

Abstract
Homozygous loss of function of the melanocortin 1 receptor (MC1R) is associated with a pheomelanotic pigment phenotype and increased melanoma risk. MC1R heterozygosity is less well studied, although individuals inheriting one loss-of-function MC1R allele are also melanoma-prone. Using the K14-Scf C57BL/6J animal model whose skin is characterized by life-long retention of interfollicular epidermal melanocytes like that of the human, we studied pigmentary, UV responses and DNA repair capacity in the skin of variant Mc1r background. Topical application of forskolin, a skin-permeable pharmacologic activator of cAMP induction to mimic native Mc1r signaling, increased epidermal eumelanin levels, increased the capacity of Mc1r-heterozygous skin to resist UV-mediated inflammation, and enhanced the skin’s ability to clear UV photolesions from DNA. Interestingly, topical cAMP induction also promoted melanin accumulation, UV resistance, and accelerated clearance in Mc1r fully-intact skin. Together, our findings suggest that heterozygous Mc1r loss is associated with an intermediately melanized and DNA repair-proficient epidermal phenotype and that topical cAMP induction enhances UV resistance in Mc1r-heterozygous or –wild type individuals by increasing eumelanin deposition and by improving nucleotide excision repair.
Keywords: melanocyte, UV radiation, melanin, melanocortin 1 receptor (MC1R), cAMP, DNA repair
Introduction
Cutaneous melanoma, the sixth most commonly diagnosed cancer in the U.S. (Cronin et al., 2018), is the deadliest of skin malignancies. Roughly 100,000 US citizens are projected to be diagnosed with the disease and almost 10,000 will die of melanoma in 2019 (Siegel, Miller, & Jemal, 2019). Skin complexion has long been appreciated to be a major inherited risk factor for melanoma, with fair-skinned UV-sensitive individuals having a much higher incidence than persons of a dark, UV-protected phenotype. One of the major genes that influences pigmentary and UV responses is the melanocortin 1 receptor (MC1R) which encodes a G protein-coupled receptor (GPCR) localized to the melanocyte plasma membrane that interacts with melanocortins such as melanocyte stimulating hormone (MSH) to transmit differentiation and UV-protective responses via cAMP second messenger signaling (reviewed in (Wolf Horrell, Boulanger, & D’Orazio, 2016)). When the MC1R signaling axis is functional, melanocytes respond to melanocortins by increasing the production and export of UV-blocking eumelanin (Suzuki et al., 1999), increasing antioxidant defenses (Kadekaro et al., 2012; Song et al., 2009) and optimizing the efficiency of the nucleotide excision repair pathway (Hauser et al., 2006), the genomic maintenance pathway that recognizes and removes UV photoproducts from UV-damaged cells. Since the majority of somatic mutations found in melanoma bear the “UV signature” defined by transitional mutations in adjacent pyrimidines (Shain et al., 2015), it follows that a strategy that reduces UV mutagenesis would attenuate melanocyte carcinogenesis and lower melanoma risk.
In humans, polymorphisms are common at the MC1R locus, with many mutations resulting in reduced capacity to transmit melanocortin signals to melanocytes (Sturm, Teasdale, & Box, 2001). Loss-of-function MC1R polymorphisms are associated with a UV-sensitive, fair-skinned (Valverde, Healy, Jackson, Rees, & Thody, 1995) and melanoma-prone phenotype (Valverde et al., 1996). While homozygous loss of MC1R function is classically associated with a “red hair color” (RHC) phenotype and a clear predisposition to melanoma (Kennedy et al., 2001), heterozygous MC1R loss is less well studied. MC1R heterozygosity, however, has been linked to melanoma even without the overt RHC pigment phenotype (Guida et al., 2015; Matichard et al., 2004; Pasquali et al., 2015). Indeed, a recent study documented that melanoma somatic mutational burden was higher in individuals with either one or two MC1R loss of function alleles than those which had two wild type MC1R alleles (Robles-Espinoza et al., 2016), suggesting that MC1R haploinsufficiency raises UV mutagenic susceptibility and melanoma risk.
We utilized the C57BL/6J K14-Scf mouse model to determine whether heterozygous inheritance of Mc1r loss-of-function mutation influences UV sensitivity and skin responses and whether topical cAMP induction would promote eumelanization and/or accelerated DNA repair in the skin. This animal model, characterized by life-long retention of interfollicular epidermal melanocytes due to constitutive expression of stem cell factor (c-Kit ligand) in the basal epidermis (Kunisada et al., 1998), was previously crossed with the recessive yellow “extension” Mc1re/e C57BL/6J mice to generate homozygous Mc1r-defective animals with a pheomelanotic epidermal skin and coat color which could be switched to a eumelanotic UV-protective phenotype by pharmacologic induction of cAMP in the skin (D’Orazio et al., 2006). The extension locus encodes a shortened and signaling-dead Mc1r due to a frameshift caused by a deletion of a single nucleotide at position 549 (Robbins et al., 1993). Here, we describe the pigment phenotype, DNA repair capacity, and UV sensitivity of K14-Scf C57BL/6J mice of variable Mc1r signaling capacity. Our results indicate that topical application of forskolin, a direct activator of adenylyl cyclase that increases cAMP levels in the skin to mimic Mc1r signaling, increased epidermal eumelanin levels, heightened UV resistance and accelerated clearance of UV photodamage in Mc1r-mutant, -heterozygous or -wild type skin. Together, these proof-of-concept studies suggest that topical cAMP stimulation may be a useful UV-protective and potentially cancer-preventive approach in MC1R-defective, -heterozygous or even wild type individuals.
Materials and Methods
Animals.
C57BL/6J K14-Scf mice, developed as previously reported (D’Orazio et al., 2006; Kunisada et al., 1998), were used in all experiments. Animals were maintained and bred at the University of Kentucky Division of Lab Animal Resources facilities in microisolator caging and the protocol was reviewed and approved by the University of Kentucky Institutional Animal Care and Use Committee (IACUC). The procedures employed herein were in compliance with the US National Research Council’s Guide for the Care and Use of Laboratory Animals, the US Public Health Service’s Policy on Humane Care and Use of Laboratory Animals, and Guide for the Care and Use of Laboratory Animals. Since K14-Scf transgenic females have fertility problems, our approach to generating Mc1rE/e K14-Scf C57BL/6J mice was by crossing Mc1rE/E K14-Scf C57BL/6J males with Mc1re/e non-transgenic females. A similar strategy was used to generate Mc1rE/E and Mc1re/e experimental animals, using parents of appropriate genotype. The presence of the K14-Scf transgene was assessed phenotypically in offspring since transgenic animals have a distinct epidermal pigmentary phenotype (darkly pigmented nose spots, paw spots and darkened genitalia and melanized generalized epidermis throughout life). Amelanotic (albino) Mc1rE/e K14-Scf C57BL/6J mice for use in cutaneous DNA repair assays (to isolate pigment-independent effects of Mc1r heterozygosity on nucleotide excision repair) were generated by crossing Mc1rE/E Tyrc2j/c2j K14-Scf C57BL/6J males with Mc1re/e Tyrc2j/c2j non-transgenic females. All offspring were Mc1rE/e Tyrc2j/c2j and the presence of the K14-Scf transgene was assessed phenotypically since K14-Scf Mc1rE/e Tyrc2j/c2j animals demonstrate darkening of the ears and tail after 6–8 weeks of life.
