

Abstract
Cerenkov radiation (CR) from radionuclides can act as a built-in light source for cancer theranostics, opening a new horizon in biomedical applications. However, considerably low tumor-targeting efficiency of existing radionuclides and radionuclide-based nanomedicines limits the efficacy of CR-induced theranostics (CRIT). It remains a challenge to precisely and efficiently supply CR energy to the tumor site. Here, we report a missile-detonation strategy, that first administers a high dose of p-SCN-Bn-Deferoxamine-porphyrin-PEG nanocomplex (Df-PPN) as a CR energy receiver/missile passively target to tumor, and then a low dose of the 89Zr-labeled Df-PPN is administrated as a CR energy donor/detonator, which can be visualized and quantified by Cerenkov energy transfer imaging (CRET), positron-emission tomography (PET) and Fluorescence imaging (FL). Based on homologous properties, the colocalization of Df-PPN and 89Zr-Df-PPN in the tumor site is maximized and enables efficient CR energy transfer that maximizes the tumor-targeted CRIT efficacy in an optimal spatiotemporal setting while also reducing adverse off-target effects from CRIT. This precise and efficient CRIT strategy caused significant tumor vascular damage and inhibited tumor growth.
Keywords: Cancer theranostics, targeted drug delivery, positron emission tomography, multimodal imaging, Cerenkov radiation
Graphical Abstract:
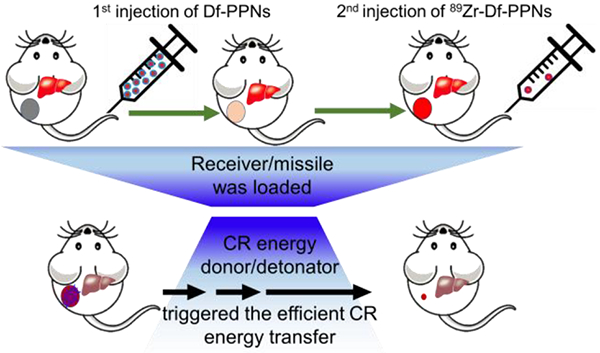
A missile-detonation strategy, by which a single dose of Df-PPN as Cerenkov radiation (CR) energy receiver/missile passively targets to tumor firstly, then 89Zr-labeled Df-PPN as a CR energy donor/detonator is administrated on-demand in a quantified optimal time window. The maximum colocalization of Df-PPN and 89Zr-Df-PPN trigger the efficient CR energy transfer to cause tumor vascular damage and inhibit tumor growth.
Cancer nanomedicine is a promising strategy for improving the targeted treatment of cancer but has seen limited translation into the clinic.[1] In many instances, the translational of nanomedicine applications was impeded by adverse toxic effects of the nanomaterial, off-target toxicity from high accumulation in the reticuloendothelial system, or low efficacy caused by limited therapeutic payloads.[2] To achieve the translation of nanomedicine, two primary concerns for effective anticancer therapy are optimized administration and controllable cell-killing efficacy.
Photodynamic therapy (PDT) is a clinically approved treatment for a variety of solid tumors.[3] PDT-induced changes in vascular permeability of tumors have been shown to enhance the delivery of therapeutic agents.[4] However, the current approach for PDT requires an external light source to trigger the generation of reactive oxygen species (ROS), which has hindered its clinical application because of the limited tissue penetration of light. Recently, the low-intensity, blue light of Cerenkov radiation (CR) has been developed as a depth-independent light source for the application of CR alone or combination with photosensitizers, such as titanium dioxide (TiO2), quantum dots, and porphyrins for molecular imaging and cancer therapy in vivo.[5] CR is considered a bridge between positron emission tomography (PET) and optical imaging, which has become attractive in clinical imaging applications.[5a] A milestone work was reported by Nalinikanth Kotagiri et al. that opened a novel paradigm for cancer therapy by utilizing CR as a light source to achieve photodynamic therapy (CRIT) by generating ROS.[5b] However, the administrated radionuclides (64Cu or 18F) had a relatively short half-life that limited the therapeutic window for CRIT. The potential combination strategy by coupling 64Cu with TiO2 might continuously generate ROS during its blood circulation which make this modality effectively a normal chemotherapeutic agent. Moreover, the administration route via intravenous injection (I.V.) typically requires more than 24 hours to achieve high tumor accumulation, which further limits possible tumor damage using short-lived radionuclides.[6] Additionally, without integrating PET and optical imaging impeded the evaluation of the treatment outcome. Moreover, the non-specific uptake of 18F-FDG and nanoparticles in the reticuloendothelial system aggravated systemic toxicity and attenuated the therapeutic effect. Effectively translating this revolutionary strategy to the clinic will require addressing tumor-targeting efficiency and off-targeting toxicity. Our group previously loaded chlorin e6 with 89Zr (higher-energy β emitters 909 keV and t1/2 = 78.4 h) in silica nanoparticles and also used magnetic targeting strategy to achieve CRIT.[7] However, the spatiotemporal colocalization of the photosensitizer with the radionuclide in these studies resulted in excellent therapeutic efficacy, but potential side effects to healthy tissues arouse from off-target effects. Thus, the development of a CRIT strategy with high-efficacy and tumor accumulation while minimizing side-effects is of major significance but remains challenging.
