemotherapeutics, such as PTX, are potent against SHMT enzymes. In our opinion, further optimization of candidate drugs, for instance by adding functional groups positioned as water molecules in our complexes has a very high chance of success. With that in mind, we believe that our results have just brought us a step closer towards a rationalized targeting of SHMTs.
Methods
Cloning, overexpression, and purification of AtSHMT2 and AtSHMT4
AtSHMT2 and AtSHMT4 were produced using a protocol similar to that for the production of Medicago truncatula SHMT37. Briefly, RNA isolated from A. thaliana leaves using the RNeasy Plant Mini Kit (Qiagen) was reverse-transcribed into the complementary DNA (cDNA) with SuperScript II reverse transcriptase (Life Technologies). The mitochondrial signal peptide of AtSHMT2 was recognized using the TargetP 1.1 server51,52; the construct was designed to yield the protein N-truncated at E41. The open reading frames coding for AtSHMT2 41–517 fragment and full-length AtSHMT4 were amplified by polymerase chain reaction. The following primers were used:
AtSHMT2-Nt41-F, TACTTCCAATCCAATGCCGAGAAATCTCGTTCTAGTTGGATTAAGCAATT; AtSHMT2-R, TTATCCACTTCCAATGTTACTACTCTTTGTATCTCATCGTCTCTTTCT; AtSHMT4-F, TACTTCCAATCCAATGCCATGGAACCAGTCTCTTCATGGGGTAA; AtSHMT4-R, TTATCCACTTCCAATGTTACTAATCCTTGTACTTCATCTCAGACATGA. Expression plasmids, based on pMCSG68 vector (Midwest Center for Structural Genomics, USA), were created by the ligase-independent cloning method53. The proteins were expressed in BL21 Gold E. coli cells (Agilent Technologies) in LB media with 150 μg/mL ampicillin. When the cultures reached A600 of 1.0, isopropyl-D-thiogalactopyranoside was added at a final concentration of 0.5 mM to chilled cultures (18 °C); overexpression was continued for 18 h. The cell pellet from the 2 L culture was centrifuged at 3,500 x g for 30 min at 4 °C, resuspended in 35 mL of binding buffer [50 mM Hepes-NaOH pH 7.5; 500 mM NaCl; 20 mM imidazole; 1 mM tris(2-carboxyethyl)phosphine (TCEP)], and stored at −80 °C.
The cells were disrupted by sonication and the lysates were cleared by centrifugation at 25,000 × g for 30 min at 4 °C. The supernatants were run through columns packed with 3 mL of HisTrap HP resin (GE Healthcare) connected to vacuum pump-VacMan setup (Promega). The resin-bound proteins were washed six times with 40 mL of the binding buffer. Afterwards, the proteins were eluted with 20 mL of elution buffer (50 mM Hepes-NaOH pH 7.5; 500 mM NaCl; 400 mM imidazole; 1 mM TCEP). Overnight dialysis (at 4 °C) lowered the imidazole concentration to 20 mM. Simultaneously, the His6-tags were cleaved with TEV protease (at the final concentration 0.1 mg/mL). A second run on the HisTrap columns was used to capture the cleaved His6-tags and the His6-tagged TEV protease. The samples were concentrated to 2.5 mL and applied on a HiLoad Superdex 200 16/60 column (GE Healthcare), equilibrated with a buffer composed of 25 mM Hepes-NaOH pH 7.5, 100 mM KCl, 50 mM NaCl, and 1 mM TCEP. The proteins were concentrated using centrifugal concentrators (Millipore): AtSHMT2 to 15 mg/mL (based on A280 extinction coefficient, ε = 49,300 M−1 × cm−1); AtSHMT4 to 19 mg/mL (ε = 54,800 M−1 × cm−1).
Kinetic tests and inhibition studies
The SHMT activity was measured with 0.2 μM enzyme samples with L-serine and THF as substrates, using a coupled assay as previously described54 at 30 °C. THF (and 5-formyl-THF; see below) was kindly provided by Merck & Cie (Schaffhausen, Switzerland). Activities were measured at different pH values (6.0, 7.0, 8.5 and 9.5). For pH 6.0, 7.0 and 8.5, the assays were performed in 20 mM phosphate buffer. Measurements at pH 9.5 were performed in a mixture of 50 mM MES, 50 mM HEPES, and 50 mM CHES and the final pH was adjusted with KOH.