Chemicals and reagents.
Forskolin for cutaneous application to the mice was prepared as described (D’Orazio et al., 2006). Briefly, Coleus forskohlii root extract (20% w⁄w forskolin) was purchased from Phytotech Extracts Pvt. Ltd (Waterloo, ON, Canada), distributed through Buckton Scott USA, Inc. (Princeton, NJ, USA) and used as a working source of forskolin. C. forskohlii extract was prepared as a 40% weight:volume solution in 70% ethanol, 30% propylene glycol (Sigma-Aldrich Chemical Corporation, St. Louis, MO, USA). Briefly, the root extract was stirred in vehicle (1h, room temperature), centrifuged (1,500 x g, 15 min) and filtered (0.22 μm cellulose acetate membrane) to remove particulate matter. Independent assay of the amount of forskolin in the 40% (w ⁄v) working solution revealed a concentration of approximately 200 mM.
Cutaneous application of topical treatments.
Mice between 4 and 8 weeks of age were used for these experiments unless otherwise noted. Topical agents were applied to the skin as previously described (Amaro-Ortiz, Vanover, Scott, & D’Orazio, 2013). Unless otherwise indicated, animals were treated once daily with 200 μL of topical agent for 5 days a week with either 200 mM, 20 mM, 2 mM or 0 mM (vehicle control; 70% ethanol, 30% propylene glycol) prepared C. forskohlii root extract to deliver 40, 4, 0.4 or 0 μmoles of forskolin per application to dorsal depilated skin.
Measurement of degree of epidermal melanization by reflective colorimetry and photography.
Skin colorimetry measurements were assessed with a CR-400 Colorimeter (Minolta Corporation, Japan) calibrated against a white standard background provided by the manufacturer. Degree of melanization was quantified on the *L axis (black-white axis) of the CIE standard color axis (Wagner, Jovel, Norton, Parra, & Shriver, 2002). Care was taken to avoid measuring areas of anagen hair regrowth, which would confound assessment of epidermal pigmentation. At least three separate colorimetry measurements were taken of each animal at each condition and time point in at least 3 distinct areas of treated skin.
Fontana-Masson staining and histologic melanin quantification.
Skin biopsies were obtained from sheared skin of animals treated as indicated using institutionally approved protocols. Samples were fixed in 4% formalin for 48 hours and subsequently placed in 70% ethanol before being paraffin embedded, sectioned (4 microns) and stained by the Markey Cancer Center Biospecimen Procurement and Translational Pathology Shared Resource Facility. Fontana–Masson stained sections (Zappi & Lombardo, 1984) were used to quantify degree of melanization (blackness) of the sub-corneal epidermis by the HALO imaging software (Indica Labs). For histological quantification of epidermal melanin levels, 4–5 representative regions of the epidermal layer (stratum basale through stratum granulosum) from images of Fontana Masson stained slides were subjected to an area quantification algorithm (HALO, Indica Labs) that quantified black pixels in the defined area. The raw count of black pixels per total area analyzed was calculated and described as “% melanin.”
cAMP quantification of the skin.
Skin samples were obtained from sheared skin of K14-Scf-transgenic Mc1rE/E, Mc1rE/e and Mc1re/e animals 60 minutes and 120 minutes after topical treatment with vehicle or 200 mM forskolin as described above (n = 3 animals per treatment condition). Dermal fat was reduced with forceps before samples were quick-frozen (dry ice) and then pulverized into a powder using a mortar and pestle. Liquid nitrogen was added as needed to keep sample frozen until homogenization was complete. Powdered tissue was transferred to a 5mL conical tube, suspended in 2mL of 0.1 N HCl and homogenized on ice with a Kinematica Polytron PT1200E Handheld Homogenizer for 2–5 minutes. Samples were then neutralized with 1mL 1 N NaOH, centrifuged to remove particulate matter and protein content was quantified by Bradford colorimetry assay. cAMP content was determined using a mouse/rat cAMP parameter assay kit (R&D Systems) according to manufacturer’s instructions.
Chemical melanin quantification.
Eumelanin and pheomelanin were quantitatively analyzed by HPLC based on the formation of pyrrole-2,3,5-tricarboxylic acid (PTCA) by permanganate oxidation of eumelanin and 4-amino-3 hydroxyphenylalanine (4-AHP) by hydriodic acid reductive hydrolysis of pheomelanin, respectively as described. Eumelanin and pheomelanin content were calculated by multiplying those of PTCA and 4-AHP by factors of 25 and 7, respectively (Ito & Wakamatsu, 2003, 2011).
UV exposure and minimal erythematous dose (MED) testing.
MED testing was carried out as described (Amaro-Ortiz et al., 2013). MED was calculated as the minimal dose of radiation (per mouse) needed to cause erythema or edema of exposed skin 24–48 hours after irradiation.
Assessment of DNA repair in the skin.
Clearance of UV photoproducts was assessed in whole skin biopsies as previously reported (Jarrett et al., 2014). Briefly, mice pre-treated (twice a day for 3 days) with vehicle or forskolin were depilated 4 days prior to radiation (7.5 kJ/m2). 1 cm2 skin biopsies were collected at the indicated time points after UV exposure, DNA was isolated by DNEasy kit (Qiagen, Germantown, MD) and quantified by Nanovue nanodrop (GE Biosciences, Pittsburgh, PA). 0.1 μg/well DNA was loaded onto nitrocellulose membranes using a slot blot apparatus. Membranes were processed as described (Wang, Wu, & Friedberg, 1991) and immunoblotted for [6,4]-photoproducts using a monoclonal antibody against this UV photolesion (Cosmo Bio, Tokyo, Japan). Band intensities were analyzed using ImageJ software.
Statistical Analysis.
Data across strains were analyzed using two way ANOVA with Bonferroni post-test (Graph Pad PRISM). Statistical comparisons between experimental cohorts were evaluated by a Tukey’s post-test. p values < 0.05 are considered statistically significant.
Results
Generation of K14-Scf Mc1r-heterozygous mice.