To optimize the administration agents, we employed a missile-detonation strategy to enhance the efficacy of tumor-targeted CRIT. This strategy involved the injection of a high dose of porphyrin-PEG nanocomplex (PPN) as a CR energy receiver/missile and then followed by the administration of a low dose of 89Zr-labeled PPN (89Zr-Df-PPN) as a CR energy donor/detonator. At an optimal time point, the 89Zr-Df-PPN detonator would ignite the PPN missile at the tumor site and efficiently kill tumor cells. However, the relative low accumulation of PPN in normal tissues circumvents the detonator payload to reduce the side effects from CRIT (Figure 1a). Based on homologous properties, the colocation of 89Zr-Df-PPNs and PPNs in the tumor site enabled CRIT in a highly efficient spatiotemporal setting to produce ROS and damage tumor cells. Moreover, CRIT significantly inhibited tumor growth and caused fragmentation of the vascular architecture in the tumor. There are several significant findings in this work. First, an optimized time frame for administrating therapeutic agents (photosensitizer and tumor-in-situ light) was visualized and quantified by multimodal imaging, specifically FL, CRET, and PET imaging. Second, a missile-detonation approach was used to reduce the adverse off-target effects and maximize the CRIT efficacy in an optimal spatiotemporal setting. Third, CRIT significantly inhibited tumor growth and caused fragmentation of the vascular architecture in the tumor, which would be a benefit for developing a selective anti-angiogenic strategy for cancer treatment in the future. In this study, a nanoplatform (an average size of 10–20 nm) that could be labeled for positron emission tomography (PET) imaging was synthesized as similarly described in our previous work (Figure S1).[8] 89Zr could be immediately chelated to the Df-PPNs as rapidly as 15 min (FigureS2). The labeling yield of 89Zr was approximate 79%. We then utilized a missile-detonation approach to reduce the toxicity of CR-induced ROS in systemic circulation as shown in Figure 1a. In this strategy, a high dose of Df-PPNs was first administered and delivered to the tumor site by the enhanced permeability and retention (EPR) effect. The second injection of a low dose of 89Zr labeled Df-PPNs (89Zr-Df-PPNs) was then performed on the third day after the first injection (Figure 2a) because Df-PPNs uptake in the tumor reached a maximum while the uptake in normal organs reached a minimum (Figure S3). The 89Zr-Df-PPNs accumulated at the tumor site in proximity to the previous injection of Df-PPNs based on their homologous properties, enabling the ignition of the Df-PPN missile by the 89Zr-Df-PPN detonator through CRET. As shown in Figure 1b, in vivo fluorescence signals for tracking Df-PPNs were very strong and lasted up to 17 days post-injection. The quantitative analysis of regions of interest (ROI) demonstrated a steady increase in the tumor uptake of Df-PPNs in the 3 days following intravenous injection (i.v.) (Figure 1e). This further confirmed the sustained circulation of Df-PPNs for 3 days and that the photosensitizer, Df-PPNs, was retained in the tumor site that enabled an extensive period before treatment with the subsequent injection. Cerenkov luminance (CL) imaging was performed to locate 89Zr-Df-PPNs and assess the signal irradiated by 89Zr-Df-PPNs in vivo. During the 3 days post-injection of 89Zr-Df-PPNs, the CL intensity remained constant because a steady-state was reached between the tumor uptake of 89Zr-Df-PPNs and the decay of 89Zr (Figure 1c and 1f). The energy-transfer phenomenon was monitored by measuring the luminescence from co-incubating Df-PPNs and 89Zr using an IVIS system, as shown in Figure S4a and S4b. In vivo radionuclide illuminated FL imaging was monitored to determine the colocalization of 89Zr-Df-PPNs and Df-PPNs. The signal at 660 nm and 720 nm, which are the primary emission peak of mTCPP, were imaged and quantified, respectively, as shown in Figure 1d and 1g. The tendency of CRET intensity was similar to the variation of CL. Since the intensity of CR is most intense in the blue light zone, the irradiation efficiency will be limited by the tissue penetration ability of the blue-weighted light produced by CR. Confocal imaging further demonstrated that 89Zr-Df-PPNs and Df-PPNs co-localized in the tumor at a distance that enabled CRIT in a highly efficient spatiotemporal manner as shown in Figure S5. Moreover, we found this missile-detonation strategy mitigated toxicity concerns compared to the coadministration method (Figure S6). Therefore, the missile-detonation strategy enabled colocalization of the photosensitizer and CR light source to achieve CR induced local photodynamic therapy in vivo.