Inhibition assays were performed using 5 μM enzyme solutions. All assays were carried out at 30 °C in 20 mM KPi buffer at either pH 8.0, in the case of AtSHMT2, or pH 7.3 in the case of AtSHMT4. Methotrexate was dissolved in pure DMSO. In all inhibition assays, the final concentration of DMSO was 5% (v/v), which did not affect enzyme activity45. Pemetrexed was dissolved in buffer.
Inhibition assays that used glycine and 5-formyl-THF were based on the spectrophotometric measurement of the quinonoid intermediate55. The intermediate develops when both ligands bind to SHMT, forming an enzyme-glycine-folate ternary complex. First, dissociation constants of glycine and 5-formyl-THF were determined by varying one ligand while keeping the other at a fixed and saturating concentration. Data were analyzed to obtain estimates of the dissociation constant (Kd) and αKd (in the presence of a saturating concentration of the other ligand) for both ligands, as reported in31.
Kinetic parameters (Table 1) were determined using Eq. 1 when L-serine was the fixed substrate and Eq. 2 when THF was the fixed substrate. In the case of data obtained with AtSHMT2 and L-serine as fixed substrate, Vmax was previously estimated with a normal Michaelis-Menten equation leaving out data obtained with THF concentrations higher than 100 μM; estimated Vmax values were then used as fixed parameters when fitting all data to Eq. 1.
| 1 |
| 2 |
Binding of MTX and PTX to SHMT was subsequently analyzed by measuring the inhibitory effect of these compounds on the formation of the quinonoid intermediate. In the assays, 5-formyl-THF was added as the last component and, after rapid manual mixing, the absorbance change was measured at 500 nm (AtSHMT4) or 505 nm (AtSHMT2). The obtained data were used to produce double reciprocal plots and fitted to linear equations. Slopes of the straight lines were plotted versus MTX or PTX concentration in secondary plots and fitted to linear equations in order to find the related inhibition constants from the intercepts to the X-axes. Fitting of data was performed with the software PRISM (GraphPad, La Jolla, CA, USA). All experiments were performed in triplicate using a Hewlett-Packard 8453 diode-array spectrophotometer (Agilent Technologies, Santa Clara, CA).
Crystallization and diffraction data collection
The AtSHMT2 crystals were grown by vapor diffusion method in hanging drops containing 3 µL of the protein solution and 2 µL of reservoir solution. The reservoir solution was composed of 90% Hampton Research Index F8 condition (0.2 M ammonium sulfate, 0.1 M Hepes pH 7.5, and 25% polyethylene glycol 3350). Crystals of AtSHMT4 were grown in 80% Hampton Research Index D9 condition (0.1 M Tris-HCl pH 8.5, and 25% polyethylene glycol 3350). Mature crystals were transferred to drops with the original screen conditions supplemented with 20% ethylene glycol, 50 mM L-serine, and 10 mM of MTX or PTX. After 24-h incubation, crystals were harvested and vitrified in liquid nitrogen. Data were collected at beamlines 22-BM and 22-ID beamlines at the Advanced Photon Source, Argonne, USA. The diffraction images were processed with XDS56. The statistics of data collection and processing are summarized in Table 3.
Table 3.
Data collection and refinement statistics.
