

Abstract
Pathological cardiac hypertrophy is a complex process and eventually develops into heart failure, in which the heart responds to various intrinsic or external stress, involving increased interstitial fibrosis, cell death and cardiac dysfunction. Studies have shown that oxidative stress is an important mechanism for this maladaptation. Cyclophilin A (CyPA) is a member of the cyclophilin (CyPs) family. Many cells secrete CyPA to the outside of the cells in response to oxidative stress. CyPA from blood vessels and the heart itself participate in a variety of signaling pathways to regulate the production of reactive oxygen species (ROS) and mediate inflammation, promote cardiomyocyte hypertrophy and proliferation of cardiac fibroblasts, stimulate endothelial injury and vascular smooth muscle hyperplasia, and promote the dissolution of extracellular matrix (ECM) by activating matrix metalloproteinases (MMPs). The events triggered by CyPA cause a decline of diastolic and systolic function and finally lead to the occurrence of heart failure. This article aims to introduce the role and mechanism of CyPA in cardiac hypertrophy and remodeling, and highlights its potential role as a disease biomarker and therapeutic target.
Keywords: Cardiac hypertrophy and remodeling, Cyclophilin A, Inflammation, Matrix metalloproteinases, Oxidative stress, Vascular injury
Introduction
Cardiac hypertrophy and remodeling occur to compensate for internal or external stress before the onset of heart failure. Although this change is initially an adaptive response to maintain cardiac function, sustained hypertrophic stimulation becomes maladaptive, and eventually develops into heart failure [1]. Cardiomyocyte hypertrophy, apoptosis, and excessive activation of cardiac fibroblasts play an important role in this process [2]. Under pressure and volume overload, as well as a variety of growth factors and hormone stimulation, pathophysiological changes such as hypertrophy and apoptosis will happen in cardiomyocytes as these are permanent cells that do not proliferate [3]. Myocardial fibroblasts are the most abundant cells in the heart [4]. Under certain stimuli, myocardial fibroblasts proliferate, undergo phenotypic conversion, and secrete large amounts of extracellular matrix (ECM). This then induces myocardial sclerosis and ventricular remodeling, which affect ventricular compliance. The changes above in cardiomyocytes and fibroblasts eventually lead to a decline in diastolic and systolic function and heart failure [5].
It is traditionally believed that the progression of pathological cardiac hypertrophy is irreversible; once it develops into severe heart failure, there are no other effective treatments besides heart transplantation [6]. It has been confirmed that reactive oxygen species (ROS) production and further activation of the inflammatory response are important mechanisms for cardiac hypertrophy and remodeling that lead to heart failure [7–14]. Excessive ROS generation triggers oxidative damage to nucleic acids, proteins, and biofilms, leading to cell dysfunction and apoptosis. However, clinical randomized trials using antioxidants such as vitamin E and β-carotene did not significantly reduce cardiovascular events [15], suggesting that the lack of specific antioxidative therapy is meaningless.
Cyclophilin A (CyPA) is a highly conserved and ubiquitous protein that was first discovered as a receptor for cyclosporin A (CsA) in cells [16]. Initially, CyPA was only detected inside cells. More in-depth studies of CyPA have determined that CyPA can be secreted outside of cells in response to ROS in various cells [17–19], and the amount of secretion increases as the degree of oxidative stress increases. This leads to vascular wall damage, proliferation and migration of cardiac fibroblasts, as well as the release of cytokines and the production of more ROS, which then induces cardiomyocyte hypertrophy and apoptosis through paracrine mechanisms; this is closely related to the occurrence and development of pathological cardiac hypertrophy [20–22].
Structural features and functions of CyPA
Cyclophilins (CyPs) belong to a group of a protein family that has peptidyl-prolyl cis–trans isomerase (PPIase) activity and is widely distributed in eukaryotes and prokaryotes [23]. CyPA was first purified from bovine thymocytes and was identified as the major cellular target for the immunosuppressive drug CsA [24]. The overall shape of CyPs is a flatted cylinder with three α-helices, eight β-strands, and six β-turns. Eight antiparallel β-strands constitute the surface, and two α-helices (α2 and α3) lie on the top and on the bottom of the cylinder, respectively (Figure 1). Seven aromatic residues (Phe8, Phe22, Phe36, Phe53, Phe112, Phe129, and Try48) and other hydrophobic residues form a hydrophobic core of the molecule within the cylinder [25]. A loop from Lys118 to His126 and four β strands (β3−β6) constitute a pocket structure, which is the binding site for CsA. Almost all the members of CyPs can bind to CsA [26,27].
Figure 1. Schematic drawing of the structure of CyPs.
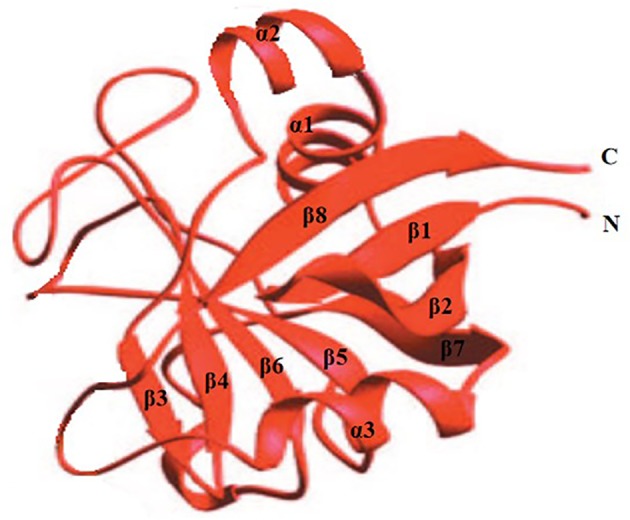
The α-helices are represented by a spiral designated as α, while β-strands are represented by an arrow marked as β. N and C denote the N and C termini of the molecule. Eight antiparallel strands (β1−β8) form a β-barrel and the two α-helices (α2 and α3) are on the top and bottom.
CyPA has several functions including PPIase activity, protein folding, protein assembly and protein trafficking in cells (such as nuclear translocation of extracellular signal-regulated kinase (ERK1/2) and apoptosis-inducing factor (AIF)) [28], and involves many biological functions including immune regulation [29], cell growth [30], tumorigenesis [31], and cardiovascular diseases (for example, CyPA can promote the development of atherosclerosis, aortic aneurysm and cardiac hypertrophy) [32]. Subsequently, an increasing number of studies have shown that cardiomyocytes, cardiac fibroblasts, vascular smooth muscle cells (VSMCs), endothelial cells (ECs), and monocytes/macrophages can secrete CyPA to the outside of cells under the action of ROS. In VSMCs, CyPA is secreted through a process of vesicle formation, vesicle transport, and fusion at the plasma membrane [17]. Extracellular CyPA has not only similar cellular functions as intracellular CypA, such as proliferation and inflammation, but also unique characteristics, such as apoptosis, migration, matrix degradation, and generation of ROS. It must be noted that the ability of extracellular CyPA to produce ROS is likely to depend on its receptor, at least from the current findings [32].