Figure 1.

Missile-detonation approach to delivering tumor in-suit light source and therapy reagent. (a) Schematic of the missile-detonation strategy, which first administered a high dose of Df-PPN as a CR energy receiver/missile passively targeted to tumor, followed by a low dose of the 89Zr-labeled Df-PPN as a CR energy donor/detonator for activated cancer therapy. (b) In vivo fluorescence (FL), (c) Cerenkov luminescence images (CL), and (d) Cerenkov radiation energy transfer (CRET) of 4T1 tumor-bearing mice taken after injection of Df-PPNs and 89Zr-Df-PPNs. Quantification analysis of in vivo (e) FL (different letters above the column mean comparing to each other p < 0.05, The different letters above the column mean that the p value is less than 0.05 while comparing to each other.), (f) CL, and (g) CRET of 4T1 tumor-bearing mice taken after injection of Df-PPNs and 89Zr-Df-PPNs.
Figure 2.

Evaluation of CRIT and in vivo biodistribution. (a) Tumor volume growth profiles of in vivo CRIT following intravenous administration of control, Df-PPNs, 89Zr-Df-PPNs, coadministration of 89Zr-Df-PPNs and Df-PPNs using the pre-targeting delivery strategy in 4T1 tumor-bearing mice. (b) The tumor size of various groups 14 days into the study. Bars with different characters indicate statistical significance (P < 0.01). The different letters above the column mean that the p value is less than 0.01 while comparing to each other. The tumor volume under cotreatment of Df-PPNs + 89Zr-Df-PPNs 1.1 mCi group was much smaller than all the other groups. (c) Biodistribution of 89Zr-Df-PPNs on the 14th-day post-injection as determined by 89Zr radioactivity measurement in various tissues and organs (n = 3, mean ± s.d.). (* p < 0.05) (d) Representative maximum intensity projection PET images were taken at various time points post-injection.
Before performing in vivo test, in vitro studies were used to confirm that CR from 89Zr could be transferred to mTCPP in Df-PPNs and produce singlet oxygen without an external light source. As shown in Figure S7 and S8, the blue light from CR can slowly trigger ROS generation when combined with Df-PPNs. The coincubation groups significantly inhibited the cancer cell growth (Figure S9) and also increased immunofluorescence signals of γ-H2AX compared to groups treated with only 89Zr or PPNs (Figure S10). The ROS levels (Figure S11) for the various treatment groups coincided with cellular nucleus damage. The tumor growth profiles for each mouse in the CRIT group (Df-PPNs + 89Zr-Df-PPNs 400 μCi or 1.1 mCi) was considerably lower than the control groups (PBS and Df-PPNs) (Figure 2a and S12). The weight of mice in treatment groups confirmed that those treatment procedures did not show significant toxicity in the tested animals (Figure S13). Moreover, mice treated with the CRIT group with a lower dose of 89Zr (Df-PPNs + 89Zr-Df-PPNs 400 μCi) also showed an effective response to CRIT, with an approximately eightfold smaller average tumor volume compared to the PBS group (Figure 2b). c. As shown in Figure 2c, the intratumor accumulation of 89Zr in the Df-PPNs + 89Zr-Df-PPNs 1.1 mCi, Df-PPNs + 89Zr-Df-PPNs 400 μCi, and 89Zr-Df-PPNs 1.1 mCi groups were relatively high with values of 20.4±1.4, 14.8±1.8, 16.0±1.4 %ID/g, respectively (n=3). PET imaging also confirmed the high tumor uptake of 89Zr-Df-PPNs, which enabled CRIT because of its long retention period (Figure 2d). These findings suggested that missile-detonation approach was successful in effectively deliver the photosensitizer and radionuclide into the tumor to inhibit tumor growth by CRIT.