As a foundation for studying UV responses and the impact of topical cAMP induction in the Mc1r-heterozygous state, we generated Mc1rE/e K14-Scf C57BL/6J mice by crossing Mc1rE/E K14-Scf C57BL/6J males with Mc1re/e (extension) non-transgenic females. The presence of the K14-Scf transgene in the F1 Mc1r-heterozygous generation was easily assessed because of the clear pigmentary phenotype on the epidermis as was previously reported in either parental strain (Vanover et al., 2009). Differences between K14-Scf transgenic and their non-transgenic Mc1rE/e counterparts were evident from the neonatal period, with transgenic animals demonstrating darkly pigmented nose spots, paw spots and darkened genitalia in infancy and accompanied by generalized pigmentation of the interfollicular epidermis including that of the dorsal skin as animals aged (Fig. 1). Images of K14-Scf transgenic and non-transgenic Mc1rE/E (Suppl. Fig. 1) or Mc1re/e mice (Suppl. Fig. 2) are included for comparison. To study pigment-independent cAMP effects such as DNA repair, amelanotic Mc1r-heterozygous animals were developed by incorporating the Tyrc2j/c2j tyrosinase deficiency on the C57BL/6J background (Suppl. Fig. 3).
Figure 1. Pigment phenotype of C57BL/6J Mc1rE/e K14-Scf transgenic and non-transgenic mice.

Mc1rE/e K14-Scf transgenic and non-transgenic mice were photographed at different ages of life. Neonatal animals between 3–8 days old are shown in panel A, weanling animals between 3–4 weeks of age are shown in panel B and adult mice between 4–6 months of age are shown in panel C. Male and female animals are each represented as indicated, with K14-Scf animals presented in the two left-most columns and non-transgenic animals shown in the two right-most columns. Note the pigmentary differences (interfollicular epidermal skin, particularly nose, genitals and paw pads) between K14-Scf and non-transgenic animals throughout life.
To measure degree of epidermal melanization, we quantified the color of depilated epidermal dorsal skin in the mice by colorimetry reading on the International Commission on Illumination (CIE) L* (black-white) color axis (Weatherall & Coombs, 1992). Epidermal pigmentation was similar between non-transgenic strains of mice, irrespective of Mc1r or Tyr status (Fig. 2), which is consistent with a lack of melanization of the epidermis in the absence of interfollicular melanocytes facilitated by the K14-Scf transgene. With incorporation of the K14-Scf transgene, however, the degree of epidermal melanization was markedly enhanced in either the wild type (Mc1rE/E, Tyrwt/wt) or heterozygous (Mc1rE/e, Tyrwt/wt) state. We also observed a trend toward a darker complexion in the pheomelanotic (extension; Mc1re/e, Tyrwt/wt) K14-Scf mice as compared to their non-transgenic counterparts (Fig. 2), consistent with our prior work documented that more pheomelanin accumulates in the skin of K14-Scf extension (Mc1re/e) mice (Vanover et al., 2009). We interpret these data to be consistent with the hypothesis that skin pigmentation is mainly regulated by eumelanin rather than pheomelanin deposition and that addition of the K14-Scf transgene results in augmentation of melanin levels in the interfollicular epidermis of tyrosinase-intact mice.
Figure 2. Skin colorimetry of K14-Scf transgenic and non-transgenic mouse strains.

Untreated C57BL/6J mice between the ages of 6 and 12 weeks of life were depilated and the color of the dorsal depilated skin was quantified by reflective colorimetry. Areas of anagen phase hair regrowth were excluded from measurements because of the confounding influence of the growing hairs on the reflectometry of the epidermal skin. Data are reported on the black-white L* CIE color axis with darker skin having lower L* scores. Mc1r and Tyr status of the strains are indicated. Colorimetry of non-transgenic (white bars) vs. K14-Scf mice (black bars) are compared. Values not sharing a common letter were significantly different as determined by one -way ANOVA; p ≤ 0.05.
Topical cAMP induction promotes melanization in the Mc1r heterozygous state.
Previously, our group reported that application of forskolin, a skin-permeable direct activator of adenylyl cyclase, promoted the accumulation of eumelanin in the epidermis of Mc1r-defective (extension), pheomelanotic K14-Scf mice which was accompanied by resistance to acute and chronic UV damage (D’Orazio et al., 2006; Spry et al., 2009). Because Mc1r-heterozygous K14-Scf mice are relatively eumelanotic in their basal state (Figs. 1, 2), we tested whether topical forskolin application could increase epidermal eumelanization, reasoning that further melanization might enhance UV resistance of the skin in the Mc1r heterozygous state. Mc1rE/e K14-Scf animals were sheared, depilated, and treated once a day for five days each week (200 μL per dose) with increasing concentrations (0.4, 4 or 40 μmoles of forskolin per application) of a root extract of the Coleus forskohlii plant, the same preparation that was previously used to induce eumelanization in fair-skinned Mc1r-defective (Mc1re/e) K14-Scf mice (D’Orazio et al., 2006). We observed progressive darkening of the skin of Mc1rE/e K14-Scf mice in a dose-dependent manner (Fig. 3A) that was characterized by melanin deposition throughout the epidermis (Suppl. Fig. 4A,B) and accumulation of eumelanin as measured by chemical analysis (Fig. 3B). We documented that topically applied forskolin increased cAMP levels in the skin irrespective of Mc1r genotype and that forskolin-induced cAMP levels were higher at 60 minutes (vs. 120 minutes) post-application. Application of forskolin caused cAMP levels to increase in the skin across Mc1r genotypes, with levels at 60 minutes higher than at 120 minutes (Suppl. Fig. 4C). In other experiments, we altered the dosing schedule from once-a-day each weekday to twice-a-day each weekday and noted robust darkening of Mc1rE/e K14-Scf mice by forskolin by day 10 (Suppl. Fig. 5), suggesting that it may be possible to more rapidly melanize the skin by increasing the dosing frequency the dosing schedule of the drug. Intriguingly, at this accelerated scheduling, each of the forskolin concentrations darkened the skin significantly (p < 0.05) when compared to control (Suppl. Fig. 5).
Figure 3. Topical forskolin promotes melanization of Mc1r-heterozygous skin in a dose-dependent manner.

Mc1rE/e K14-Scf animals were depilated, and treated with increasing concentrations of forskolin as indicated once a day for five days each week (200 μL per dose; 0, 0.4, 4 or 40 μmoles of forskolin per application. A) Skin color was recorded weekly and quantified on the black-white L* CIE color axis with darker skin having lower L* scores. * indicates statistical significance (p < 0.05) as compared to control-treated animals. B) Eumelanin and pheomelanin quantification of skin biopsies at the end of the experiment (day 21). Values not sharing a common letter were significantly different as determined by one -way ANOVA; p ≤ 0.05.