| AtSHMT2-MTX | AtSHMT4-MTX | AtSHMT2-PTX | |
|---|---|---|---|
| Data collection | |||
| Beamline | APS 22-BM | APS 22-ID | APS 22-BM |
| Wavelength (Å) | 1.0000 | 1.0000 | 1.0000 |
| Space group | P212121 | P212121 | P212121 |
| Unit cell parameters | |||
| a, b, c (Å) | 115.4, 130.7, 151.3 | 118.8, 120.9, 131.8 | 115.4, 130.8, 151.2 |
| Resolution (Å) | 80.0–1.63 (1.73–1.63) | 80.0–2.12 (2.25–2.12) | 80.0–1.54 (1.64–1.54) |
| Unique reflections | 279297 (44247) | 106878 (17038) | 330027 (52224) |
| Multiplicity | 8.2 (7.8) | 4.2 (4.3) | 8.1 (7.7) |
| Completeness (%) | 99.8 (98.5) | 98.8 (98.7) | 99.7 (98.2) |
| Rmeas a(%) | 9.3 (124.1) | 10.8 (76.0) | 8.8 (133.8) |
| <I/σ(I)> | 18.3 (2.0) | 11.6 (1.9) | 18.88 (1.8) |
| Refinement | |||
| Rfree reflections | 1396 | 1068 | 1320 |
| No. of atoms (non-H) | 17397 | 15779 | 17445 |
| protein | 15078 | 14583 | 15198 |
| ligands | 397 | 231 | 311 |
| solvent | 1922 | 965 | 1936 |
| Rwork/Rfree (%) | 12.5/17.7 | 17.4/23.4 | 12.9/16.6 |
| Average B-factor (Å2) | |||
| protein | 24.8 | 38.6 | 23.8 |
| ligands | 47.7 | 57.8 | 45.6 |
| solvent | 41.3 | 39.3 | 40.5 |
| rmsd from ideal geometry | |||
| bond lengths (Å) | 0.005 | 0.006 | 0.005 |
| bond angles (°) | 0.81 | 0.90 | 0.79 |
| Ramachandran statistics (%) | |||
| favored | 97.9 | 96.7 | 97.6 |
| allowed | 2.1 | 3.4 | 2.4 |
| outliers | 0.0 | 0.0 | 0.0 |
| PDB ID | 6smn | 6smr | 6smw |
Values in parentheses correspond to the highest resolution shell.
aRmeas = redundancy independent R-factor.
Determination and refinement of the crystal structures
Crystal structures were solved using molecular replacement in Phaser57 with M. truncatula SHMT3 as the search probe (PDB ID: 6cd07). The models were built using Phenix AutoBuild58, and were placed inside the unit cells with the ACHESYM server59. COOT60 was used for manual fitting in the electron density maps between rounds of model refinement in Phenix.refine61. The refinement statistics are listed in Table 3. After a thorough quality validation with Molprobity62, the structures were deposited in the PDB63.
Other software used
Molecular figures were created with UCSF Chimera64. Surface electrostatic potential was calculated using PDB2PQR and APBS servers65,66. Flat scheme of H-bonding network was created in LigPlot+67. Signal peptides were predicted with the TargetP51 webserver.
Accession numbers
PDB IDs: AtSHMT2-MTX complex, 6smn; AtSHMT4, MTX complex, 6smr; AtSHMT2-PTX complex, 6smw.
Supplementary information
Acknowledgements
This project was supported by the Intramural Research Program of the NCI Center for Cancer Research. Agnieszka Ruszkowska was supported by NIH Grant R00GM111430 (to Jessica A. Brown, University of Notre Dame). Diffraction data were collected at the SER-CAT beamline 22-ID and 22-BM at the Advanced Photon Source, Argonne National Laboratory, supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract W-31-109-Eng-38. Kinetic and inhibition measurements were supported by a senior “Anna Tramontano” grant of Istituto Pasteur Italia-Fondazione Cenci Bolognetti (to Roberto Contestabile).
Author contributions
M.R. designed the project. M.R., B.S. and A.R. produced AtSHMT2 and AtSHMT4 proteins and did the structural studies. R.C., I.N. and S.A. performed kinetic experiments. A.S. provided medical background of antifolate therapy. Z.D. supervised the work. M.R. wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-56043-4.
