

Abstract
Complex cellular and molecular events occur in the neurovascular unit after stroke, such as blood–brain barrier (BBB) dysfunction and inflammation that contribute to neuronal death, neurological deterioration and mortality. Caveolin-1 (Cav-1) has distinct physiological functions such as caveolae formation associated with endocytosis and transcytosis as well as in signaling pathways. Cav-1 has been proposed to be involved in BBB dysfunction after brain injury; however, its precise role is poorly understood. The goal of this study was to characterize the expression and effect of Cav-1 deletion on outcome in the first week in a transient Middle Cerebral Artery Occlusion stroke model. We found increased Cav-1 expression in new blood vessels in the lesion and in reactive astrocytes in the peri-lesion areas. In Cav-1 KO mice, the lesion volume was larger and the behavioral outcome worse than in WT mice. Cav-1 KO mice exhibited reduced neovascularization and modified astrogliosis, without formation of a proper glial scar around the lesion at three days post injury, coinciding with aggravated outcomes. Altogether, these results point towards a potential protective role of endogenous Cav-1 in the first days after ischemia by promoting neovascularization, astrogliosis and scar formation.
Keywords: Caveolin-1, ischemic stroke, astrogliosis, neovascularization, neuroprotection
Introduction
Stroke is the third cause of death worldwide and the first cause of acquired disability in adults.1 Overall, ischemic strokes represent approximately 85% of strokes.2 Intravenous administration of recombinant tissue plasminogen activator is approved for the treatment of ischemic stroke. More recently, it has been demonstrated that patients with ischemic stroke caused by a proximal occlusion in the anterior circulation benefit from endovascular thrombectomy.3 Despite these improvements, an important number of patients cannot benefit from these treatments and remain disabled. Research aimed at discovering alternative therapies needs to be continued and neuroprotective agents re-examined. An emerging concept is that protection may be achieved by influencing the molecular mechanisms not only in neurons after stroke but also in other cell types likely to participate in neuronal survival or repair mechanisms.4 As such, endothelial cells and astrocytes, as part of the neurovascular unit (NVU)5 could play a major role in preserving or recovering brain functions after stroke. The term NVU emphasises close physical and functional connection between brain tissue and blood vessels. Brain vasculature is anatomically distinct, consisting of pericytes and astrocyte endfeet in addition to endothelial cells, with microglia and nerve endings contacting brain vessels to collectively form the NVU. The blood–brain barrier (BBB) restricts passage of molecules from the blood circulation to the brain tissue as well as coordinating the exchange of ions, molecules and cells between the blood and brain and the presence and maintenance of barrier properties by the NVU are critical for brain homeostasis and neuronal functioning.6 After stroke, brain tissue follows a remodeling process with neo-vascularization in the lesion core and formation of a glial scar around the core of the lesion.7,8 Therefore, NVU function is likely central for brain tissue remodeling and recovery.
Caveolins are transmembrane proteins involved in the formation of caveolae (plasma membrane invaginations or vesicles of 50 to 100 nm in size that serve as membrane organizing centers, macromolecular transport and permeability, as well as signal transduction). There are three members of the caveolin (Cav) family, Cav-1, Cav-2 and Cav-3, with highly conserved sequences across species.9 In the brain, Cav-1 was initially believed to be restricted to endothelial cells; however, it was then found in astrocytic cultures10,11 and juvenile rat astrocytes after traumatic brain injury (TBI)12 and neurons.7 Caveolin proteins are key modulators of a variety of intracellular signaling pathways.13 In caveolae, Cav-1 is the major multifunctional scaffolding protein providing a docking site to anchor various proteins.14 Caveolae are present in most cell types but are particularly abundant in endothelial cells, smooth and striated muscle cells, adipocytes and astrocytes and neurons.14 Recently, new evidence showed a beneficial role of caveolins in kidney, hind limb and cardiac ischemia models, indicating a ubiquitous role in ischemic cell death.7 Caveolins regulate signaling pathways by oligomerization or via their scaffolding domain.15 Cav-1 for example, down-regulates the endothelial nitric oxide synthase (eNOS) pathway resulting in inhibition of nitric oxide (NO) production.16 Also, Cav-1 activates matrix metalloproteinases (MMPs), especially MMP9 and MMP2, involved in BBB opening after injury.17,18 However, the exact role of Cav-1 in modulating BBB integrity after brain injury is still controversial. Notably, the expression of Cav-1 in brain microvessels is downregulated at different time points after ischemia and reperfusion.17 On the contrary, other studies revealed an increase in Cav-1 expression in ischemic rat brains or in the number of endothelial caveolae in a mouse model of cerebral ischemia.19,20 Moreover, Cav-1 deficiency increased cerebral ischemic injury in a permanent distal MCAO model.19 Therefore, Cav-1 is likely to play an important role in NVU function but this probably depends on the injury model. Furthermore, it is now well established that the same protein can have several roles depending on its location with respect to the lesion core and the time after the injury.14,21 Here, we address the role of Cav-1 using histological and immunohistochemical analysis of the NVU in the lesion core and perilesional tissue (penumbra). We provide a detailed behavioral evaluation and investigate Cav-1 in astrocytes after stroke in Cav-1 KO and wild type (WT) mice from 6 h to seven days post-injury in a transient middle cerebral artery occlusion (tMCAO) model.