Since we had previously established that topically applied forskolin promoted darkening of complexion and eumelanin deposition in the skin of Mc1r-defective animals, we included Mc1re/e K14-Scf mice as a positive control to ensure function of the forskolin preparation. We also incorporated homozygous wild type Mc1r K14-Scf animals in our studies to determine if melanin content of fully Mc1r-functional skin is maximal or whether melanin can also be induced in the Mc1r-wild type background. Topical application of the forskolin extract darkened the skin of K14-Scf extension (Mc1re/e) mice and enhanced eumelanin levels (Fig. 4A,B), as previously reported (D’Orazio et al., 2006). Application of the extract to tyrosinase-null animals had no impact on skin color or melanin levels (Fig. 4A,B), confirming that synthesis of either eumelanin or pheomelanin is tyrosinase-dependent. Melanin quantification across the different strains of mice also confirmed that there is a diluted eumelanin phenotype in the Mc1r-heterozygous state, suggesting that eumelanin production may be proportional to Mc1r gene dosage. Importantly, we found that topical forskolin increased eumelanin levels even in the Mc1r-homozygous wild type state (Fig. 4A,B), suggesting that melanocyte pigment synthetic capacity can be up-regulated pharmacologically even with two unmutated Mc1r alleles, which is consistent with the concept of Mc1r-mediated adaptive tanning after UV injury (Cui et al., 2007). In the Mc1r-heterozygotes, forskolin not only rescued the diluted eumelanin phenotype (as compared with homozygous Mc1r animals) but raised eumelanin levels much higher than in forskolin-treated Mc1re/e mice and to roughly the same degree as in forskolin-treated Mc1rE/E mice (Fig. 4A,B). Fontana Masson-stained images and histologic melanin quantification are shown for each of the four pigment strains (Suppl. Fig. 6). Together, these data suggest that pharmacologic induction of cAMP in the skin can promote eumelanization of the skin in tyrosinase-intact individuals, irrespective of Mc1r function and that topical cAMP induction results in the accumulation of similar levels of eumelanin in the Mc1r homozygous or heterozygous state.
Figure 4. Topical forskolin promotes melanization of tyrosinase-intact K14-Scf mice irrespective of Mc1r status.

A) Wild type (Mc1rE/E Tyrwt/wt), heterozygote (Mc1rE/e Tyrwt/wt), extension (Mc1re/e Tyrwt/wt), or albino extension (Mc1re/e Tyrc2j/c2j) K14-Scf animals were depilated, and treated with vehicle (dotted lines) or forskolin (solid lines) as indicated once a day for five days each week (40 μmoles of forskolin per application) through 21 days. Skin color was recorded weekly and quantified on the black-white L* CIE color axis with darker skin having lower L* scores. B) Eumelanin and pheomelanin quantification of skin biopsies at the end of the experiment (day 21). * indicates statistical significance (p < 0.05) as compared to control-treated animals within the same strain.
cAMP-induced melanin protects Mc1r heterozygous mice from UV-mediated inflammation.
We next determined whether forskolin-induced melanization could increase cutaneous resistance to UV-induced inflammation by quantifying the minimal erythematous dose (MED) of animals treated with vehicle control or with forskolin. The MED is the least amount of UV required to cause either erythema or edema of the skin, and we have used this measure in the C57BL/6J K14-Scf animal model to assess UV sensitivity (Amaro-Ortiz et al., 2013). Mc1rE/e K14-Scf mice were pre-treated with increasing doses of forskolin as described above (Fig. 3A) before being subjected to MED testing. We found a dose-dependent forskolin-mediated protective effect (Fig. 5A). In the animals treated with 40 μmoles of the drug per application, we noted a 5.6-fold degree of protection (MED from 3.4 ± 0.7 kJ/m2 in the control-treated mice to 19.0 ± 4.6 kJ/m2 in 40 μmoles forskolin-treated animals).
Figure 5. Pharmacologic cutaneous cAMP induction protects against UV-mediated inflammation.

A) Mc1rE/e K14-Scf animals were depilated, and treated with increasing concentrations of forskolin as indicated once a day for five days each week (200 μL per dose; 0, 0.4, 4 or 40 μmoles of forskolin per application. Animals were then exposed to varying doses of UV-B and MED was calculated per mouse. Mean ± SEM MEDs are shown for each forskolin dose group. * indicates statistical significance (p < 0.05) as compared to the other groups. B) MED testing was extended to wild type (Mc1rE/E Tyrwt/wt), heterozygote (Mc1rE/e Tyrwt/wt), extension (Mc1re/e Tyrwt/wt), or albino extension (Mc1re/e Tyrc2j/c2j) K14-Scf animals treated (21d, vehicle vs. forskolin (40 μmoles per application) as indicated. Animals were then exposed to varying doses of UV-B and MED was calculated per mouse. Mean ± SEM MEDs are shown for each strain; values not sharing a common letter were significantly different as determined by one -way ANOVA; p ≤ 0.05.
We next compared the ability of topical forskolin to impact UV sensitivity among strains with fully functional (Mc1rE/E) or null (Mc1re/e) Mc1r status. We documented that in control-treated mice, the MED ± SEM for Mc1re/e K14-Scf mice was 1.0 ± 0.0 kJ/m2 while the MED ± SEM for Mc1rE/e K14-Scf mice was 3.4 ± 0.7 kJ/m2 (Fig. 5B). We interpret this difference to be due to the increased amount of eumelanin in vehicle-treated skin of Mc1r-heterozygous mice as compared to the pheomelanotic skin of vehicle-treated Mc1r-defective counterparts. Consistent with prior findings (Amaro-Ortiz et al., 2013; Spry et al., 2009), pre-treatment of extension K14-Scf (Mc1re/e) mice protected the animals against UV-induced inflammation, as evidenced by an increase in MED from 1.0 ± 0.0 kJ/m2 to 10.7 ± 0.7 kJ/m2 in control vs. forskolin-treated animals (10.7-fold protection). Application of forskolin to K14-Scf wild type (Mc1rE/E) mice also showed a protective effect, increasing the MED ± SEM from 1.7 ± 0.6 kJ/m2 to 23.3 ± 5.8 kJ/m2 (Fig. 5B). Importantly, forskolin had no effect on the MED of amelanotic animals, showing that the protective effect of cAMP stimulation against UV-mediated inflammation is tyrosinase (and presumably melanin)-dependent (Fig. 5B). Together, these data indicate that topical forskolin protects against UV-induced inflammation in a pigment-dependent manner irrespective of Mc1r signaling ability.
cAMP increases the efficiency of nucleotide excision repair in Mc1r heterozygous mice.