Receptor for CyPA in cardiovascular diseases
Yurchenko et al. first identified extracellular matrix metalloproteinase inducer (EMMPRIN, also known as CD147), which is encoded by basigin (Bsg), as a receptor for extracellular CyPA [33]. CyPA-induced chemotaxis and signaling pathways are achieved in two ways: one is through its PPIase activity and the other is through binding to EMMPRIN [34]. The mature form of EMMPRIN is a highly glycosylated transmembrane protein, which contains a short cytoplasmic domain, a transmembrane domain, and an extracellular domain [35]. EMMPRIN is highly expressed in many types of cells, including inflammatory cells and tumor cells. It is considered a matrix metalloproteinases (MMPs) inducing factor on the surface of tumor cells, which is involved in tumor invasion and metastasis [36]. In addition to the expression on tumor cells, EMMPRIN is also widely expressed on cardiovascular-related cells [37]. CyPA itself does not up-regulate the expression of EMMPRIN on the cell membrane, but after silencing EMMPRIN, the ERK1/2, nuclear factor κappa B (NK-κB) and Protein Kinase B (Akt) signaling pathways activated by CyPA are significantly attenuated [38,39]. These signaling pathways are involved in the development of pathological cardiac hypertrophy, including hypertrophic gene expression [40], fibrosis-related cell activation, migration and proliferation [41], regulation of the expression of various pro-inflammatory factors [42], and activation of MMPs [43]. Clinical studies have found that serum levels of EMMPRIN were significantly increased in patients with heart failure and were closely related to long-term survival [44]. After performing transverse aortic constriction (TAC) in Bsg−/− (knockout) mice and in wild-type (WT) mice, Bsg−/− mice showed reduced oxidative stress and MMPs activity in the left ventricle, less cardiac hypertrophy and cardiac interstitial fibrosis, as well as significantly improved long-term survival. Angiotensin II (AngII) or mechanical stretch induced cardiac fibroblasts express EMMPRIN. Up-regulation of EMMPRIN stimulated proliferation of cardiac fibroblasts by activating Akt and mitogen-activated protein kinases (MAPK) signaling pathways [45]. These data suggest that EMMPRIN plays a crucial role in the pathogenesis of cardiac hypertrophy, fibrosis, and heart failure, and may be a new biomarker and therapeutic target for pathological cardiac hypertrophy.
CyPA in cardiac hypertrophy and remodeling
Pathological cardiac hypertrophy is often secondary to a variety of heart diseases, including hypertension, valvular heart disease, cardiomyopathy, and myocardial infarction. It is generally believed that the excessive production of ROS is an important mechanism for cardiac hypertrophy and remodeling [7–14]. ROS are generated by several mechanisms (Figure 2), including nicotinamide–adenine dinucleotide phosphate (NADPH) oxidases, xanthine oxidase (XO), the mitochondrial respiratory chain, and nitric oxide synthases (NOS) [10,13,14]. ROS formation can be stimulated by mechanical forces, (such as stretch, pressure, and shear stress), environmental factors (such as hypoxia), secreted factors coupled with tyrosine kinase receptors (such as platelet-derived growth factor (PDGF)), and secreted factors coupled with G-protein-coupled receptors (such as AngII, α, and β adrenergic receptor (αAR and βAR)) [46–49]. The human body produces a certain amount of ROS to control the normal redox reaction of physiological signaling pathways. Excessive ROS can cause cell dysfunction, lipid peroxidation, protein oxidation inactivation and DNA mutagenesis, leading to irreversible cell damage or death [10,13,14].
Figure 2. The possible mechanism of ROS production.

Low level of ROS is thought to play a role in controlling the normal redox reaction of physiological signaling pathways. High level plays a role in cardiac hypertrophy, apoptosis, ventricular remodeling, systolic, and diastolic dysfunction.
CyPA is a pathogenic protein that mediates oxidative stress-induced cardiovascular dysfunction, such as atherosclerosis [50], aortic dissection [15], and cardiac hypertrophy [51]. The production of CyPA and ROS is a vicious cycle. Under the condition of oxidative stress, excessive ROS stimulate the secretion of CyPA and activate related signaling pathways, further increasing the production of ROS [17–19]. In vitro and in vivo experiments have confirmed that CyPA can cause cardiac hypertrophy, and ventricular remodeling, and promote pathological cardiac hypertrophy [51–56]. The main mechanisms by which CyPA promotes cardiac hypertrophy and remodeling are characterized as follows: (1) CyPA produces ROS to promote oxidative damage, which mediates an inflammatory response. (2) CyPA directly promotes cardiomyocyte hypertrophy, myocardial fibroblast proliferation, and migration. (3) CyPA stimulates vascular endothelial injury and vascular smooth muscle hyperplasia. (4) CyPA activates MMPs via EMMPRIN to promote ECM dissolution (Figure 3).
Figure 3. The main mechanism by which CyPA promotes cardiac hypertrophy and remodeling.

(1) CyPA is secreted in response to ROS, and then produces ROS to promote oxidative damage, mediates inflammatory response. (2) CyPA promotes the proliferation of cardiac fibroblasts and has a direct hypertrophic effect on cardiomyocytes. (3) CyPA stimulates vascular injury. (4) CyPA activates MMPs via EMMPRIN to promote ECM dissolution.
CyPA promotes oxidative stress and inflammatory response
Studies have shown that the concentration of CyPA and oxidative stress in plasma of patients with heart failure caused by various factors is significantly increased [52,55,56], suggesting that CyPA is involved as a pro-inflammatory and pro-oxidative factor in the process from cardiac hypertrophy and remodeling to heart failure. Animal experiments showed that after four weeks of Ang II infusion, the cardiac hypertrophy index increased significantly in both Ppia−/− (CyPA knockout) and WT mice; however, there were no significant differences in the degree of cardiac hypertrophy between Ppia−/- and WT mice [51]. Satoh hypothesized that the role of CyPA in cardiac hypertrophy requires a situation in which ROS production or inflammation is increased. The hearts of apolipoprotein E (ApoE−/−) mice have been shown to exhibit increased ROS production; moreover, CyPA is highly expressed in atherosclerotic plaques of ApoE−/- mice, which has a pro-inflammatory effect on endothelial cells, leading to plaque progression [57]. When researchers treated ApoE−/− mice and ApoE−/−Ppia−/− mice with Ang II infusion, four weeks later they found that the cardiac weight, interventricular septum thickness, and cardiac myocyte size of ApoE−/−Ppia−/− mice were significantly improved compared with ApoE−/− mice. In addition, the cells that proliferate and migrate to the perivascular area are significantly reduced in ApoE−/−Ppia−/− mice, as well as collagen content and fibrosis in the perivascular area. The production of ROS in the hearts and blood vessels of ApoE−/−Ppia−/− mice was significantly reduced [51]. These data suggest that CyPA is secreted in the context of increased oxidative stress and inflammation, and acts as a determinant of ROS production to promote cardiac hypertrophy, remodeling, and vascular fibrosis in ApoE−/− mice. Nigro et al. confirmed that CyPA is involved in the development of atherosclerosis by promoting oxidative stress and inflammation. Researchers simultaneously fed ApoE−/− mice and ApoE−/−Ppia−/− mice a high-fat diet. After 16 weeks, they found that the ApoE−/−Ppia−/− mice developed significantly reduced atherosclerosis compared with ApoE−/− mice, and CyPA deficiency was associated with decreased vascular cell adhesion molecule 1 (VCAM-1) expression, ROS, inflammation, ECs activation and apoptosis [57]. In a mouse model of Ang II-induced aortic dissection, CyPA is secreted in response to ROS produced by ECs and mediates ECs expression of adhesion molecule-1, activation of MMPs and inflammatory cell infiltration. Secreted CyPA activates VSMCs by ERK1/2 phosphorylation, enhances ROS production in VSMCs and promotes vascular inflammatory responses, thereby up-regulating the susceptibility of structural components of the aortic wall to aortic dissection [15]. These findings suggest that inhibition of CyPA expression in oxidative stress and inflammation-related cardiovascular disease may improve the progression of the disease.