References
- 1.Schirch V, Szebenyi DM. Serine hydroxymethyltransferase revisited. Curr Opin Chem Biol. 2005;9:482–487. doi: 10.1016/j.cbpa.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 2.Florio R, di Salvo ML, Vivoli M, Contestabile R. Serine hydroxymethyltransferase: a model enzyme for mechanistic, structural, and evolutionary studies. Biochim Biophys Acta. 2011;1814:1489–1496. doi: 10.1016/j.bbapap.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 3.Ducker GS, Rabinowitz JD. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017;25:27–42. doi: 10.1016/j.cmet.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson DD, Quintero CM, Stover PJ. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc Natl Acad Sci USA. 2011;108:15163–15168. doi: 10.1073/pnas.1103623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ducker GS, et al. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016;23:1140–1153. doi: 10.1016/j.cmet.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Sun K, Sandoval FJ, Santiago K, Roje S. One-carbon metabolism in plants: characterization of a plastid serine hydroxymethyltransferase. Biochem J. 2010;430:97–105. doi: 10.1042/BJ20100566. [DOI] [PubMed] [Google Scholar]
- 7.Ruszkowski M, Sekula B, Ruszkowska A, Dauter Z. Chloroplastic Serine Hydroxymethyltransferase From Medicago truncatula: A Structural Characterization. Front Plant Sci. 2018;9:584. doi: 10.3389/fpls.2018.00584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauwe H, Hagemann M, Fernie AR. Photorespiration: players, partners and origin. Trends Plant Sci. 2010;15:330–336. doi: 10.1016/j.tplants.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Maurino VG, Peterhansel C. Photorespiration: current status and approaches for metabolic engineering. Curr Opin Plant Biol. 2010;13:249–256. doi: 10.1016/j.pbi.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Kikuchi G, Motokawa Y, Yoshida T, Hiraga K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc Jpn Acad Ser B Phys Biol Sci. 2008;84:246–263. doi: 10.2183/pjab.84.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douce R, Bourguignon J, Neuburger M, Rebeille F. The glycine decarboxylase system: a fascinating complex. Trends Plant Sci. 2001;6:167–176. doi: 10.1016/S1360-1385(01)01892-1. [DOI] [PubMed] [Google Scholar]
- 12.Rebeille F, Neuburger M, Douce R. Interaction between glycine decarboxylase, serine hydroxymethyltransferase and tetrahydrofolate polyglutamates in pea leaf mitochondria. Biochem J. 1994;302(Pt 1):223–228. doi: 10.1042/bj3020223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ros R, Munoz-Bertomeu J, Krueger S. Serine in plants: biosynthesis, metabolism, and functions. Trends Plant Sci. 2014;19:564–569. doi: 10.1016/j.tplants.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Jain M, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7:1248–1258. doi: 10.1016/j.celrep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 16.Meyer LM, Miller FR, Rowen MJ, Bock G, Rutzky J. Treatment of Acute Leukemia with Amethopterin (4-Amino, 10-Methyl Pteroyl Glutamic Acid) Acta Haematologica. 1950;4:157–167. doi: 10.1159/000203749. [DOI] [PubMed] [Google Scholar]
- 17.Benedek TG. Methotrexate: from its introduction to non-oncologic therapeutics to anti-TNF-alpha. Clinical and Experimental Rheumatology. 2010;28:S3–S8. [PubMed] [Google Scholar]
- 18.Prodduturi P, Bierman PJ. Current and emerging pharmacotherapies for primary CNS lymphoma. Clin Med Insights Oncol. 2012;6:219–231. doi: 10.4137/CMO.S7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angelov L, et al. Blood-brain barrier disruption and intra-arterial methotrexate-based therapy for newly diagnosed primary CNS lymphoma: a multi-institutional experience. J Clin Oncol. 2009;27:3503–3509. doi: 10.1200/JCO.2008.19.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown PM, Pratt AG, Isaacs JD. Mechanism of action of methotrexate in rheumatoid arthritis, and the search for biomarkers. Nat Rev Rheumatol. 2016;12:731–742. doi: 10.1038/nrrheum.2016.175. [DOI] [PubMed] [Google Scholar]
- 21.Manegold C. Pemetrexed (Alimta, MTA, multitargeted antifolate, LY231514) for malignant pleural mesothelioma. Seminars in Oncology. 2003;30:32–36. doi: 10.1016/S0093-7754(03)00283-5. [DOI] [PubMed] [Google Scholar]