Having established that topically applied forskolin increases melanization and resistance to UV-mediated inflammation of the skin in Mc1r-heterozygous mice, we next asked whether cutaneous cAMP induction could influence DNA repair of UV photodamage. We previously documented improved recovery of UV-induced photodamage in Mc1re/e K14-Scf mice by cutaneous cAMP induction (Jarrett et al., 2014). To understand whether topically-applied forskolin could similarly accelerate nucleotide excision repair in the Mc1r-heterozygous state, we used tyrosinase-null animals to isolate pigment-independent effects of cAMP on the DNA repair process. Mc1rE/e Tyrc2j/c2j K14-Scf mice were pre-treated twice a day for three days with either vehicle or 40 μmoles forskolin prior to UV exposure (7.5 kJ/m2) and skin samples were biopsied over time for quantification of [6,4]-photoproducts, a known mutagenic UV lesion. We found that forskolin accelerated repair efficiency in the Mc1r heterozygous state (Fig. 6A). As a positive control, we verified a similar benefit to nucleotide excision repair in Mc1r-defective (Mc1re/e Tyrc2j/c2j K14-Scf) mice (Fig. 6B), confirming our prior observations (Jarrett et al., 2014). Importantly, we also observed cAMP-enhanced nucleotide excision repair in Mc1r wild type (Mc1rE/E Tyrc2j/c2j K14-Scf) mice (Fig. 6C), suggesting that the ability of the skin to remove mutagenic UV photolesions can be enhanced by pharmacologic cAMP induction irrespective of Mc1r status. Together, these proof-of-principle experiments suggest that topical cAMP induction protects skin of any Mc1r genotype against UV injury by enhancing epidermal melanization, reducing UV-mediated inflammation and increasing the capacity of the skin to recover from UV-induced DNA damage.
Figure 6. Topical forskolin accelerates clearance of UV photodamage in the skin.
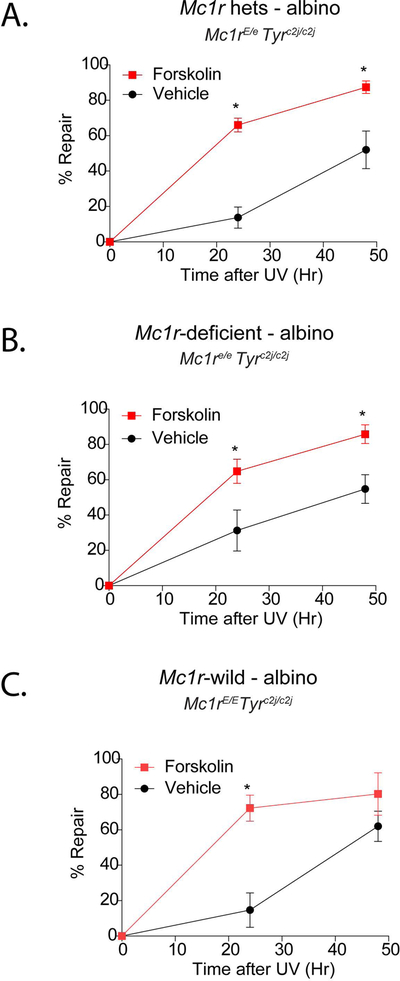
Mc1rE/e Tyrc2j/c2j K14-Scf (A), Mc1re/e Tyrc2j/c2j K14-Scf (B) or Mc1rE/E Tyrc2j/c2j K14-Scf (C) mice were depilated and pre-treated (twice a day × 2 days; 0 vs. 40 μmoles forskolin) before exposure to 7.5 kJ/m2 UV-B. Skin was biopsied at the indicated time points, DNA was extracted, and quantification of [6,4]-photoproducts was accomplished by Southwestern blotting. Data are expressed as relative to un-irradiated mice. * indicates statistical significance (p < 0.05) between vehicle and forskolin-treated animals at the given time point. Mean time ± SEM to repair 50% of UV damage is indicated as RT50.
Discussion
In this study, we examined the pigmentation and DNA repair responses of Mc1r-heterozygous mice, which because of incorporation of the K14-Scf transgene, maintain interfollicular epidermal melanocytes in the skin throughout life (Kunisada et al., 1998) and thus mimic the MC1R-heterozygous human. Using this model but in the fully Mc1r-deficient state, we previously observed that on the C57BL/6J background, Mc1r loss led to a profound loss of eumelanin in the epidermis and was essentially replaced by pheomelanin instead (D’Orazio et al., 2006). Reasoning that topical administration of forskolin, a skin-permeable activator of adenylyl cyclase, might mimic native Mc1r signaling and rescue eumelanization of the skin of Mc1re/e K14-Scf C57BL/6J mice, we previously observed robust accumulation of eumelanin (and UV protection) in a time- and dose-dependent manner (D’Orazio et al., 2006), and further reported that the efficiency of DNA repair of UV photoproducts was up-regulated as well (Jarrett et al., 2014). Each of these effects would be expected to diminish UV-dependent photodamage and mutagenesis and theoretically reduce melanoma risk.
The rationale for extending our studies into the Mc1r heterozygous state is that while homozygous MC1R loss is a well-documented UV sensitivity and melanoma risk factor, it is now evident that genetic depletion of even a single MC1R allele leads to a hyper mutagenic state in melanocytes (Robles-Espinoza et al., 2016) and increased melanoma propensity (Guida et al., 2015; Matichard et al., 2004; Pasquali et al., 2015). It is unclear how MC1R loss-of-function mutations elevate melanoma risk even as heterozygotes. It is unknown whether MC1R haploinsufficiency might limit eumelanin synthesis and/or DNA repair and if so, what the mechanism of this might be. Indeed, MC1R protein expression is typically low, with an estimated 700 protein units expressed per melanocyte and somewhat higher numbers on melanoma cells (Donatien et al., 1992; Roberts, Newton, Beaumont, Helen Leonard, & Sturm, 2006). Reducing the MC1R gene dosage (in the heterozygous state) might decrease MC1R signaling below a critical threshold to attenuate melanin synthesis and nucleotide excision repair. Alternatively, other mechanisms might be involved including epigenetic silencing wherein some melanocytes only express the mutant allele while others express the wild type allele; more work is needed to clarify these possibilities.
Our goal was to characterize the baseline pigmentary and DNA repair capacity of Mc1r-heterozygous mice and to determine whether topical cAMP induction would result in enhanced UV resistance. Crossing Mc1r-intact (Mc1rE/E) and Mc1r-deficient (Mc1re/e) K14-Scf breeders, we generated Mc1r-heterozygous (Mc1rE/e) K14-Scf animals and tested the capacity of topically applied forskolin to alter melanin composition and UV resistance of these mice. We noted a time- and dose-dependent darkening of the epidermal skin in Mc1rE/e K14-Scf animals. Indeed, when the highest dose of forskolin was used, the amount of darkening and eumelanin induction in Mc1rE/e K14-Scf animals was similar to that of those harboring two wild type Mc1r (Mc1rE/E) alleles. Pre-treating the animals with forskolin improved their UV resistance, as judged by MED testing which measures the ability of the skin to resist UV-induced inflammation. We interpret MED differences between strains and conditions to be mainly influenced by the amount of eumelanin in the interfollicular epidermis which attenuates UV penetration into the skin.