Material and methods
Animal experiments and care complied with the Federal regulations and guidelines of the Swiss Veterinary Office and were approved by the Animal Care and Use Committee (license VD2017.5). Animal reporting was according to ARRIVE guidelines.
Animal groups
Cav-1 KO mice in a C57Bl/6J background from Jackson Laboratory (JAX stock #007083) were bred on site. Male C57Bl/6J WT mice (6 weeks) were from Charles River. Animals were housed for at least one week in a temperature-controlled animal facility on a 12-h light-dark cycle with ad libitum access to food and water. Cages contained standard bedding and enrichment material.
We performed two sets of experiments with 25 WT and 28 Cav-1 KO mice (see Table 1):
Behavioral evaluation and lesion size measurements after tMCAO with n = 9 WT and n = 12 Cav-1 KO. Sham experiments were conducted in parallel with n = 5 WT and n = 5 Cav-1 KO mice. Animals were randomly assigned to sham or tMCAO procedure. Results of sham animals are in supplementary information.
Immunofluorescence experiments with n = 14 WT and n = 17 Cav-1 KO animals, two to three different samples at three different time points (sham and tMCAO at 6 h, one and three days post injury, dpi).
Table 1.
Summary of the animal used in the two sets of experiments with a total of 25 WT and 28 Cav-1 KO mice.
| WT |
Cav-1 KO |
|||
|---|---|---|---|---|
| tMCAO | Sham | tMCAO | Sham | |
| Total | 17 | 8 | 21 | 7 |
| Sample collection: Total | 8 | 3 | 9 | 2 |
| Completed | 8 | 3 | 7 | 2 |
| Terminal endpoint reached | 0 | 0 | 2 | 0 |
| Behavioral and lesion size study: Total | 9 | 5 | 12 | 5 |
| Completed | 7 | 5 | 3 | 5 |
| Terminal endpoint reached | 1 | 0 | 6 | 0 |
| Found dead | 1 | 0 | 3 | 0 |
tMCAO: transient middle cerebral artery occlusion; WT: wild type.
We tested animals in behavioral tests daily after tMCAO and evaluated lesion size at 7 dpi. For immunofluorescence, animals were perfused (see below) at 6 h, 1 and 3 dpi. Our veterinary authority requested the following humane termination endpoints: loss of righting reflex from 24 h post-injury, status epilepticus, body weight loss of more than 25%. In our experiments, we terminated nine mice (1 WT, 8 Cav-1 KO).