The recruitment and transportation of leukocytes are important aspects of the inflammatory process [58]. Although chemokines are thought to be a major factor regulating cell trafficking, extracellular CyPA has recently been shown to have a direct chemical attraction to human leukocytes [59]. CyPA which is detected at high levels in tissues with ongoing inflammation, is secreted by a variety of cells [60]. EMMPRIN has been identified as the major signaling receptor for CyPA on human leukocytes. Interestingly, the expression of EMMPRIN is elevated on leukocytes from inflamed tissues, indicating a strong correlation among the presence of extracellular CyPA, EMMPRIN expression, and inflammatory responses [61]. The pro-inflammatory effect of CyPA is specifically directing chemotactic leukocytes, which can induce aggregation of neutrophils, eosinophils, T lymphocytes, and monocytes [34], activate the inflammatory signaling pathways, and promote the production of more ROS, leading to irreversible cell damage or death. CyPA promotes inflammation mainly through the following cascades: (1) CyPA binds to its receptor EMMPRIN to chemotactic inflammatory cells. Yurchenko et al. demonstrated that EMMPRIN is an essential component in the CyPA initiation signaling cascade [33]. After blocking EMMPRIN, chemotaxis of monocytes, expression of related inflammatory factors, and activation of signaling pathways induced by CyPA are inhibited [62], suggesting that the CyPA-EMMPRIN interaction is an important pro-inflammatory signal. (2) After CyPA binds to EMMPRIN, it activates the NK-κB, Akt, and MAPK signaling pathways [38,39,45] and stimulates ECs to express VCAM-1, intercellular adhesion molecule 1 (ICAM-1), and E-selectin [63]. VCAM-1 on the surface of ECs is a ligand of integrin α4β1 (VLA-4) on the surface of lymphocytes and monocytes. VCAM-1 interacts with VLA-4 and promotes the rolling and adhesion of leukocytes [64]. After inflammatory cells aggregate, they further pass through the gap of ECs and reach the subendothelium and differentiate, secreting cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-8 (IL-8), further promoting ROS production and inducing ECs activation and apoptosis [65]. (3) Activation of the CyPA/EMMPRIN pathway induces secretion of TNF-α, IL-6, and ROS, and enhances activation of MMPs [66]. ROS that come from targeted interactions with critical cysteines in the propeptide autoinhibitory domain activate MMPs post-translationally, which are normally secreted in an inactive form [67]. At the same time, inflammatory mediators, such as TNF-α and IL-6, and ROS also activate the NK-κB pathway [68]. Activation of this inflammatory signaling pathway promotes more ROS production, which forms a vicious cycle of ROS-CyPA-ROS and accelerates the progression of the inflammatory response. Activation of the inflammatory signaling pathway increases the expression of pro-hypertrophic and pro-fibrotic genes [69]. Cardiac inflammation is an important component of the pathogenesis of cardiovascular disease and an effective mediator of ROS-induced myocardial fibrosis and hypertrophy. Seizer has demonstrated that CyPA and EMMPRIN were up-regulated in patients with inflammatory cardiomyopathy and suggested that CyPA and its ligands can serve as novel diagnostic markers for oxidative stress and inflammation [70].
CyPA promotes the proliferation of cardiac fibroblasts and has a direct hypertrophic effect on cardiomyocytes
In vitro cell experiments have shown that culture medium prepared from AngII-stimulated cardiac fibroblasts, which were isolated from ApoE−/− mice, were used to culture cardiomyocytes, and resulted in an increased myocardial hypertrophy index, such as cardiomyocyte protein synthesis and embryonic gene expression. Further experiments confirmed that cardiac fibroblasts isolated from ApoE−/− mice secreted a large amount of CyPA in response to Ang II, stimulated the proliferation and migration of cardiac fibroblasts by enhancing ROS production, and the migration increased in a concentration-dependent manner. Researchers also tried to study whether cardiomyocytes secrete CyPA under the stimulation of Ang II. However, the required serum-free medium caused a large number of cardiomyocytes to die. Direct stimulation of cardiomyocytes with recombinant CyPA can lead to cardiomyocyte hypertrophy [51]. The observations above suggest that CyPA induces cardiac hypertrophy and remodeling through autocrine mechanisms in cardiac fibroblasts and paracrine mechanisms in cardiomyocytes (Figure 4), but the specific signaling pathway remains to be studied. Tian et al. confirmed that Ang II interacts with angiotensin type 2 receptor (AT2R) to stimulate ROS production in H9C2 cells and up-regulated CyPA expression [53]; Su et al. found that melatonin reduces Ang II-induced ROS production and cardiac hypertrophy in H9C2 cells by inhibiting the CyPA/EMMPRIN pathway [54]. This suggests that there may also be events in cardiomyocytes similar to the secretion of CyPA by cardiac fibroblasts, induction of oxidative stress, and cardiac hypertrophy. However, CyPA secreted by cardiomyocytes under hypoxia/reoxygenation reduced oxidative stress and apoptosis by activating the Akt pathway [71], suggesting that CyPA may play the opposite role in acute and chronic oxidative stress events. Therefore, it is necessary to study how the downstream signals of the CyPA/EMMPRIN pathway specifically regulate oxidative stress and cardiac hypertrophy, to find suitable therapeutic targets in different cardiovascular events.
Figure 4. The general mechanism of CyPA-mediated cardiac fibrosis and hypertrophy.

CyPA promotes cardiac fibrosis activation and proliferation, which is mediated by cell surface EMMPRIN. CyPA activates certain signaling pathways and induces oxidative stress, and inflammatory cytokines, resulting in MMPs activation. ROS promotes the release of CyPA, which interacts with EMMPRIN again through autocrine mechanisms in cardiac fibroblasts and paracrine mechanisms in cardiomyocytes.
ROS is essential for acetylation of CyPA and its secretion [72], while acetylated CyPA secreted extracellularly has stronger proinflammatory and oxidative stress than unmodified CyPA [73]. Inhibiting the acetylation of CyPA may improve the prognosis of patients with oxidative stress-related cardiovascular disease.
CyPA promotes endothelial injury and vascular smooth muscle hyperplasia
In addition to cardiomyocytes and cardiac fibroblasts, mammalian myocardial tissues also include ECs and VSMCs [74]. Activation and apoptosis of vascular walls may lead to stenosis and occlusion, and further lead to excessive cardiac load, myocardial ischemia [75], cardiac hypertrophy, remodeling, and even heart failure [50].