Anti-angiogenic therapy is considered as a promising and well-known strategy to treat several solid tumor types seen in the clinic.[9] However, variable clinical experience has revealed a serious of severe off-target adverse events associated with anti-angiogenic agents across different cancer types.[10] However, experimental evidence indicates that in many cases, anti-angiogenic drugs might inhibit anticancer drug transport into the tumor by reducing vascular density, which is not a suitable outcome.[11] Thus, developing an anti-angiogenic strategy to treat tumors is a critical goal in oncology. As shown in Figure 3a, we found that the tumor tissues and the blood vessel around/in the tumor site were damaged after cotreatment. Bruises were found at the surface of the tumor and its surrounding tissues 10 days after cotreatment with Df-PPNs and 89Zr-Df-PPNs (Figure 3b and S14). As shown in Figure 3c, the results indicated that most of the tumor tissue in the cotreatment group of Df-PPNs and 89Zr-Df-PPNs were necrotic, while the tumor tissue in the other three control groups generally retained their normal morphology (Figure S15). The residual tumor tissues indicated severe necrotic centers and fragmentary architecture. As shown in Figure 3d1, the red fluorescence of mTCPP in Df-PPNs (indicated by arrow 1) was distributed well around the blue nucleus (DAPI) in the residual tumor tissue, indicating the efficient uptake of Df-PPNs in the tumor. The gap indicated by arrow 2 was found both in H&E and confocal fluorescence images. This gap was believed to be due to a blood vessel and confirmed by CD31 immunostaining (Figure 3d2, 3d3, S16, and S17) to show the destroyed vessel architecture. The red line and dots found in H&E images (arrows 3 and 4) in the edge of the tumor are attributed to the aggregation of Df-PPNs in tumor. We also found the noteworthy red node shown in Figure 3d4 (arrow 4), which was also due to the aggregation of Df-PPNs. Therefore, cotreatment of CR-induced PDT significantly inhibited tumor growth and caused fragmented the tumor vasculature. As shown in Figure S18, the possible mechanisms for aggregation of Df-PPNs in the tumor may due to the cross-linking of Df-PPNs and further attach with mixture of dead cells and released contents from dying cells during local PDT.
Figure 3.

(a) Schematic demonstrating the missile-detonation strategy of the photosensitizer (Df-PPNs) into the tumor for activation from CR produced by 89Zr-Df-PPNs. (b) Representative photographs of tumors from cotreatment of Df-PPNs and 89Zr-Df-PPNs. The tumor is indicated by the yellow circle. (c) H&E-stained tumor slices collected 14 days after cotreatment of Df-PPNs and 89Zr-Df-PPNs. (d) Confocal images of tumor tissues collected 14 days after cotreatment of Df-PPNs and 89Zr-Df-PPNs (scale bar 50 μm). The areas indicated by arrows with the same numbers represent similar pathology in H&E and confocal images.
In summary, we reported a missile-detonation strategy that separately delivered nanophotosensitizer (Df-PPNs) and 89Zr-Df-PPNs to limit systemic toxicity, but colocalized in tumor site to maximize the utility of the weak CR for CRIT in a spatiotemporal manner. The spatial proximity of photosensitizer (mTCPP) and radionuclide (89Zr) endowed excellent integration of optical and PET imaging. CR from 89Zr was transferred to mTCPP in Df-PPNs and produced singlet oxygen without an external light source. Moreover, this CRIT significantly inhibited tumor growth and caused fragmentary vascular architecture in the tumor. During CRIT, the aggregation of Df-PPNs with 89Zr-Df-PPNs in the tumor produced ROS that damaged cells in the tumor site. Our results offer a rational design to harness weak CR as an efficient cancer theranostic therapy by combining multimodal imaging with damaging tumor vascular. One limitation of this study was the limited hematology assays. Herein, only the two primary groups (co-delivery method and pre-delivery strategy) were performed to demonstrate the reduction of side effects using the missile-detonation strategy. Further research is needed to determine the potential toxicity of Df-PPNs with various concentrations and activities in living subjects. In addition, anti-angiogenic therapy is a well-known and promising strategy to treat solid tumors. Some molecularly-targeted anti-angiogenic agents inhibited tumor metastasis and tumor angiogenesis at specific doses but promoted these processes at higher doses. A novel finding is the damage to the tumor blood vessels caused by the local photodynamic therapy. Another limitation of this study was the limited analysis of tumor metastasis and inflammation using the proposed therapy strategy. Further studies are needed to determine the advantages and disadvantages of our strategy that damages tumor vessels.