Transient middle cerebral artery occlusion
We used the intraluminal suture tMCAO model as previously described.22 Briefly, mice were anesthetized with isoflurane (3% induction, 1.5–2% maintenance). Body temperature was maintained throughout surgery at 37 ± 0.5℃ with a heating pad (DC Temperature Controller, FHC). We monitored cerebral blood flow (CBF) by Doppler flowmetry (Perimed) using a flexible probe fixed onto the skull (1 mm posterior, 6 mm lateral to bregma). Baseline CBF was obtained before carotid exposure. We induced ischemia by introducing an 11-mm silicone-coated 8–0 filament (Doccol Corp, USA) from the left common into the internal carotid artery, advancing until we felt resistance. We left the filament in place for 35 min. Successful ischemia was obtained if CBF during tMCAO was below 20% of baseline and reperfusion at least 50%. Two WT animals and one Cav-1 KO did not reperfuse. We performed sham surgery under anesthesia by placing the Doppler probe onto the skull and dissecting the carotid arteries without filament insertion. After surgery, animals were maintained overnight in an incubator at 28℃. We assessed the coat, eyes and nose, neurological deficit, epileptic seizures, body weight loss and dehydration of all animals daily.
Behavioral assessment
Neuroscore
We assessed the neuroscore daily with a modified Bederson scale23: no observable deficit: 0; failure to extend the right forepaw: 1; circling: 2; humane endpoint reached or death: 3. Intermittent circling was graded 1.5.24
Rotarod test
We assessed post-stroke behavior using modified tests from Balkaya et al.25
For rotarod test, mice were placed on an accelerating rotating cylinder (UgoBasile), and the longest latency to fall in three trials of maximum 900 s was recorded.24 We trained mice three days before injury and tested on 1, 3, 5 and 7 dpi. We expressed performance as percentage of the best performance in training. Mice excluded before the end of the experiment due to sacrifice for humane reasons were allocated a minimum score of 0 s thereafter to limit survival bias.
Adhesive removal test
Animals were placed in a transparent Perspex box for 60 s for habituation. We then applied a small piece of adhesive tape (rectangular 3 × 4 mm cut from Time®Tape, TimeMed Inc) under the forepaws while restraining the mouse. Placement order (right or left forepaw first) was alternated for each trial. Directly after placing adhesives, the experimenter pressed both forelimbs simultaneously to minimize bias. We then placed the mouse back into the Perspex box for 120 s and the time to contact and remove each adhesive tape was blindly recorded and scored using the Observer software (Noldus). Contact occurred when the paw was shaken or the mouth used to touch the adhesive. We performed three consecutive trials for each mouse daily. Mice were trained three days before injury and tested at 2, 4 and 6 dpi. SG and CB scored blindly time to contact and remove adhesive on ipsilateral and contralateral paws and pooled data. We gave the maximum score of 120 s to mice excluded before the end of the experiment because of death or sacrifice to limit survival bias.
Lesion volume measurement
We froze brains in liquid nitrogen vapor. We collected 20 µm-thick coronal cryostat (Leica CM3050) sections on Superfrost-Plus® slides. Sections were stained with cresyl violet, then scanned with a stereomicroscope (Nikon SMZ 25) at 5× magnification and analyzed with ImageJ/FIJI software. CB quantitated infarction volumes, blinded to the group, on 12 720 µm-distant coronal sections. Infarction volumes were calculated by multiplying the sum of the infarcted areas on each section by the spacing distance.
Immunofluorescence staining and image analysis
For tissue collection, mice were transcardially perfused first with PBS and then 4% PFA at 4℃, 10 mL/min. Following overnight post-fixation in PFA, we cryoprotected brains in PBS with 30% sucrose for 48 h, then froze them in isopentane on dry ice. We collected 25 µm-thick coronal cryostat sections into cryoprotectant solution. Immunofluorescence staining was performed on free-floating sections with antigen retrieval with cold 33% acetic acid + 66% ethanol, blocked with 1% bovine serum albumin (BSA, Sigma Aldrich) + 5% horse serum + 0.1% Triton X100 solution for 1 h at room temperature. Primary antibodies used were: Rabbit anti-Cav-1 (1:500, Abcam, cat # ab2910), Rat anti-CD31 (1:100, BD Biosciences, cat # 550274), Rabbit anti-GFAP (1:2000, Millipore Merck, cat # AB5804), Mouse anti-GFAP (1:2000, Millipore Merck, cat # MAB3402), Mouse anti-GS (1:1000, Millipore Merck, cat # MAB302), Mouse anti-MAP-2 (1:500, Millipore Merck, cat # MAB3418), Rabbit anti-Ki67 (1:200, Thermofisher, cat # RM-9106-R7). Primary antibodies were incubated in antibody solution (1% BSA + 0.3% Triton X100 in PBS) overnight at 4℃. Alexa Fluor® labeled (Invitrogen) secondary antibodies were incubated in antibody solution with DAPI as nuclear counterstain for 1 h at room temperature. Sections were mounted on SuperFrost-Plus® slides (Fischer Scientific) with FluorSave medium (Calbiochem) and coverslipped.