EC dysfunction is a key event in the development and progression of vascular disease [76]. Jin et al. studied the pro-inflammatory effects of CyPA on ECs. Recombinant CyPA activated MAPK signaling pathways in cultured human umbilical vein endothelial cells, including ERK1/2, JNK and p38, and stimulated IκB-α phosphorylation and NF-κB activation. This induces the expression of adhesion molecules, including VCAM-1 and E-selectin [18]. ECs usually resist the attachment of leukocytes that flow through them. After activation, they express chemokines and adhesion molecules, leading to leukocyte recruitment [64]. The bound leukocytes migrate into the intima and monocytes (the most recruited cells) mature into macrophages, which secrete inflammatory cytokines and ROS, and activate MMPs [62]. Studies have also shown that the effect of CyPA on ECs may depend on its concentration. At low concentrations (10−100 ng/ml), CyPA increases EC proliferation, migration, invasive capacity, tubulogenesis, and the secretion of active MMP-2, which is a mediator of cell migration and angiogenesis. At high concentrations (such as 2 μg/ml), CyPA has opposite effects, reducing EC migration and viability, possibly related to the induction of Toll-like receptor-4 expression, as detected by immunocytochemistry and flow cytometry [77]. Progress in pathological cardiac hypertrophy has been considered irreversible. However, recent clinical observations and experimental studies have confirmed the reversal of pathological cardiac hypertrophy, and angiogenesis is essential in this process [6]. The pro-angiogenic effect of low concentration of CyPA in ECs suggests that it may also play a role in reversing cardiac hypertrophy and remodeling.
Abnormal VSMC growth contributes to the pathogenesis of blood vessels [75]. CyPA was first identified as a redox-sensitive medium secreted by VSMCs, activating ERK1/2, promoting DNA replication, and mediating VSMCs proliferation and hypertrophy [78]. CyPA can also promote NADPH oxidase such as p47phox translocation to the plasma membrane, produce ROS, and indirectly promote cell protein synthesis, cell growth, inflammation, and VSMC proliferation [79]. Satoh et al. found that VSMC-derived intracellular and extracellular CypA are required for the production of ROS, inflammatory cytokine expression, and activation of MMPs. Inflammation and matrix degradation in blood vessels are critical for vascular remodeling [80]. At the same time, CyPA and ROS produced by the vascular system may act on cardiomyocytes and cardiac fibroblasts, promoting the process anywhere from cardiac hypertrophy and remodeling to heart failure.
CyPA promotes ECM dissolution
Cardiac fibroblasts are the main source of the ECM. Activated cardiac fibroblasts secrete a large amount of ECM, which promotes collagen accumulation in the myocardial interstitium (i.e, fibrosis), changes the passive stiffness properties of the myocardium, and affects ventricular compliance [81]. There are highly diverse proteolytic enzymes in the ECM, and MMPs are the most widely studied. Activated MMPs directly or indirectly proteolytically process the ECM, while certain MMP types may enhance profibrotic signaling, leading to unstable structures and abnormal functions of the ECM, and further impairing both systolic and diastolic functions [82]. There are currently about 23 types of MMPs expressed in humans, and different types of MMPs are up-regulated in different stimulation-induced cardiac hypertrophy models and exhibit diverse biological effects [83]. For example, the expression and activity of MMP-2 increase in the context of cardiac pressure overload [84], whereas in inflammatory cardiomyopathy, MMP-9 plays a key role in the poor remodeling process after myocardial injury [85]. EMMPRIN is considered to be an MMP-inducing factor involved in vascular and ventricular remodeling in cardiovascular disease. In vascular cells, cardiac fibroblasts, and monocytes, CyPA binds to EMMPRIN and induces activation of MMPs by activating signaling pathways such as the ERK1/2 and NF-κB [18,45,62,78].
The expression of CyPA and EMMPRIN in plasma of patients with acute myocardial infarction (AMI) increases. The change in CyPA concentration affects ventricular remodeling and cardiac function after AMI, which may be related to CyPA-mediated MMP-2 and MMP-9 activation [56,86]. In an animal model of AMI, intracoronary administration of CsA to inhibit CyPA reduces inflammation, down-regulates MMP expression, and reduces left ventricular remodeling [87]. The results above suggest that activation of the CyPA/EMMPRIN pathway induces secretion of inflammatory cytokines and production of ROS. The synergistic effects of inflammatory cytokines and ROS promote collagen deposition and activate MMPs, which contribute to ventricular remodeling after myocardial infarction.
Based on the up-regulation of CyPA and EMMPRIN in myocardial tissue of patients with inflammatory cardiomyopathy [70], Seizer and his team further studied the role of CyPA and EMMPRIN in mice with coxsackievirus-induced myocarditis. The results showed that compared with WT mice, inflammatory cell recruitment, MMP-9 expression, myocardial fibrosis, and contractile dysfunction were significantly decreased in Ppia−/- mice, while MMP-2 expression was not significantly different between the two groups [88]. A study by Yuan showed that CyPA-EMMPRIN interaction activates MAPK/NF-κB and promotes inflammation and MMP-9 expression in monocytes [62]. MMP-9 is therefore considered to be a key proteolytic enzyme for CyPA in mediating ventricular remodeling secondary to inflammatory cardiomyopathy.
In VSMCs, CyPA binds to EMMPRIN to promote MMP-2 expression and mediate migration and proliferation of VSMCs [72]. The expression and activity of MMP-2 in plasma increase in patients with left ventricular pressure overload such as hypertension and aortic stenosis, and the level of MMP-2 is positively correlated with the development of left ventricular hypertrophy and heart failure [89,90]. While it induces local ECM degradation, MMP-2 also promotes ECM synthesis by altering intracellular signaling cascades to enhance profibrotic signaling pathways [91]. Whether CyPA promotes ventricular remodeling by up-regulating myocardial MMP-2 expression in a stress overload-induced cardiac hypertrophy model remains unclear.
CyPA as a therapeutic target for cardiac hypertrophy and remodeling
Clinical studies have found that CyPA is significantly elevated in the plasma of patients with ventricular remodeling and heart failure caused by various diseases such as hypertension, AMI, and myocarditis [52,55,56]. A follow-up study of patients with ST-segment elevation myocardial infarction (STEMI) found that CyPA levels were positively correlated with inflammatory factors, such as IL-6 and high-sensitivity C-reactive protein (CRP), and activated MMPs. Decreased CyPA concentration at a one-month follow-up after STEMI predicted improvement of left ventricular function at 6 months [56]. CRP can only be used as an inflammatory marker to assess the level of plasma inflammation in patients with AMI. Continuously increased plasma circulating CyPA levels after STEMI may reflect a state of high oxidative stress and inflammation. ROS and inflammation further up-regulate cardiac hypertrophy and profibrotic gene expression [69]. This suggests that CyPA can be used as a marker of oxidative stress and inflammation for patients with cardiac hypertrophy and remodeling to conduct heart failure risk assessment and prognosis analysis, and provide new therapeutic targets for drugs that prevent and improve cardiac remodeling.
Early studies have shown that the Ca2+-calmodulin (CaM)-calcineurin (CN)-nuclear factor of activated T cell (NFAT) signaling pathway plays a crucial role in the development of cardiac hypertrophy [92]. CsA, which is an inhibitor of CN, binds to CyPA to form a dimeric complex and then binds to CN and inhibits its activity [93]. The use of CsA in animal models can effectively block or reduce cardiac hypertrophy caused by Ang II, β-AR and pressure load [94–96], but simply knocking out CN expression does not effectively improve cardiac hypertrophy [97], suggesting that CyPA may be a more important factor in promoting cardiac hypertrophy. However, CsA is immunosuppressive and therefore is not an ideal therapeutic drug [93]. NIM811, a non-immunosuppressive CsA analog, efficiently binds CyPA inside and outside the cell [98], and may bring some adverse reactions due to the inhibition of intracellular CyPA. Thus it is necessary to develop a new type of CyPA inhibitor, which is non-immunosuppressive, does not penetrate the cell membrane, and can specifically bind to extracellular CyPA (especially acetylated CyPA).