Forskolin is a direct activator of adenylyl cyclase enzymes, however it is important to point out that the degree of cAMP signaling downstream of forskolin-induced signaling may be influenced by the presence of activated Gs alpha, which synergistically potentiates binding of the drug to the enzyme (Alewijnse et al., 1997; Sutkowski, Tang, Broome, Robbins, & Seamon, 1994). Therefore it is possible that topically-applied forskolin may result in different levels of cAMP induction in the skin depending on expression and function of Mc1r in melanocytes. Although forskolin-mediated cAMP induction appeared to be similar across K14-Scf transgenic mice of Mc1rE/E, Mc1rE/e and Mc1re/e genotypes (Suppl. Fig. 4C), our observations that forskolin has a limited effect on the eumelanin content of Mc1re/e K14-Scf mice (Fig. 4), unlike the more robust induction of eumelanin seen in Mc1rE/E or Mc1re/e counterparts, might be consistent with a differential effect of the drug based on Mc1r status, or alternatively a cAMP-independent effect of Mc1r such as recently-described AKT-mediated MC1R effects (Castejon-Grinan, Herraiz, Olivares, Jimenez-Cervantes, & Garcia-Borron, 2018).
While degree of eumelanization of the epidermis is certainly important to UV resistance, pigmentation is not the only determinant of UV injury and mutagenesis. Rather, the capacity of the skin to recover from UV injury is also critical to long-term UV toxicities including carcinogenesis. We and others documented that MC1R function and cAMP signaling regulate the efficiency of nucleotide excision repair (Abdel-Malek et al., 2009; Hauser et al., 2006; Jagirdar et al., 2013; Jarrett, Carter, Shelton, & D’Orazio, 2017; Jarrett, Wolf Horrell, Boulanger, & D’Orazio, 2015; Jarrett et al., 2014; Swope et al., 2014; Yin, Sturm, & Smith, 2014). Nucleotide excision repair is the genomic maintenance pathway responsible for recognizing and clearing of mutagenic lesions that distort the architecture of the double helix as UV photoproducts do (reviewed in (Kusakabe et al., 2019)). Thus, we explored whether DNA repair could be up-regulated by topical cAMP induction in Mc1r-heterozygous skin. By measuring retention of [6,4]-photoproducts over time in the skin, we observed that pre-treatment of the skin with topically-applied forskolin enhanced the clearance of UV photoproducts. Interestingly, we observed that eumelanin accumulation, resistance to UV inflammation and nucleotide excision repair capacity were all improved by topical cAMP stimulation irrespective of Mc1r status, since forskolin improved each of these parameters in Mc1r homozygously defective, heterozygously defective or homozygously Mc1r-intact animals.
One interesting observation from our studies is that the basal clearance rate of UV photoproducts was similar between Mc1rE/E, Mc1rE/e and Mc1re/e mice, which might be surprising in the light of the significant constitutive activity of wild type (but not extension) Mc1r. However, these observations may be explained by the fact that most of the epidermis, even in K14-Scf mice, is made up of keratinocytes which do not express Mc1r and that cumulative repair of photodamage in the skin represents clearance of damage in both Mc1r-expressing and Mc1r-negative cells. Since keratinocytes greatly outnumber melanocytes, it is not surprising that basal repair kinetics should be similar between genotypes. Pharmacologic induction of cAMP by forskolin would raise cAMP levels in cells independently of Mc1r expression, thus our data suggest that cAMP stimulation may augment DNA repair in both melanocytes and keratinocytes and imply that topical cAMP induction might be useful to prevent both melanomas and keratinocyte-derived malignancies (BCC, SCC).
An important caveat when considering our findings is that we used a crude root extract from the Coleus forskohlii plant as a forskolin source for topical application to the mice. This was done in these proof-of-concept studies because of the cost-prohibitive nature of using purified forskolin for in vivo animal experiments. Although we previously validated our findings with purified forskolin in the C57BL/6J K14-Scf animal model (D’Orazio et al., 2006), we cannot eliminate the potential contribution of other plant-derived compounds present in the root extract. Nonetheless, the preparation that was used was highly enriched for forskolin content (20% of the total weight including insoluble content), therefore we are confident that the UV-protective responses observed were due to adenylyl cyclase activation and cAMP induction in the skin. Indeed, we showed that topical application of the root extract robustly increased cAMP levels in K14-Scf mice of any Mc1r genotype. Since eumelanin induction was previously reported in K14-Scf Mc1re/e mice by topical application of Rolipram, a phosphodiesterase 4 inhibitor that raised cutaneous cAMP levels by preventing its degradation (Khaled, Levy, & Fisher, 2010), it might be possible to replicate our findings using an alternative pharmacologic agents (e.g. Rolipram or other skin-permeable phosphodiesterase inhibitors) to raise cAMP levels in the skin.
It is also important to note that the Mc1r e allele encodes for a complete loss-of-function, truncated form of the receptor protein which may be retained in an intracellular compartment rather than trafficked to the cell surface. Accordingly, the e allele may not exactly mimic many human loss-of-function MC1R alleles which may be correctly trafficked but functionally impaired. Indeed, receptor variants with massive intracellular retention may behave as dominant negative mutants (Beaumont et al., 2007; Sanchez-Laorden et al., 2006). Moreover, although mouse and human MC1R proteins are similar in structure, there may be functional differences between the two that limit the direct translatability of our findings to humans. For example, there is evidence that MAPK signaling responses may differ downstream of cAMP induction (Herraiz et al., 2011). Therefore, cAMP-mediated responses can be different in mouse versus human skin.
Taken together, our data indicate that pharmacologic induction of cAMP signaling in the skin enhances eumelanin accumulation and augments nucleotide excision repair in any type of Mc1r background. Each of these properties result in enhance UV resistance through attenuating penetration of UV photons into the epidermis by eumelanin deposition and through accelerated recovery of DNA photolesions after UV injury to improve melanocyte genomic stability. These findings suggest that it may be possible for MC1R-heterozygous (and even MC1R-intact) individuals, like their MC1R-homozygously defective counterparts, to resist acute and chronic UV toxicities through cutaneous cAMP induction. Our data suggest that topical cAMP induction may be of benefit against UV-induced damage and mutagenesis in persons of any MC1R genotype.
Supplementary Material
Significance.
Inherited signaling defects in the melanocortin 1 receptor (MC1R) are a known risk factor for UV sensitivity and melanoma. Using a suitable mouse model, we tested whether pharmacologic cutaneous cAMP induction could impact pigmentary and UV responses in the Mc1r-heterozygous or –homozygous state. This is relevant to human disease because many melanomas occur in individuals whose melanocytes retain partial MC1R signaling capacity. We found that cAMP induction increased epidermal eumelanin levels, reduced UV-mediated inflammation and accelerated clearance of mutagenic UV photolesions by DNA repair in the Mc1r-heterozygous or –homozygous state, suggesting that cutaneous cAMP induction might be a useful melanoma-preventive strategy irrespective of MC1R status.