We captured images of coronal whole-brain sections using a slide-scanner (Zeiss AxioScan Z1) at 10× magnification. We acquired higher magnification images in ipsilateral and contralateral striatum to the lesion with a confocal microscope (Zeiss LSM 710 Quasar) at 63× magnification.
Images for the vessel density analysis and quantification of Ki67 were acquired with the same confocal microscope at 40× magnification. We performed vessel density analysis using the vessel density plugin from Fiji. The quantification of Ki67 positive nuclei was done on averaged z-projections using Fiji on stacks of 15 to 17 images with 1 µm-spacing on 3 images per animal, 3 animals per group (WT and Cav-1 KO).
Stacks of images for the morphological analysis were acquired with an epifluorescence microscope (Zeiss Imager Axio Z1) at 40× magnification. Astrocyte morphology analysis was performed by TC using Fiji’s bandpass and unsharp mask filtering, binarization, skeletonization and analysis as described in Morrison et al.26 on the averaged z-projections on n = 4 areas per animal, three animals per group. Quantification of Ki67-positive nuclei and astrocyte morphology analysis were performed blinded.
Western blots
Proteins were prepared from frozen WT Sham and tMCAO cryostat sections separating ischemic from contralateral non-ischemic hemispheres. Samples were homogenized in cold lysis buffer containing protease and phosphatase inhibitors as previously.27 We quantified proteins using the Bradford Kit (Bio-Rad); 20 μg of proteins were separated by gel electrophoresis (NuPAGE 10% Bis-Tris Gel, cat # NP0302BOX) and transferred to PVDF membranes. Membranes were placed in blocking solution (Li-Cor Odyssey Blocking buffer, No:927-4000) for 1 h before incubation with Cav-1 (1:1000, Abcam, cat # ab2910) and α-tubulin (alpha-tubulin (TU-02), 1:2000, Santa Cruz Biotech, cat # sc-8035) primary antibodies overnight at 4℃. Afterwards, membranes were washed in PBS + 0.05% Tween and incubated 1 h at room temperature with IR-conjugated secondary antibodies (Odyssey IRDye® Goat anti-Mouse 800 CW and Goat anti-Rabbit 680LT, Li-Cor). The Li-Cor system detected bound primary antibodies and quantification was performed using Fiji, taking the mean gray value of each band. After normalizing to alpha tubulin, we compared using one-way ANOVA.
Statistical analysis
We performed measurements and analyses blinded using Prism software (GraphPad). We represented data as box and whiskers plots, showing the median, minimal and maximal values as well as individual data points. Bar graphs are expressed as mean ± standard deviation (SD) and individual data points are displayed. We compared the survival of the two groups with the Mantel–Cox test. We carried out two-group comparisons with the Student unpaired t-test after testing both samples for Gaussian distribution and similar SD. We analyzed behavioral tests performed on the same groups over several days with two-way ANOVA for repeated measures with Holm–Sidak multiple comparison post hoc test. Statistical significance was set at *p < 0.05, **p < 0.01, ***p < 0.001 or ****p < 0.0001.