Statins such as simvastatin can inhibit oxidative stress-induced CyPA secretion in a dose-dependent manner in VSMCs likely via decreased isoprenylation of small GTPases [17]. Statins have been proven to have antioxidant effects that inhibit the development of hypertension and cardiac hypertrophy and have beneficial effects on cardiovascular changes [99]. It has been suggested that statins may play a role in improving oxidative stress and ventricular remodeling by inhibiting the secretion of CyPA.
The renin–angiotensin–aldosterone system (RAAS) plays an important role in the progression of left ventricular remodeling and heart failure after myocardial infarction. A large amount of clinical evidence indicates drugs that inhibit RAAS, such as angiotensin converting enzyme inhibitors (ACEI) and angiotensin II receptor blockers (ARB), can slow left ventricular remodeling and improve the prognosis of patients with heart failure [100]. Studies have shown that RAAS is a ROS inducer that promotes CyPA secretion [101]. Inhibition of RAAS can attenuate oxidative stress, left ventricular remodeling, and possibly reduce CyPA levels. In a rabbit model of myocardial infarction, ACEI can significantly reduce the concentration of CyPA in the left ventricular wall and improve left ventricular remodeling and left ventricular function [102].
Conclusions and perspectives
This article describes the main mechanisms by which CyPA promotes cardiac hypertrophy and remodeling, including the promotion of oxidative stress and inflammation and the role of CyPA in the main cells (cardiomyocytes, cardiac fibroblasts, ECs, and VSMCs) in myocardial tissue, and summarizes the effects of several drugs for the treatment of heart failure on CyPA. We suggest that CyPA and its receptor EMMPRIN can be used as new therapeutic targets for preventing and improving pathological cardiac hypertrophy. However, specific signaling pathways by which the CyPA/EMMPRIN axis regulates oxidative stress and inflammation, thereby inducing cardiac hypertrophy and remodeling, are not yet known. In pathological cardiac hypertrophy caused by various factors, CyPA may activate different types of MMPs to mediate myocardial fibrosis and remodeling. At present, most of the viewpoints suggest that CyPA plays a role of promoting inflammation and oxidative stress in the development of pathological cardiac hypertrophy, but the pro-angiogenic effect of low concentration of CyPA in ECs suggests that it may have an advantage in promoting the regression of pathological cardiac hypertrophy. Further basic science and clinical trials are required to elucidate the specific mechanisms by which CyPA/EMMPRIN promotes pathological cardiac hypertrophy under different pathophysiological conditions, which will be of great significance for the development of new treatments that prevent or reverse cardiac remodeling.
Abbreviations
- ACEI
angiotensin converting enzyme inhibitor
- ARB
angiotensin II receptor blocker
- CyPA
Cyclophilin A
- CyP
cyclophilin
- ECM
extracellular matrix
- MMP
matrix metalloproteinase
- RAAS
renin–angiotensin–aldosterone system
- ROS
reactive oxygen species
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Tham Y.K., Bernardo B.C., Ooi J.Y. et al. (2015) Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch.Toxicol. 89, 1401–1438 10.1007/s00204-015-1477-x [DOI] [PubMed] [Google Scholar]
- 2.Porrello E.R., Mahmoud A.I., Simpson E. et al. (2011) Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 10.1126/science.1200708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sobirin M.A., Kinugawa S., Takahashi M. et al. (2012) Activation of natural killer T cells ameliorates postinfarct cardiac remodeling and failure in mice. Circ. Res. 111, 1037–1047 10.1161/CIRCRESAHA.112.270132 [DOI] [PubMed] [Google Scholar]
- 4.Frangogiannis Nikolaos G. (2019) Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 65, 70–99 10.1016/j.mam.2018.07.001 [DOI] [PubMed] [Google Scholar]
- 5.Furtado M.B., Nim H.T., Boyd S.E. et al. (2016) View from the heart: cardiac fibroblasts in development, scarring and regeneration. Development 143, 387–397 10.1242/dev.120576 [DOI] [PubMed] [Google Scholar]
- 6.Hou J. and Kang Y.J. (2012) Regression of pathological cardiac hypertrophy: signaling pathways and therapeutic targets. Pharmacol. Ther. 135, 337–354 10.1016/j.pharmthera.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mcmurray J., Chopra M., Abdullah I. et al. (1993) Evidence of oxidative stress in chronic heart failure in humans. Eur. Heart J. 14, 1493–1498 10.1093/eurheartj/14.11.1493 [DOI] [PubMed] [Google Scholar]
- 8.Keith M., Geranmayegan A., Sole M.J. et al. (1998) Increased oxidative stress in patients with congestive heart failure. J. Am. Coll. Cardiol. 31, 1352–1356 10.1016/S0735-1097(98)00101-6 [DOI] [PubMed] [Google Scholar]
- 9.Hare J.M. and Stamler J.S. (2005) NO/redox disequilibrium in the failing heart and cardiovascular system. J. Clin. Invest. 115, 509–517 10.1172/JCI200524459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murdoch C.E., Zhang M., Cave A.C. et al. (2006) NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 71, 208–215 10.1016/j.cardiores.2006.03.016 [DOI] [PubMed] [Google Scholar]
- 11.Mallat Z., Philip I., Lebret M. et al. (1998) Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 97, 1536–1539 10.1161/01.CIR.97.16.1536 [DOI] [PubMed] [Google Scholar]
- 12.Valgimigli M., Merli E., Malagutti P. et al. (2004) Hydroxyl radical generation, levels of tumor necrosis factor-alpha, and progression to heart failure after acute myocardial infarction. J. Am. Coll. Cardiol. 43, 2000–2008 10.1016/j.jacc.2004.01.036 [DOI] [PubMed] [Google Scholar]
- 13.Sawyer D.B., Siwik D.A., Xiao L. et al. (2002) Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell Cardiol. 34, 379–388 10.1006/jmcc.2002.1526 [DOI] [PubMed] [Google Scholar]
- 14.Giordano F.J. (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 115, 500–508 10.1172/JCI200524408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan L.M., Douglas G., Bendall J.K. et al. (2014) Endothelial cell-specific reactive oxygen species production increases susceptibility to aortic dissection. Circulation 129, 2661–2672 10.1161/CIRCULATIONAHA.113.005062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handschumacher R.E., Harding M.W., Rice J., Drugge R.J. et al. (1984) Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226, 544–547 10.1126/science.6238408 [DOI] [PubMed] [Google Scholar]
- 17.Suzuki J., Jin Z.-G., Meoli D.F. et al. (2006) Cyclophilin A is secreted by a vesicular pathway in vascular smooth muscle cells. Circ. Res. 98, 811–817 10.1161/01.RES.0000216405.85080.a6 [DOI] [PubMed] [Google Scholar]