- 22.Cohen MH, Justice R, Pazdur R. Approval Summary: Pemetrexed in the Initial Treatment of Advanced/Metastatic Non-Small Cell Lung Cancer. Oncologist. 2009;14:930–935. doi: 10.1634/theoncologist.2009-0092. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Zhang P, Ma Q, Wang D, Zhou T. Cisplatin-based chemoradiotherapy with 5-fluorouracil or pemetrexed in patients with locally advanced, unresectable esophageal squamous cell carcinoma: A retrospective analysis. Mol Clin Oncol. 2017;6:743–747. doi: 10.3892/mco.2017.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chattopadhyay S, Moran RG, Goldman ID. Pemetrexed: biochemical and cellular pharmacology, mechanisms, and clinical applications. Molecular Cancer Therapeutics. 2007;6:404–417. doi: 10.1158/1535-7163.Mct-06-0343. [DOI] [PubMed] [Google Scholar]
- 25.Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by Cytotoxic Metabolite Accumulation: Pemetrexed Causes ZMP Accumulation, AMPK Activation, and Mammalian Target of Rapamycin Inhibition. Cancer Research. 2009;69:5467–5474. doi: 10.1158/0008-5472.Can-08-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daidone F, et al. In silico and in vitro validation of serine hydroxymethyltransferase as a chemotherapeutic target of the antifolate drug pemetrexed. Eur J Med Chem. 2011;46:1616–1621. doi: 10.1016/j.ejmech.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Sliwoski G, Kothiwale S, Meiler J, Lowe EW., Jr. Computational methods in drug discovery. Pharmacol Rev. 2014;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scaletti E, Jemth AS, Helleday T, Stenmark P. Structural basis of inhibition of the human serine hydroxymethyltransferase SHMT2 by antifolate drugs. FEBS Lett. 2019;593:1863–1873. doi: 10.1002/1873-3468.13455. [DOI] [PubMed] [Google Scholar]
- 29.Raju TN. The Nobel chronicles. 1988: James Whyte Black, (b 1924), Gertrude Elion (1918-99), and George H Hitchings (1905–98) Lancet. 2000;355:1022. doi: 10.1016/s0140-6736(05)74775-9. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Wu J, Gao Y, Chen H, Zhou J. Scaffold Repurposing of Old Drugs Towards New Cancer Drug Discovery. Curr Top Med Chem. 2016;16:2107–2114. doi: 10.2174/1568026616666160216155556. [DOI] [PubMed] [Google Scholar]
- 31.Tramonti A, et al. Human Cytosolic and Mitochondrial Serine Hydroxymethyltransferase Isoforms in Comparison: Full Kinetic Characterization and Substrate Inhibition Properties. Biochemistry. 2018;57:6984–6996. doi: 10.1021/acs.biochem.8b01074. [DOI] [PubMed] [Google Scholar]
- 32.Amornwatcharapong W, Maenpuen S, Chitnumsub P, Leartsakulpanich U, Chaiyen P. Human and Plasmodium serine hydroxymethyltransferases differ in rate-limiting steps and pH-dependent substrate inhibition behavior. Arch Biochem Biophys. 2017;630:91–100. doi: 10.1016/j.abb.2017.07.017. [DOI] [PubMed] [Google Scholar]
- 33.Liebschner D, et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Cryst. D. 2017;73:148–157. doi: 10.1107/S2059798316018210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Renwick SB, Snell K, Baumann U. The crystal structure of human cytosolic serine hydroxymethyltransferase: a target for cancer chemotherapy. Structure. 1998;6:1105–1116. doi: 10.1016/s0969-2126(98)00112-9. [DOI] [PubMed] [Google Scholar]
- 35.Hou ZJ, Matherly LH. Oligomeric Structure of the Human Reduced Folate Carrier Identification of Homo-Oligomers and Dominant-Negative Effects on Carrier Expression and Function. Journal of Biological Chemistry. 2009;284:3285–3293. doi: 10.1074/jbc.M807206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matherly LH, Hou ZJ. Structure and Function of the Reduced Folate Carrier: A Paradigm of a Major Facilitator Superfamily Mammalian Nutrient Transporter. Folic Acid and Folates. 2008;79:145–184. doi: 10.1016/S0083-6729(08)00405-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho RC, et al. Effects of folate and folylpolyglutamyl synthase modulation on chemosensitivity of breast cancer cells. Mol Cancer Ther. 2007;6:2909–2920. doi: 10.1158/1535-7163.MCT-07-0449. [DOI] [PubMed] [Google Scholar]
- 38.Liani E, et al. Loss of folylpoly-gamma-glutamate synthetase activity is a dominant mechanism of resistance to polyglutamylation-dependent novel antifolates in multiple human leukemia sublines. International Journal of Cancer. 2003;103:587–599. doi: 10.1002/ijc.10829. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto, T., Shikano, K., Nanki, T. & Kawai, S. Folylpolyglutamate synthase is a major determinant of intracellular methotrexate polyglutamates in patients with rheumatoid arthritis. Sci Rep6, 10.1038/srep35615 (2016). [DOI] [PMC free article] [PubMed]
- 40.Muller IB, et al. Personalized medicine in rheumatoid arthritis: methotrexate polyglutamylation revisited. Expert Review of Precision Medicine and Drug Development. 2018;3:331–334. doi: 10.1080/23808993.2018.1517025. [DOI] [Google Scholar]