Results
Expression of caveolin-1 in endothelial cells and reactive astrocytes
We investigated the expression and cellular localization of Cav-1 in ipsilateral and contralateral hemispheres to the lesion using Western blotting and double-immunostaining with anti-CD31 (endothelial cell marker), anti-Glutamine Synthetase (GS, pan-astrocyte marker) or anti-Glial Fibrillary Acidic Protein (GFAP, reactive astrocyte marker) and anti-Cav-1 in WT coronal brain sections 3 dpi (Figure 1). Quantification of Cav-1 protein by Western blot (Figure 1(a)) revealed higher levels in the ipsilateral compared to the contralateral side of the lesion using one-way ANOVA (F = 11.45, p = 0.0090). We found co-localization of Cav-1 and CD31 immunolabeling, in the striatum bilaterally (Figure 1(c) and (d)). Cav-1 also co-localized with GS positive astrocytes in the ipsilateral striatum and to a lesser extent on the contralateral side to the lesion (Figure 1(e) and (f)). In the peri-lesion area of the ipsilateral hemisphere (Figure 1(g)), we observed Cav-1 in some reactive astrocytes, labeled by GFAP. In the contralateral hemisphere (Figure 1(h)), only few astrocytes in the striatum were GFAP-positive, and a few co-localized with Cav-1.
Figure 1.

(a) Western blot showing caveolin-1 protein levels (Cav-1, 22 kDa band) in WT sham and tMCAO ipsilateral (tMCAO Ipsi) and contralateral (tMCAO Contra) side to the lesion. α-Tubulin (50 kDa band) was used as loading control. Quantification was done using mean grey values and normalized against the mean value for Sham. n = 3 animals per condition. (b) Image showing the locations where pictures were taken for Cav-1 and cell-marker expression in relation to the lesion (dotted line showing loss of neuronal MAP-2 staining) on a coronal section in immunofluorescence analysis after tMCAO. (c–d) Cav-1 (green) co-localizes with CD31-labeled (red) endothelial cells in the striatum ipsilateral (c) and contralateral (d) to the ischemic lesion at 3 dpi. (e–f) In the ipsilateral (e) and in the contralateral hemisphere (f), Cav-1 (green) was also observed (arrowheads) in GS-positive astrocytes (red). (g–h) In the cortex, Cav-1 (green) was found to co-localize (arrows) in reactive astrocytes stained with GFAP (red). Respective control staining was done using the same markers on Cav-1 KO tissue and is available in Supplementary Figure 1. Scale bar = 20 µm.
Negative controls showing absence of Cav-1 labeling in Cav-1 KO samples are available in Supplementary Figure 1. We did not see co-localization of Cav-1 with microglial or neuronal markers in the lesion. However, in the peri-lesion, some co-localization of Cav-1 and NeuN is seen (shown by yellow arrows in Supplementary Figure 2) confirming previously reported expression of Cav-1 in neurons.7
These results provide evidence of the modification of Cav-1 expression in the NVU after stroke. More specifically, we saw enhanced Cav-1 in endothelial cells in the lesion and interestingly, its presence in reactive astrocytes in WT animals after stroke.
Outcome assessment
Assessment of survival and lesion size
We used Cav-1 KO mice to investigate the impact of the absence of Cav-1 on outcome after tMCAO.
Cav-1 KO mice showed a significantly decreased survival rate after 35 min tMCAO (found dead or early sacrifice), compared to WT (3/12 vs. 7/9) quantified using a Kaplan–Meier curve (Figure 2(a), Mantel-Cox test (χ2 = 4.603, p = 0.0319)). Of note, there is no difference in mortality between WT and Cav-1 KO sham groups exposed to sham injuries. Sham animals survived the planned 7 dpi (Supplementary Figure 3). Likewise, as shown in Supplementary Figure 3, there was no difference in behavioral outcome after sham surgery between groups, indicating that worse outcome in knock-outs after tMCAO is related to the ischemic injury.
Figure 2.

Survival and lesion size assessment after 35 min tMCAO in WT and Cav-1 KO mice. (a) Kaplan–Meier survival plot and (b) lesion size at time of sacrifice measured on cresyl violet-stained sections (not shown). Box plots show the mean lesion volumes ± min/max for WT and Cav-1 KO mice after 35 min tMCAO and the dots correspond to individual animals with the time of sacrifice specified on the x-axis. (c) Immunofluorescence staining using MAP-2 (neuronal marker) on coronal slices of WT and Cav-1 KO mice after tMCAO or sham surgery collected 6 h, 24 h and three days after stroke, with lesion delimited by the yellow dotted line. Scale bar = 1 mm.