- 18.Jin Z.-G., Lungu A.O., Xie L. et al. (2004) Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler. Thromb. Vasc. Biol. 24, 1186–1191 10.1161/01.ATV.0000130664.51010.28 [DOI] [PubMed] [Google Scholar]
- 19.Ramachandran S., Venugopal A., Sathisha K. et al. (2012) Proteomic profiling of high glucose primed monocytes identifies cyclophilin A as a potential secretory marker of inflammation in type 2 diabetes. Proteomics 12, 2808–2821 10.1002/pmic.201100586 [DOI] [PubMed] [Google Scholar]
- 20.Bang C., Antoniades C., Antonopoulos A.S. et al. (2015) Intercellular communication lessons in heart failure. Eur. J. Heart Fail. 17, 1091–1103 10.1002/ejhf.399 [DOI] [PubMed] [Google Scholar]
- 21.Takeda N. and Manabe I. (2011) Cellular Interplay between Cardiomyocytes and Nonmyocytes in Cardiac Remodeling. Int. J. Inflam. 2011, 535241 10.4061/2011/535241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viereck J., Bang C., Foinquinos A. et al. (2014) Regulatory RNAs and paracrine networks in the heart. Cardiovasc. Res. 102, 290–301 10.1093/cvr/cvu039 [DOI] [PubMed] [Google Scholar]
- 23.Nigro P., Pompilio G. and Capogrossi M.C. (2013) Cyclophilin A: a key player for human disease. Cell Death Dis. 4, e888 10.1038/cddis.2013.410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tropschug M., Barthelmess I.B. and Neupert W. (1989) Sensitivity to cyclosporin A is mediated by cyclophilin in Neurospora crassa and Saccharomyces cerevisiae. Nature 342, 953–955 10.1038/342953a0 [DOI] [PubMed] [Google Scholar]
- 25.Wang P. and Heitman J. (2005) The cyclophilins. Genome Biol. 6, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kallen J., Spitzfaden C., Zurini M.G. et al. (1991) Structure of human cyclophilin and its binding site for cyclosporin A determined by X-ray crystallography and NMR spectroscopy. Nature 353, 276–279 10.1038/353276a0 [DOI] [PubMed] [Google Scholar]
- 27.Ke H.M., Zydowsky L.D., Liu J. et al. (1991) Crystal structure of recombinant human T-cell cyclophilin A at 2.5 A resolution. Proc. Natl. Acad. Sci. U.S.A. 88, 9483–9487 10.1073/pnas.88.21.9483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffmann H. and Schiene-Fischer C. (2014) Functional aspects of extracellular cyclophilins. Biol. Chem. 395, 721–735 10.1515/hsz-2014-0125 [DOI] [PubMed] [Google Scholar]
- 29.Li J., Chen C., Wong G. et al. (2016) Cyclophilin A protects mice against infection by influenza A virus. Sci. Rep. 6, 28978 10.1038/srep28978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obchoei S., Weakley S.M., Wongkham S. et al. (2011) Cyclophilin A enhances cell proliferation and tumor growth of liver fluke-associated cholangiocarcinoma. Mol. Cancer. 10, 102 10.1186/1476-4598-10-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volker S.E., Hedrick S.E., Feeney Y.B. et al. (2018) Cyclophilin A Function in Mammary Epithelium Impacts Jak2/Stat5 Signaling, Morphogenesis, Differentiation, and Tumorigenesis in the Mammary Gland. Cancer Res. 78, 3877–3887 10.1158/0008-5472.CAN-17-2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue C., Sowden M.P. and Berk B.C. (2018) Extracellular and Intracellular Cyclophilin A, Native and Post-Translationally Modified, Show Diverse and Specific Pathological Roles in Diseases. Arterioscler. Thromb. Vasc. Biol. 38, 986–993 10.1161/ATVBAHA.117.310661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yurchenko V., Zybarth G., O’Connor M. et al. (2002) Active site residues of cyclophilin A are crucial for its signaling activity via CD147. J. Biol. Chem. 277, 22959–22965 10.1074/jbc.M201593200 [DOI] [PubMed] [Google Scholar]
- 34.Song F., Zhang X., Ren X.B. et al. (2011) Cyclophilin A(CyPA) induces chemotaxis independent of its peptidylprolyl cis-trans isomerase activity: direct binding between CyPA and the ectodomain of CD147. J. Biol. Chem. 286, 8197–8203 10.1074/jbc.C110.181347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muramatsu T. and Miyauchi T. (2003) Basigin (CD147): a multifunctional transmembrane protein involved in reproduction, neural function, inflammation and tumor invasion. Histol. Histopathol. 18, 981–987 [DOI] [PubMed] [Google Scholar]
- 36.Toole Bryan P. (2003) EMMPRIN (CD147), a cell surface regulator of matrix metalloproteinase production and function. Curr. Top. Dev. Biol. 54, 371–389 10.1016/S0070-2153(03)54015-7 [DOI] [PubMed] [Google Scholar]
- 37.Seizer P., Gawaz M., May A.E. et al. (2014) Cyclophilin A and EMMPRIN (CD147) in cardiovascular diseases. Cardiovasc. Res. 102, 17–23 10.1093/cvr/cvu035 [DOI] [PubMed] [Google Scholar]
- 38.Bahmed K., Henry C., Holliday M. et al. (2012) Extracellular cyclophilin A stimulates ERK1/2 phosphorylation in a cell-dependent manner but broadly stimulates nuclear factor κappa B. Cancer Cell Int. 12, 19 10.1186/1475-2867-12-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seizer P., Ungern-Sternberg S.N., Schönberger T. et al. (2015) Extracellular cyclophilin A activates platelets via EMMPRIN (CD147) and PI3K/Akt signaling, which promotes platelet adhesion and thrombus formation in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 35, 655–663 10.1161/ATVBAHA.114.305112 [DOI] [PubMed] [Google Scholar]
- 40.Nomura S., Satoh M., Fujita T. et al. (2018) Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 9, 4435 10.1038/s41467-018-06639-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu L., Jin X., Hu C.F. et al. (2018) Amphiregulin enhances cardiac fibrosis and aggravates cardiac dysfunction in mice with experimental myocardial infarction partly through activating EGFR-dependent pathway. Basic Res. Cardiol. 113, 12 10.1007/s00395-018-0669-y [DOI] [PubMed] [Google Scholar]
- 42.Ma Y., Chiao Y.A., Clark R. et al. (2015) Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc. Res. 106, 421–431 10.1093/cvr/cvv128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka T., Ogawa M., Suzuki J. et al. (2012) Inhibition of IκB phosphorylation prevents load-induced cardiac dysfunction in mice. Am. J. Physiol. Heart Circ. Physiol. 303, H1435–H1445 10.1152/ajpheart.00290.2012 [DOI] [PubMed] [Google Scholar]
- 44.Spinale F.G., Coker M.L., Heung L.J. et al. (2000) A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation 102, 1944–1949 10.1161/01.CIR.102.16.1944 [DOI] [PubMed] [Google Scholar]
- 45.Suzuki K., Satoh K., Ikeda S. et al. (2016) Basigin Promotes Cardiac Fibrosis and Failure in Response to Chronic Pressure Overload in Mice. Arterioscler. Thromb. Vasc. Biol. 36, 636–646 10.1161/ATVBAHA.115.306686 [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z.Z., Wang W., Jin H.Y. et al. (2017) Apelin Is a Negative Regulator of Angiotensin II-Mediated Adverse Myocardial Remodeling and Dysfunction. Hypertension 70, 1165–1175 10.1161/HYPERTENSIONAHA.117.10156 [DOI] [PubMed] [Google Scholar]