Acknowledgments
This work was supported by the following NIH grants: R01CA131075, T32CA160003, P30CA177558 and P30ES026529. We thank the Regina Drury Pediatric Research Endowed Chair Fund, the Markey Cancer Foundation, the Melanoma Research Alliance and the DanceBlue Golden Matrix Fund for their support. We acknowledge the critical contribution of the Biospecimen Procurement and Translational Pathology Shared Resource Facility of the University of Kentucky Markey Cancer Center. We thank James Sheppard III and Michael Jax of the University of Kentucky Department of Surgery for assistance with photography and image analysis. We thank Dr. Hong Pu for technical assistance. This manuscript’s contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Statement
All authors declare that they lack any ethical or financial conflicts of interest with the contents of this article. The content is entirely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or any other funding agency.
References:
- Abdel-Malek ZA, Ruwe A, Kavanagh-Starner R, Kadekaro AL, Swope V, Haskell-Luevano C, … Knittel JJ (2009). alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes. Pigment Cell Melanoma Res, 22(5), 635–644. doi: 10.1111/j.1755-148X.2009.00598.x [DOI] [PubMed] [Google Scholar]
- Alewijnse AE, Smit MJ, Rodriguez Pena MS, Verzijl D, Timmerman H, & Leurs R (1997). Modulation of forskolin-mediated adenylyl cyclase activation by constitutively active G(S)-coupled receptors. FEBS Lett, 419(2–3), 171–174. [DOI] [PubMed] [Google Scholar]
- Amaro-Ortiz A, Vanover JC, Scott TL, & D’Orazio JA (2013). Pharmacologic induction of epidermal melanin and protection against sunburn in a humanized mouse model. J Vis Exp(79). doi: 10.3791/50670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont KA, Shekar SN, Newton RA, James MR, Stow JL, Duffy DL, & Sturm RA (2007). Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles. Hum Mol Genet, 16(18), 2249–2260. doi: 10.1093/hmg/ddm177 [DOI] [PubMed] [Google Scholar]
- Castejon-Grinan M, Herraiz C, Olivares C, Jimenez-Cervantes C, & Garcia-Borron JC (2018). cAMP-independent non-pigmentary actions of variant melanocortin 1 receptor: AKT-mediated activation of protective responses to oxidative DNA damage. Oncogene, 37(27), 3631–3646. doi: 10.1038/s41388-018-0216-1 [DOI] [PubMed] [Google Scholar]
- Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, … Jemal A (2018). Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer, 124(13), 2785–2800. doi: 10.1002/cncr.31551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, … Fisher DE (2007). Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell, 128(5), 853–864. doi:S0092-8674(07)00185-7 [pii] [DOI] [PubMed] [Google Scholar]
- D’Orazio JA, Nobuhisa T, Cui R, Arya M, Spry M, Wakamatsu K, … Fisher DE (2006). Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature, 443(7109), 340–344. doi:nature05098 [pii] [DOI] [PubMed] [Google Scholar]
- Donatien PD, Hunt G, Pieron C, Lunec J, Taieb A, & Thody AJ (1992). The expression of functional MSH receptors on cultured human melanocytes. Arch Dermatol Res, 284(7), 424–426. [DOI] [PubMed] [Google Scholar]
- Guida S, Bartolomeo N, Zanna PT, Grieco C, Maida I, De Summa S, … Guida G (2015). Sporadic melanoma in South-Eastern Italy: the impact of melanocortin 1 receptor (MC1R) polymorphism analysis in low-risk people and report of three novel variants. Arch Dermatol Res. doi: 10.1007/s00403-015-1552-4 [DOI] [PubMed] [Google Scholar]
- Hauser JE, Kadekaro AL, Kavanagh RJ, Wakamatsu K, Terzieva S, Schwemberger S, … Abdel-Malek ZA (2006). Melanin content and MC1R function independently affect UVR-induced DNA damage in cultured human melanocytes. Pigment Cell Res, 19(4), 303–314. doi:PCR315 [pii] [DOI] [PubMed] [Google Scholar]
- Herraiz C, Journe F, Abdel-Malek Z, Ghanem G, Jimenez-Cervantes C, & Garcia-Borron JC (2011). Signaling from the human melanocortin 1 receptor to ERK1 and ERK2 mitogen-activated protein kinases involves transactivation of cKIT. Mol Endocrinol, 25(1), 138–156. doi:me.2010-0217 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, & Wakamatsu K (2003). Quantitative analysis of eumelanin and pheomelanin in humans, mice, and other animals: a comparative review. Pigment Cell Res, 16(5), 523–531. [DOI] [PubMed] [Google Scholar]
- Ito S, & Wakamatsu K (2011). Diversity of human hair pigmentation as studied by chemical analysis of eumelanin and pheomelanin. J Eur Acad Dermatol Venereol, 25(12), 1369–1380. doi: 10.1111/j.1468-3083.2011.04278.x [DOI] [PubMed] [Google Scholar]
- Jagirdar K, Yin K, Harrison M, Lim W, Muscat GE, Sturm RA, & Smith AG (2013). The NR4A2 nuclear receptor is recruited to novel nuclear foci in response to UV irradiation and participates in nucleotide excision repair. PLoS ONE, 8(11), e78075. doi: 10.1371/journal.pone.0078075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Carter KM, Shelton BJ, & D’Orazio JA (2017). The melanocortin signaling cAMP axis accelerates repair and reduces mutagenesis of platinum-induced DNA damage. Sci Rep, 7(1), 11708. doi: 10.1038/s41598-017-12056-5 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Jarrett SG, Wolf Horrell EM, Boulanger MC, & D’Orazio JA (2015). Defining the Contribution of MC1R Physiological Ligands to ATR Phosphorylation at Ser435, A Predictor of DNA Repair in Melanocytes. J Invest Dermatol. doi: 10.1038/jid.2015.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett SG, Wolf Horrell EM, Christian PA, Vanover JC, Boulanger MC, Zou Y, & D’Orazio JA (2014). PKA-mediated phosphorylation of ATR promotes recruitment of XPA to UV-induced DNA damage. Mol Cell, 54(6), 999–1011. doi: 10.1016/j.molcel.2014.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kadekaro AL, Chen J, Yang J, Chen S, Jameson J, Swope VB, … Abdel-Malek Z (2012). Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes. Mol Cancer Res, 10(6), 778–786. doi:1541-7786.MCR-11-0436 [pii] [DOI] [PubMed] [Google Scholar]
- Kennedy C, ter Huurne J, Berkhout M, Gruis N, Bastiaens M, Bergman W, … Bavinck JN (2001). Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color. J Invest Dermatol, 117(2), 294–300. [DOI] [PubMed] [Google Scholar]