In agreement with the decreased survival rate, the lesion volume measured on cresyl violet-stained sections at the time of sacrifice (3 to 7 dpi) was significantly larger in the Cav-1 group compared to WT (65.73 ± 27.33 mm3 (mean ± SD) versus 33.33 ± 18.67 mm3, respectively Figure 2(b), unpaired t-test (t = 2.44, p = 0.0326)). The increased volume also persisted when taking into account the edema; the size of the ischemic lesion compared to the total size of the brain.
The evolution of the lesion over time after reperfusion at 6 h, 24 h and three days was evaluated in WT and Cav-1 KO mice using microtubule-associated protein 2 (MAP-2) staining of neurons (Figure 2(c)). Decreased MAP-2-labeling revealed the lesion core, highlighted by a yellow dotted line in Figure 2(c). The area of decreased MAP-2 staining appeared already larger in Cav-1 KO sections compared to WT as early as 6 h post-injury. In the Cav-1 KO sections, we observed decreased MAP-2 labeling in the striatum as well as in a large part of the cortex at 6 h after reperfusion, while in the WT group at 6 h after tMCAO, the decrease in MAP-2 staining was limited to the striatum.
Overall, these results suggest that the presence of Cav-1 attenuated and/or delayed the development of the ischemic lesion.
Assessment of sensorimotor deficits
We determined general neurological status using a modified version of the Bederson neuroscore.23 The neuroscore was assessed daily after the stroke (see Supplementary Figure 4); however, only the scores obtained at 1, 4 and 7 dpi are shown in Figure 3(a) for clarity. As outlined in the methods, behavioral performances were analyzed statistically with ANOVA, using either days or experimental groups as variables. Globally, there was a significantly higher proportion of animals exhibiting neuroscore improvement over time after injury in WT compared to KO mice (two-way ANOVA with repeated measures, percentage of total variation: 8.061%, p < 0.0001). The Cav-1 KO mice showed the opposite trend, with a higher number of animals developing a more severe score than the WT group (Figure 3(a)). There was a signifcant difference between experimental groups: WT and Cav-1 KO (percentage of variation: 13.02%, p = 0.0367) as well as on the interaction between experimental groups and day variables (percentage of variation: 6.88%, p = 0.0001).
Figure 3.

Behavioral assessment (a) Neuroscore assessed on a scale from 0 (no deficit) to 3 (sacrifice/death) at 1, 4 and 7 dpi. (b) Latency to fall from the Rotarod apparatus assessed at one, three, five and seven days post-injury (dpi) and expressed as percent of the best baseline performance value (mean ± SD). (c) Time before contact and (d) before removal of the adhesive placed under each paw of the mouse assessed at 2, 4 and 6 dpi and expressed as the difference between best baseline performance and best test time for each mouse (mean ± SD).
The time spent on the rotarod before falling is presented as percentage of the best performance (or longest latency to fall) measured before stroke for each animal in Figure 3(b). We recorded rotarod performance at 1, 3, 5 and 7 dpi (Figure 3(b)). At 1 dpi, both WT and Cav-1 KO animals performed below 50% of their best baseline. At 3 dpi, WT animals improved to 73.14 ± 34% (mean ± SD), whereas Cav-1 KO animals only improved to 45.43 ± 45%. WT animals performed similarly at 5 and 7 dpi, while Cav-1 KO performance decreased due to the increased mortality. The difference between experimental groups (WT versus Cav-1 KO animals) was significant by two-way ANOVA with repeated measures (percentage of total variation: 13.79%, p = 0.0117).
We used the adhesive removal test to assess fine sensorimotor skills involving mouth and digits. We present the time to contact or remove the adhesive as the difference between the testing time and the best baseline time measured before stroke onset for each animal, with results from the paw ipsilateral to the lesion pooled with results of the contralateral paw. The mean time to contact (Figure 3(c)) and remove (Figure 3(d)) the adhesive was increased for both groups at 3 dpi and Cav1-KO mice on average took longer to contact and remove the adhesive than WT mice even at this time-point. The ANOVA with repeated measures for contact times was significant between days of testing post-stroke (percentage of variation: 16.24%, p < 0.0001) and between WT and Cav-1 KO groups (percentage variation 6.031%, p = 0.0293) but also the interaction between days and experimental groups was significantly different (percentage of variation: 4.117%, p = 0.0166). Significant differences for the removal time were also seen for the effect of days (percentage of variation: 3.987%, p = 0.0202) and experimental groups (percentage of variation: 15.48%, p = 0.0018) but not for the interaction of the two variables.