- 47.Matsushima S., Kuroda J., Ago T. et al. (2013) Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ. Res. 112, 651–663 10.1161/CIRCRESAHA.112.279760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osadchii O.E. (2007) Cardiac hypertrophy induced by sustained beta-adrenoreceptor activation: pathophysiological aspects. Heart Fail. Rev. 12, 66–86 10.1007/s10741-007-9007-4 [DOI] [PubMed] [Google Scholar]
- 49.Jelenik T., Flögel U., Álvarez-Hernández E. et al. (2018) Insulin Resistance and Vulnerability to Cardiac Ischemia. Diabetes 67, 2695–2702 10.2337/db18-0449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohtsuki T., Satoh K., Omura J. et al. (2017) Prognostic Impacts of Plasma Levels of Cyclophilin A in Patients With Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 37, 685–693 10.1161/ATVBAHA.116.308986 [DOI] [PubMed] [Google Scholar]
- 51.Satoh K., Nigro P., Zeidan A. et al. (2011) Cyclophilin A promotes cardiac hypertrophy in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 31, 1116–1123 10.1161/ATVBAHA.110.214601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sunamura S., Satoh K., Kurosawa R. et al. (2018) Different roles of myocardial ROCK1 and ROCK2 in cardiac dysfunction and postcapillary pulmonary hypertension in mice. Proc. Natl. Acad. Sci. U.S.A. 115, 7129–7138 10.1073/pnas.1721298115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian H., Yu D., Hu Y. et al. (2018) Angiotensin II upregulates cyclophilin A by enhancing ROS production in rat cardiomyocytes. Mol. Med. Rep. 18, 4349–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Su H., Li J., Chen T. et al. (2016) Melatonin attenuates angiotensin II-induced cardiomyocyte hypertrophy through the CyPA/CD147 signaling pathway. Mol. Cell. Biochem. 422, 85–95 10.1007/s11010-016-2808-9 [DOI] [PubMed] [Google Scholar]
- 55.Su H., Chen T., Li J. et al. (2017) Correlations of Serum Cyclophilin A and Melatonin Concentrations with Hypertension-induced Left Ventricular Hypertrophy. Arch. Med. Res. 48, 526–534 10.1016/j.arcmed.2017.12.003 [DOI] [PubMed] [Google Scholar]
- 56.Huang C.H., Chang C.C., Kuo C.L. et al. (2015) Decrease in plasma cyclophilin A concentration at 1 month after myocardial infarction predicts better left ventricular performance and synchronicity at 6 months: a pilot study in patients with ST elevation myocardial infarction. Int. J. Biol. Sci. 11, 38–47 10.7150/ijbs.10271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nigro P., Satoh K., O’Dell M.R. et al. (2011) Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E-deficient mice. J. Exp. Med. 208, 53–66 10.1084/jem.20101174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wright R.D. and Cooper D. (2014) Glycobiology of leukocyte trafficking in inflammation. Glycobiology 24, 1242–1251 10.1093/glycob/cwu101 [DOI] [PubMed] [Google Scholar]
- 59.Malesevic M., Gutknecht D., Prell E. et al. (2013) Anti-inflammatory effects of extracellular cyclosporins are exclusively mediated by CD147. J. Med. Chem. 56, 7302–7311 10.1021/jm4007577 [DOI] [PubMed] [Google Scholar]
- 60.Dawar F.U., Xiong Y., Khattak M.N.K. et al. (2017) Potential role of cyclophilin A in regulating cytokine secretion. J. Leukoc. Biol. 102, 989–992 10.1189/jlb.3RU0317-090RR [DOI] [PubMed] [Google Scholar]
- 61.von Ungern-Sternberg S.N.I., Zernecke A. and Seizer P. (2018) Extracellular Matrix Metalloproteinase Inducer EMMPRIN (CD147) in Cardiovascular Disease. Int. J. Mol. Sci. 19, 507 10.3390/ijms19020507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan W., Ge H. and He B. (2010) Pro-inflammatory activities induced by CyPA-EMMPRIN interaction in monocytes. Atherosclerosis 213, 415–421 10.1016/j.atherosclerosis.2010.09.033 [DOI] [PubMed] [Google Scholar]
- 63.Payeli S.K., Schiene-Fischer C., Steffel J. et al. (2008) Cyclophilin A differentially activates monocytes and endothelial cells: role of purity, activity, and endotoxin contamination in commercial preparations. Atherosclerosis 197, 564–571 10.1016/j.atherosclerosis.2007.08.025 [DOI] [PubMed] [Google Scholar]
- 64.Klemke M., Weschenfelder T., Konstandin M.H. et al. (2007) High affinity interaction of integrin alpha4beta1 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) enhances migration of human melanoma cells across activated endothelial cell layers. J. Cell. Physiol. 212, 368–374 10.1002/jcp.21029 [DOI] [PubMed] [Google Scholar]
- 65.Wong M.M., Chen Y., Margariti A. et al. (2014) Macrophages control vascular stem/progenitor cell plasticity through tumor necrosis factor-α-mediated nuclear factor-κB activation. Arterioscler. Thromb. Vasc. Biol. 34, 635–643 10.1161/ATVBAHA.113.302568 [DOI] [PubMed] [Google Scholar]
- 66.Siwik D.A., Kuster G.M., Brahmbhatt J.V. et al. (2008) EMMPRIN mediates beta-adrenergic receptor-stimulated matrix metalloproteinase activity in cardiac myocytes. J. Mol. Cell. Cardiol. 44, 210–217 10.1016/j.yjmcc.2007.07.054 [DOI] [PubMed] [Google Scholar]
- 67.Siwik D.A. and Colucci W.S. (2004) Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail. Rev. 9, 43–51 10.1023/B:HREV.0000011393.40674.13 [DOI] [PubMed] [Google Scholar]
- 68.Sorescu D. and Griendling K.K. (2002) Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest. Heart Fail. 8, 132–140 10.1111/j.1527-5299.2002.00717.x [DOI] [PubMed] [Google Scholar]
- 69.Mian M.O.R., He Y., Bertagnolli M. et al. (2019) TLR (Toll-Like Receptor) 4 Antagonism Prevents Left Ventricular Hypertrophy and Dysfunction Caused by Neonatal Hyperoxia Exposure in Rats. Hypertension 74, 843–853 10.1161/HYPERTENSIONAHA.119.13022 [DOI] [PubMed] [Google Scholar]
- 70.Seizer P., Geisler T., Bigalke B. et al. (2013) EMMPRIN and its ligand cyclophilin A as novel diagnostic markers in inflammatory cardiomyopathy. Int. J. Cardiol. 163, 299–304 10.1016/j.ijcard.2011.06.049 [DOI] [PubMed] [Google Scholar]
- 71.Cheng F., Yuan W., Cao M. et al. (2019) Cyclophilin A Protects Cardiomyocytes against Hypoxia/Reoxygenation-Induced Apoptosis via the AKT/Nox2 Pathway. Oxid. Med. Cell Longev. 10.1155/2019/2717986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soe N.N., Sowden M., Baskaran P. et al. (2014) Acetylation of cyclophilin A is required for its secretion and vascular cell activation. Cardiovasc. Res. 101, 444–453 10.1093/cvr/cvt268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xue C., Sowden M. and Berk B.C. (2017) Extracellular Cyclophilin A, Especially Acetylated, Causes Pulmonary Hypertension by Stimulating Endothelial Apoptosis, Redox Stress, and Inflammation. Arterioscler. Thromb. Vasc. Biol. 37, 1138–1146 10.1161/ATVBAHA.117.309212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brown R.D., Ambler S.K., Mitchell M.D. et al. (2005) THE CARDIAC FIBROBLAST: Therapeutic Target in Myocardial Remodeling and Failure. Annu. Rev. Pharmacol. Toxicol. 45, 657–687 10.1146/annurev.pharmtox.45.120403.095802 [DOI] [PubMed] [Google Scholar]