Supplementary Material
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81601605, 21571147) and the Postdoctoral Science Foundation of China (2016M600670). This work was also partly supported by the Natural Science Foundation of SZU (Grant No. 827-000143), Shenzhen Peacock Plan (KQTD2016053112051497), and Shenzhen Basic Research Program (JCYJ20170302151858466). This work was supported, in part, by the University of Wisconsin–Madison, the National Institutes of Health (P30CA014520), and the Brazilian Science without Borders Program (SwB-CNPq). All animal studies were conducted under the protocol approved by the University of Wisconsin Institutional Animal Care and Use Committee.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Bo Yu, National-Regional Key Technology Engineering Laboratory for Medical Ultrasound, Guangdong Key Laboratory for Biomedical Measurements and Ultrasound Imaging, School of Biomedical Engineering, Health Science Center, Shenzhen University, Shenzhen 518060, China.; Departments of Radiology and Medical Physics, University of Wisconsin–Madison, Madison, WI 53705, USA. Key Laboratory for Green Chemical Process of Ministry of Education, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, Wuhan 430073, China.
Dalong Ni, Departments of Radiology and Medical Physics, University of Wisconsin–Madison, Madison, WI 53705, USA..
Zachary T. Rosenkrans, Departments of Radiology and Medical Physics, University of Wisconsin–Madison, Madison, WI 53705, USA.
Todd E. Barnhart, Departments of Radiology and Medical Physics, University of Wisconsin–Madison, Madison, WI 53705, USA.
Hao Wei, Departments of Radiology and Medical Physics, University of Wisconsin–Madison, Madison, WI 53705, USA.; Dept of Nuclear Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430073, China.
Carolina A. Ferreira, Departments of Radiology and Medical Physics, University of Wisconsin–Madison, Madison, WI 53705, USA.
Xiaoli Lan, Dept of Nuclear Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430073, China..
Jonathan W. Engle, Departments of Radiology and Medical Physics, University of Wisconsin–Madison, Madison, WI 53705, USA.
Qianjun He, National-Regional Key Technology Engineering Laboratory for Medical Ultrasound, Guangdong Key Laboratory for Biomedical Measurements and Ultrasound Imaging, School of Biomedical Engineering, Health Science Center, Shenzhen University, Shenzhen 518060, China..
Faquan Yu, Key Laboratory for Green Chemical Process of Ministry of Education, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, Wuhan 430073, China..
Weibo Cai, Departments of Radiology and Medical Physics, University of Wisconsin–Madison, Madison, WI 53705, USA..