In summary, Cav-1 KO mice exhibit worse functional outcome than WT mice in accordance with the larger lesion volumes observed. All together, these results suggest a protective role for Cav-1 protein after stroke.
Assessment of neovascularization
In Figure 4(a) and (b), we show CD31-labeling (red) with brighter staining in the lesion compared to the contralateral area at 3 dpi in both WT and Cav-1 KO mice, suggesting an increase in blood vessel CD31-reactivity (Figure 4(a) and (b)). This enhanced labeling could reflect neovascularization so we used Ki67, a marker of cell proliferation in combination with CD31 to test this. Coronal brain sections showed Ki67 staining in the ischemic lesion in both groups (Figure 4(c) and (d)). Interestingly, the overall number of Ki67-positive nuclei appeared higher in WT animals (Figure 4(c)) compared to Cav-1 KO animals (Figure 4(d)). We confirmed this on images at 40× magnification where we saw a significantly higher number of CD31-positive cells with Ki67-labeled nuclei (Figure 4(e) and (g)), in WT than in Cav-1 KO mice, suggesting reduced cell proliferation in Cav-1 KO after stroke. On quantification (Figure 4(g)), the number of Ki67-positive nuclei per area was significantly higher in WT with 49.33 ± 12.1 Ki67-positive nuclei (mean ± SD) compared to Cav-1 KO animals, 21.56 ± 6.15 Ki67-positive nuclei (unpaired t-test, t = 6 and p < 0.0001) in images at 20x magnification. Similarly, the number of Ki67-positive nuclei in endothelial cells was significantly higher with 21.1 ± 4.5 positive nuclei per area for the WT and 13.44 ± 3.82 (Mean ± SD) for the Cav-1 KO (t-test, t = 2.498, p = 0.0238). In the vessel density analysis (Figure 4(f)), more vessel area was found in the WT compared to the Cav-1 KO images (unpaired t-test: t = 3.485, p = 0.0019).
Figure 4.

Neovascularization in the lesion. (a–d) Immunofluorescence staining with CD31 (red) and Ki67 (green) in WT (a, c) and Cav-1 KO (b, d) mice at three days post-MCAO. Scale bar = 1 mm. (e) Immunofluorescence staining with CD31 (red), Ki67 (green) and DAPI counterstaining (blue) in the striatum ipsilateral to the lesion in the WT (top image) and in the Cav-1 KO (bottom image). Scale bar = 20 µm. (f) Quantification of the vessel density showing the ratio of vasculature area to the total image area (individual points, mean and SD) in WT and Cav-1 KO mice (g) Quantification (individual points, mean and SD) of the total number of nuclei stained with Ki67 per area (left plot) and the number of nuclei labeled by Ki67 in vessels per area (right plot) in WT and Cav-1 KOs. n = 3 areas per animal with three animals per group, 20× sections.
Our results show that the absence of Cav-1 affects the number of proliferating endothelial cells in the lesion and this may impair the process of neovascularization observed in the core of the lesion after stroke.