- 41.Raz S, Stark M, Assaraf YG. Folylpoly-gamma-glutamate synthetase: A key determinant of folate homeostasis and antifolate resistance in cancer. Drug Resist Updat. 2016;28:43–64. doi: 10.1016/j.drup.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 42.Zhao R, Titus S, Gao F, Moran RG, Goldman ID. Molecular analysis of murine leukemia cell lines resistant to 5, 10-dideazatetrahydrofolate identifies several amino acids critical to the function of folylpolyglutamate synthetase. J Biol Chem. 2000;275:26599–26606. doi: 10.1074/jbc.M002580200. [DOI] [PubMed] [Google Scholar]
- 43.Shih C, et al. LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Research. 1997;57:1116–1123. [PubMed] [Google Scholar]
- 44.Stark M, Wichman C, Avivi I, Assaraf YG. Aberrant splicing of folylpolyglutamate synthetase as a novel mechanism of antifolate resistance in leukemia. Blood. 2009;113:4362–4369. doi: 10.1182/blood-2008-08-173799. [DOI] [PubMed] [Google Scholar]
- 45.Paiardini A, et al. Screening and in vitro testing of antifolate inhibitors of human cytosolic serine hydroxymethyltransferase. ChemMedChem. 2015;10:490–497. doi: 10.1002/cmdc.201500028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marani M, et al. A pyrazolopyran derivative preferentially inhibits the activity of human cytosolic serine hydroxymethyltransferase and induces cell death in lung cancer cells. Oncotarget. 2016;7:4570–4583. doi: 10.18632/oncotarget.6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwertz G, et al. Potent Inhibitors of Plasmodial Serine Hydroxymethyltransferase (SHMT) Featuring a Spirocyclic Scaffold. ChemMedChem. 2018;13:931–943. doi: 10.1002/cmdc.201800053. [DOI] [PubMed] [Google Scholar]
- 48.Witschel MC, et al. Inhibitors of plasmodial serine hydroxymethyltransferase (SHMT): cocrystal structures of pyrazolopyrans with potent blood- and liver-stage activities. J Med Chem. 2015;58:3117–3130. doi: 10.1021/jm501987h. [DOI] [PubMed] [Google Scholar]
- 49.Schwertz G, et al. Antimalarial Inhibitors Targeting Serine Hydroxymethyltransferase (SHMT) with in Vivo Efficacy and Analysis of their Binding Mode Based on X-ray Cocrystal Structures. J Med Chem. 2017;60:4840–4860. doi: 10.1021/acs.jmedchem.7b00008. [DOI] [PubMed] [Google Scholar]
- 50.Schwertz G, et al. Conformational Aspects in the Design of Inhibitors for Serine Hydroxymethyltransferase (SHMT): Biphenyl, Aryl Sulfonamide, and Aryl Sulfone Motifs. Chemistry. 2017;23:14345–14357. doi: 10.1002/chem.201703244. [DOI] [PubMed] [Google Scholar]
- 51.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 52.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 53.Kim Y, et al. High-throughput protein purification and quality assessment for crystallization. Methods. 2011;55:12–28. doi: 10.1016/j.ymeth.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schirch V, Hopkins S, Villar E, Angelaccio S. Serine hydroxymethyltransferase from Escherichia coli: purification and properties. J Bacteriol. 1985;163:1–7. doi: 10.1128/jb.163.1.1-7.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schirch L, Ropp M. Serine transhydroxymethylase. Affinity of tetrahydrofolate compounds for the enzyme and enzyme-glycine complex. Biochemistry. 1967;6:253–257. doi: 10.1021/bi00853a039. [DOI] [PubMed] [Google Scholar]
- 56.Kabsch W. Xds. Acta Cryst. D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Cryst. D. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Terwilliger TC, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst. D. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kowiel M, Jaskolski M, Dauter Z. ACHESYM: an algorithm and server for standardized placement of macromolecular models in the unit cell. Acta Cryst. D. 2014;70:3290–3298. doi: 10.1107/S1399004714024572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. D. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pettersen EF, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 65.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 68.Shen J, et al. Organelle pH in the Arabidopsis endomembrane system. Mol Plant. 2013;6:1419–1437. doi: 10.1093/mp/sst079. [DOI] [PubMed] [Google Scholar]
- 69.Szebenyi DM, Liu X, Kriksunov IA, Stover PJ, Thiel DJ. Structure of a murine cytoplasmic serine hydroxymethyltransferase quinonoid ternary complex: evidence for asymmetric obligate dimers. Biochemistry. 2000;39:13313–13323. doi: 10.1021/bi000635a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.