References
- [1].a) Tavares AJ, Poon W, Zhang YN, Dai Q, Besla R, Ding D, Ouyang B, Li A, Chen J, Zheng G, Robbins C, Chan WCW, Proc. Natl. Acad. Sci. U. S. A 2017, 114, E10871; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim KY, Nanomed. 2007, 3, 103; [Google Scholar]; c) Bai J, Jia X, Zhen W, Cheng W, Jiang X, J. Am. Chem. Soc 2018, 140, 106; [DOI] [PubMed] [Google Scholar]; d) Zhen W, Liu Y, Lin L, Bai J, Jia X, Tian H, Jiang X, Angew. Chem. Int. Ed. Engl 2018, 57, 10309. [DOI] [PubMed] [Google Scholar]
- [2].a) Song G, Cheng L, Chao Y, Yang K, Liu Z, Adv. Mater 2017, 29; [DOI] [PubMed] [Google Scholar]; b) Dai Q, Wilhelm S, Ding D, Syed AM, Sindhwani S, Zhang Y, Chen YY, MacMillan P, Chan WCW, ACS Nano 2018, 12, 8423; [DOI] [PubMed] [Google Scholar]; c) Chan WCW, Khademhosseini A, Parak WJ, Weiss PS, ACS Nano 2017, 11, 8528. [DOI] [PubMed] [Google Scholar]
- [3].a) Seshadri M, Spernyak JA, Mazurchuk R, Camacho SH, Oseroff AR, Cheney RT, Bellnier DA, Clin. Cancer Res 2005, 11, 4241; [DOI] [PubMed] [Google Scholar]; b) Ma Z, Zhang M, Jia X, Bai J, Ruan Y, Wang C, Sun X, Jiang X, Small 2016, 12, 5477; [DOI] [PubMed] [Google Scholar]; c) Ma Z, Jia X, Bai J, Ruan Y, Wang C, Li J, Zhang M, Jiang X, Adv. Funct. Mater 2017, 27, 1604258. [Google Scholar]
- [4].Zhen Z, Tang W, Chuang YJ, Todd T, Zhang W, Lin X, Niu G, Liu G, Wang L, Pan Z, Chen X, Xie J, ACS Nano 2014, 8, 6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Shaffer TM, Pratt EC, Grimm J, Nat. Nanotechnol 2017, 12, 106; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kotagiri N, Sudlow GP, Akers WJ, Achilefu S, Nat. Nanotechnol 2015, 10, 370; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kotagiri N, Cooper ML, Rettig M, Egbulefu C, Prior J, Cui G, Karmakar P, Zhou MZ, Yang XX, Sudlow G, Marsala L, Chanswangphuwana C, Lu L, Habimana-Griffin L, Shokeen M, Xu XM, Weilbaecher K, Tomasson M, Lanza G, DiPersio JF, Achilefu S, Nat. Commun 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Yu B, Goel S, Ni D, Ellison PA, Siamof CM, Jiang D, Cheng L, Kang L, Yu F, Liu Z, Barnhart TE, He Q, Zhang H, Cai W, Adv. Mater 2018, 30, e1704934; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ni DL, Ehlerding EB, Cai WB, Angew. Chem. Int. Ed. Engl 2019, 58, 2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Kamkaew A, Cheng L, Goel S, Valdovinos HF, Barnhart TE, Liu Z, Cai W, ACS Appl. Mater. Interfaces 2016, 8, 26630; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ni D, Ferreira CA, Barnhart TE, Quach V, Yu B, Jiang D, Wei W, Liu H, Engle JW, Hu P, Cai W, J. Am. Chem. Soc 2018, 140, 14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yu B, Wei H, He Q, Ferreira CA, Kutyreff CJ, Ni D, Rosenkrans ZT, Cheng L, Yu F, Engle JW, Lan X, Cai W, Angew. Chem. Int. Ed. Engl 2018, 57, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Volz J, Mammadova-Bach E, Gil-Pulido J, Nandigama R, Remer K, Sorokin L, Zernecke A, Abrams SI, Ergun S, Henke E, Nieswandt B, Blood 2019; [DOI] [PubMed] [Google Scholar]; b) Elice F, Rodeghiero F, Thromb. Res 2012, 129 Suppl 1, S50. [DOI] [PubMed] [Google Scholar]
- [10].a) Potiron V, Clement-Colmou K, Jouglar E, Pietri M, Chiavassa S, Delpon G, Paris F, Supiot S, Cancer Lett. 2019, 457, 1; [DOI] [PubMed] [Google Scholar]; b) A. Hulin, J. Stocco, M. Bouattour, Clin. Pharmacokinet 2019. [DOI] [PubMed] [Google Scholar]
- [11].a) Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN, Hendrikse NH, Eriksson J, Windhorst AD, Postmus PE, Verheul HM, Serne EH, Lammertsma AA, Smit EF, Cancer Cell 2012, 21, 82; [DOI] [PubMed] [Google Scholar]; b) Rohrig F, Vorlova S, Hoffmann H, Wartenberg M, Escorcia FE, Keller S, Tenspolde M, Weigand I, Gatzner S, Manova K, Penack O, Scheinberg DA, Rosenwald A, Ergun S, Granot Z, Henke E, Oncogene 2017, 36, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Arjaans M, Oude Munnink TH, Oosting SF, Terwisscha van Scheltinga AG, Gietema JA, Garbacik ET, Timmer-Bosscha H, Lub-de Hooge MN, Schroder CP, de Vries EG, Cancer Res 2013, 73, 3347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.