- Khaled M, Levy C, & Fisher DE (2010). Control of melanocyte differentiation by a MITF-PDE4D3 homeostatic circuit. Genes Dev, 24(20), 2276–2281. doi:24/20/2276 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisada T, Lu SZ, Yoshida H, Nishikawa S, Nishikawa S, Mizoguchi M, … Longley BJ (1998). Murine cutaneous mastocytosis and epidermal melanocytosis induced by keratinocyte expression of transgenic stem cell factor. J Exp Med, 187(10), 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe M, Onishi Y, Tada H, Kurihara F, Kusao K, Furukawa M, … Sugasawa K (2019). Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ, 41, 2. doi: 10.1186/s41021-019-0119-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matichard E, Verpillat P, Meziani R, Gerard B, Descamps V, Legroux E, … Soufir N (2004). Melanocortin 1 receptor (MC1R) gene variants may increase the risk of melanoma in France independently of clinical risk factors and UV exposure. J Med Genet, 41(2), e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali E, Garcia-Borron JC, Fargnoli MC, Gandini S, Maisonneuve P, Bagnardi V, … Group MSS (2015). MC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: a pooled-analysis from the M-SKIP project. Int J Cancer, 136(3), 618–631. doi: 10.1002/ijc.29018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins LS, Nadeau JH, Johnson KR, Kelly MA, Roselli-Rehfuss L, Baack E, … Cone RD (1993). Pigmentation phenotypes of variant extension locus alleles result from point mutations that alter MSH receptor function. Cell, 72(6), 827–834. [DOI] [PubMed] [Google Scholar]
- Roberts DW, Newton RA, Beaumont KA, Helen Leonard J, & Sturm RA (2006). Quantitative analysis of MC1R gene expression in human skin cell cultures. Pigment Cell Res, 19(1), 76–89. doi:PCR286 [pii] [DOI] [PubMed] [Google Scholar]
- Robles-Espinoza CD, Roberts ND, Chen S, Leacy FP, Alexandrov LB, Pornputtapong N, … Adams DJ (2016). Germline MC1R status influences somatic mutation burden in melanoma. Nat Commun, 7, 12064. doi: 10.1038/ncomms12064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Laorden BL, Sanchez-Mas J, Martinez-Alonso E, Martinez-Menarguez JA, Garcia-Borron JC, & Jimenez-Cervantes C (2006). Dimerization of the human melanocortin 1 receptor: functional consequences and dominant-negative effects. J Invest Dermatol, 126(1), 172–181. doi:5700036 [pii] [DOI] [PubMed] [Google Scholar]
- Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, … Bastian BC (2015). The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med, 373(20), 1926–1936. doi: 10.1056/NEJMoa1502583 [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, & Jemal A (2019). Cancer statistics, 2019. CA Cancer J Clin, 69(1), 7–34. doi: 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- Song X, Mosby N, Yang J, Xu A, Abdel-Malek Z, & Kadekaro AL (2009). alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment Cell Melanoma Res, 22(6), 809–818. doi:PCR615 [pii] [DOI] [PubMed] [Google Scholar]
- Spry ML, Vanover JC, Scott T, Abona-Ama O, Wakamatsu K, Ito S, & D’Orazio JA (2009). Prolonged treatment of fair-skinned mice with topical forskolin causes persistent tanning and UV protection. Pigment Cell Melanoma Res, 22(2), 219–229. doi:PCR536 [pii] [DOI] [PubMed] [Google Scholar]
- Sturm RA, Teasdale RD, & Box NF (2001). Human pigmentation genes: identification, structure and consequences of polymorphic variation. Gene, 277(1–2), 49–62. doi:S0378111901006941 [pii] [DOI] [PubMed] [Google Scholar]
- Sutkowski EM, Tang WJ, Broome CW, Robbins JD, & Seamon KB (1994). Regulation of forskolin interactions with type I, II, V, and VI adenylyl cyclases by Gs alpha. Biochemistry, 33(43), 12852–12859. [DOI] [PubMed] [Google Scholar]
- Suzuki I, Im S, Tada A, Scott C, Akcali C, Davis MB, … Abdel-Malek Z (1999). Participation of the melanocortin-1 receptor in the UV control of pigmentation. J Investig Dermatol Symp Proc, 4(1), 29–34. [DOI] [PubMed] [Google Scholar]
- Swope V, Alexander C, Starner R, Schwemberger S, Babcock G, & Abdel-Malek ZA (2014). Significance of the melanocortin 1 receptor in the DNA damage response of human melanocytes to ultraviolet radiation. Pigment Cell Melanoma Res, 27(4), 601–610. doi: 10.1111/pcmr.12252 [DOI] [PubMed] [Google Scholar]
- Valverde P, Healy E, Jackson I, Rees JL, & Thody AJ (1995). Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat Genet, 11(3), 328–330. doi: 10.1038/ng1195-328 [DOI] [PubMed] [Google Scholar]
- Valverde P, Healy E, Sikkink S, Haldane F, Thody AJ, Carothers A, … Rees JL (1996). The Asp84Glu variant of the melanocortin 1 receptor (MC1R) is associated with melanoma. Hum Mol Genet, 5(10), 1663–1666. [DOI] [PubMed] [Google Scholar]
- Vanover JC, Spry ML, Hamilton L, Wakamatsu K, Ito S, & D’Orazio JA (2009). Stem cell factor rescues tyrosinase expression and pigmentation in discreet anatomic locations in albino mice. Pigment Cell Melanoma Res, 22(6), 827–838. doi:PCR617 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JK, Jovel C, Norton HL, Parra EJ, & Shriver MD (2002). Comparing quantitative measures of erythema, pigmentation and skin response using reflectometry. Pigment Cell Res, 15(5), 379–384. doi:2o042 [pii] [DOI] [PubMed] [Google Scholar]
- Wang ZG, Wu XH, & Friedberg EC (1991). Nucleotide excision repair of DNA by human cell extracts is suppressed in reconstituted nucleosomes. J Biol Chem, 266(33), 22472–22478. [PubMed] [Google Scholar]
- Weatherall IL, & Coombs BD (1992). Skin color measurements in terms of CIELAB color space values. J Invest Dermatol, 99(4), 468–473. [DOI] [PubMed] [Google Scholar]
- Wolf Horrell EM, Boulanger MC, & D’Orazio JA (2016). Melanocortin 1 Receptor: Structure, Function, and Regulation. Front Genet, 7, 95. doi: 10.3389/fgene.2016.00095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin K, Sturm RA, & Smith AG (2014). MC1R and NR4A receptors in cellular stress and DNA repair: implications for UVR protection. Exp Dermatol, 23(7), 449–452. doi: 10.1111/exd.12420 [DOI] [PubMed] [Google Scholar]
- Zappi E, & Lombardo W (1984). Combined Fontana-Masson/Perls’ staining. Am J Dermatopathol, 6 Suppl, 143–145. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.