- 75.Bennett M.R. (1999) Apoptosis of vascular smooth muscle cells in vascular remodelling and atherosclerotic plaque rupture. Cardiovasc. Res. 41, 361–368 10.1016/S0008-6363(98)00212-0 [DOI] [PubMed] [Google Scholar]
- 76.Yuan W., Yan J., Liang Y. et al. (2013) Antiapoptotic and proapoptotic signaling of cyclophilin A in endothelial cells. Inflammation 36, 567–572 10.1007/s10753-012-9578-7 [DOI] [PubMed] [Google Scholar]
- 77.Kim S.H., Lessner S.M., Sakurai Y. et al. (2004) Cyclophilin A as a novel biphasic mediator of endothelial activation and dysfunction. Am. J. Pathol. 164, 1567–1574 10.1016/S0002-9440(10)63715-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Melaragno M.I., Lambeth J.D., Berk B.C. et al. (2000) Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ. Res. 87, 789–796 10.1161/01.RES.87.9.789 [DOI] [PubMed] [Google Scholar]
- 79.Soe N.N., Sowden M., Baskaran P. et al. (2013) Cyclophilin A is required for Angiotensin II-induced p47phox translocation to caveolae in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 33, 2147–2153 10.1161/ATVBAHA.113.301894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Satoh K., Nigro P., Matoba T. et al. (2009) Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat. Med. 15, 649–656 10.1038/nm.1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fomovsky G.M., Thomopoulos S. and Holmes J.W. (2010) Contribution of extracellular matrix to the mechanical properties of the heart. J. Mol. Cell. Cardiol. 48, 490–496 10.1016/j.yjmcc.2009.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spinale F.G., Janicki J.S. and Zile M.R. (2013) Membrane-associated matrix proteolysis and heart failure. Circ. Res. 112, 195–208 10.1161/CIRCRESAHA.112.266882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gao Y.A., Agnihotri R., Vary C.P. et al. (2004) Expression and characterization of recombinant osteopontin peptides representing matrix metalloproteinase proteolytic fragments. Matrix Biol. 23, 457–466 10.1016/j.matbio.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 84.Skrbic B., Engebretsen K.V., Strand M.E. et al. (2015) Lack of collagen VIII reduces fibrosis and promotes early mortality and cardiac dilatation in pressure overload in mice. Cardiovasc. Res. 106, 32–42 10.1093/cvr/cvv041 [DOI] [PubMed] [Google Scholar]
- 85.Pan H.Y., Sun H.M., Xue L.J. et al. (2014) Ectopic trypsin in the myocardium promotes dilated cardiomyopathy after influenza A virus infection. Am. J. Physiol. Heart Circ. Physiol. 307, H922–H932 10.1152/ajpheart.00076.2014 [DOI] [PubMed] [Google Scholar]
- 86.Seizer P., Fuchs C., Ungern-Sternberg S.N. et al. (2016) Platelet-bound cyclophilin A in patients with stable coronary artery disease and acute myocardial infarction. Platelets 27, 155–158 [DOI] [PubMed] [Google Scholar]
- 87.Yin Q., Pei Z., Wang H. et al. (2014) Cyclosporine A-nanoparticles enhance the therapeutic benefit of adipose tissue-derived stem cell transplantation in a swine myocardial infarction model. Int. J. Nanomed. 9, 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Seizer P., Klingel K., Sauter M. et al. (2012) Cyclophilin A affects inflammation, virus elimination and myocardial fibrosis in coxsackievirus B3-induced myocarditis. J. Mol. Cell. Cardiol. 53, 6–14 10.1016/j.yjmcc.2012.03.004 [DOI] [PubMed] [Google Scholar]
- 89.Marchesi C., Dentali F., Nicolini E. et al. (2012) Plasma levels of matrix metalloproteinases and their inhibitors in hypertension. J. Hypertens. 30, 3–16 10.1097/HJH.0b013e32834d249a [DOI] [PubMed] [Google Scholar]
- 90.Zile M.R., Desantis S.M., Baicu C.F. et al. (2011) Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ. Heart Fail. 4, 246–256 10.1161/CIRCHEARTFAILURE.110.958199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Newby A.C. (2015) Metalloproteinases promote plaque rupture and myocardial infarction: A persuasive concept waiting for clinical translation. Matrix Biol. 44-46, 157–166 10.1016/j.matbio.2015.01.015 [DOI] [PubMed] [Google Scholar]
- 92.Ritter O., Hack S., Schuh K. et al. (2002) Calcineurin in human heart hypertrophy. Circulation 105, 2265–2269 10.1161/01.CIR.0000016044.19527.96 [DOI] [PubMed] [Google Scholar]
- 93.Colgan J., Asmal M., Yu B. et al. (2005) Cyclophilin A-deficient mice are resistant to immunosuppression by cyclosporine. J. Immunol. 174, 6030–6038 10.4049/jimmunol.174.10.6030 [DOI] [PubMed] [Google Scholar]
- 94.Sussman M.A. (1998) Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science 281, 1690–1693 10.1126/science.281.5383.1690 [DOI] [PubMed] [Google Scholar]
- 95.Jordan M.C., Quednau B.D., Roos K.P. et al. (2002) Cyclosporin A regulates sodium-calcium exchanger (NCX1) gene expression in vitro and cardiac hypertrophy in NCX1 transgenic mice. Ann. N.Y. Acad. Sci. 976, 259–267 10.1111/j.1749-6632.2002.tb04748.x [DOI] [PubMed] [Google Scholar]
- 96.Goldspink P.H., McKinney R.D., Kimball V.A. et al. (2001) Angiotensin II induced cardiac hypertrophy in vivo is inhibited by cyclosporin A in adult rats. Mol. Cell. Biochem. 226, 83–88 10.1023/A:1012789819926 [DOI] [PubMed] [Google Scholar]
- 97.Kreusser M.M., Lehmann L.H., Keranov S. et al. (2014) Cardiac CaM Kinase II genes δ and γ contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation 130, 1262–1273 10.1161/CIRCULATIONAHA.114.006185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zulian A., Rizzo E., Schiavone M. et al. (2014) NIM811, a cyclophilin inhibitor without immunosuppressive activity, is beneficial in collagen VI congenital muscular dystrophy models. Hum. Mol. Genet. 23, 5353–5363 10.1093/hmg/ddu254 [DOI] [PubMed] [Google Scholar]
- 99.Rieck ÅE, Cramariuc D., Boman K. et al. (2012) Hypertension in aortic stenosis: implications for left ventricular structure and cardiovascular events. Hypertension 60, 90–97 10.1161/HYPERTENSIONAHA.112.194878 [DOI] [PubMed] [Google Scholar]
- 100.Solomon S.D., Shin S.H., Shah A. et al. (2011) Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur. Heart J. 32, 1227–1234 10.1093/eurheartj/ehq522 [DOI] [PubMed] [Google Scholar]
- 101.Satoh K., Godo S., Saito H. et al. (2014) Dual roles of vascular-derived reactive oxygen species-with a special reference to hydrogen peroxide and cyclophilin A. J. Mol. Cell. Cardiol. 73, 50–56 10.1016/j.yjmcc.2013.12.022 [DOI] [PubMed] [Google Scholar]
- 102.Chen C.Y., Lee B.C., Hsu H.C. et al. (2008) A proteomic study of the effects of ramipril on post-infarction left ventricular remodelling in the rabbit. Eur. J. Heart Fail. 10, 740–748 10.1016/j.ejheart.2008.06.001 [DOI] [PubMed] [Google Scholar]