Cav-1 and astrocyte reactivity
We observed for the first time Cav-1 in reactive astrocytes after stroke (Figure 1(g)). We performed double labeling with MAP-2 (green) to delineate the lesion area and GFAP (red) to stain reactive astrocytes on coronal brain sections from WT and KO animals collected at 6 h, 24 h and 3 dpi, to establish a time-course of astrocyte changes in relation to the ischemic lesion (Figure 5(a)). We observed reactive astrocytes with increased GFAP staining in the peri-lesion areas in WT samples, with an obvious increase at 3 dpi compared to 6 and 24 h. Interestingly, the Cav-1 KO mice did not display such a noticeable increase in GFAP labeling in the ischemic peri-lesion at 3 dpi nor at earlier time points; however, on quantification, the total number of GFAP-positive astrocytes was higher in the Cav-1 KO group compared to the WT (Figure 5(d), t = 2.196, p = 0.0372). This does not confirm the visual impression that may reflect morphological differences with altered processes (see below). Double immunostaining with GFAP and Ki67 identified new GFAP-positive cells, indicating proliferation of reactive astrocytes in the peri-lesion in WT and KOs (Figure 5(b)). We analyzed possible morphological differences in GFAP-positive astrocytes and their processes between the two groups on images at higher magnification (40×) (Figure 5(c)) and skeletonized. In the Cav-1 KO mice, the GFAP-labelled reactive astrocytes showed a distinct morphology from the WT group, with shorter processes (arrowheads) and less branches. We confirmed this striking visual impression by morphology analysis (Figure 5(d)). The number of endpoints of the astrocytes showed a tendency to decrease in Cav-1 KO images compared to WT (t = 1.893, p = 0.0695). Similarly, the total length of astrocyte branches (272 ± 42.77 for WT vs. 239 ± 30.5 for Cav-1 KO) and the maximum branch-length (48.66 ± 6.25 for WT vs. 41.21 ± 6.86 for Cav-1 KO) were significantly decreased in the Cav-1 KO animals (t = 2.498, p = 0.0192 and t = 2.897, p = 0.0076, respectively, n = 4 images per animal in n = 3 animals per group).
Figure 5.

Astrocyte reactivity and morphology. (a) Astrocyte reactivity time-course on coronal brain sections from WT and Cav-1 KO mice stained with GFAP and MAP-2. Sections were collected from sham animals and 6 h, one and three days after tMCAO. Scale bar = 1 mm (b) Immunofluorescence staining with proliferating cells-marker Ki67 (green), reactive astrocytes marker GFAP (red) and DAPI nuclear counterstaining (blue) in the striatum ipsilateral to the lesion in the WT (top image) and in the Cav-1 KO (bottom image). Scale bar = 20 µm (c) High magnification (40×) panels from sections obtained at 3 dpi illustrating GFAP-positive reactive astrocytes (astrocytes white on black background) in the striatal peri-lesion in WT (top image) and Cav-1 KO (bottom image). Scale bar = 20 µm. Inserts with zoom on a single astrocyte from each group. (d) Analysis of astrocyte number and astrocytic morphology by skeletonization: plot of the number of GFAP-positive cells, number of endpoints, total length of the segment and the maximum branch length in WT and Cav-1 KO astrocytes in the peri-lesion. n = 4 areas per animal with three animals per group.
All together, these results point towards a change in the pattern and the distribution of reactive astrocytes with morphological differences, perhaps indicating impaired astrogliosis following ischemic injury in the absence of Cav-1.
Discussion
The NVU has been proposed as a key element for future development of new treatments for stroke and other brain disorders.5 We and others previously showed changes of Cav-1 expression in the NVU in various brain disorders12,19,28; however, the changes in Cav-1 levels and the role of the protein in stroke are not clear at present.7 To address this question, we studied changes in Cav-1 levels in the NVU over time after tMCAO and compared functional outcome as well as changes at the cellular level in WT and genetically modified Cav-1 KO mice. Overall, in this study, the absence of Cav-1 is associated with (1) increased mortality and larger lesion volumes after tMCAO, (2) a more severe functional outcome with sensorimotor deficits assessed by the neuroscore, rotarod and adhesive removal behavioral tests, (3) reduced proliferation of endothelial cells, (4) altered reactive astrocyte morphology and distribution in the peri-lesion.
We believe that the presence of Cav-1 is important for limiting the progression of the pathology after stroke onset by facilitating (1) neovascularization in the ischemic lesion and (2) astrogliosis and scar formation in the perilesional tissue, not so far addressed after stroke (Figure 6). Our work suggests a decisive role for Cav-1 in the two processes contributing to tissue repair and highlights novel findings regarding the behavior and role of astrocytes in Cav-1 deficient mice after ischemic stroke.
Figure 6.
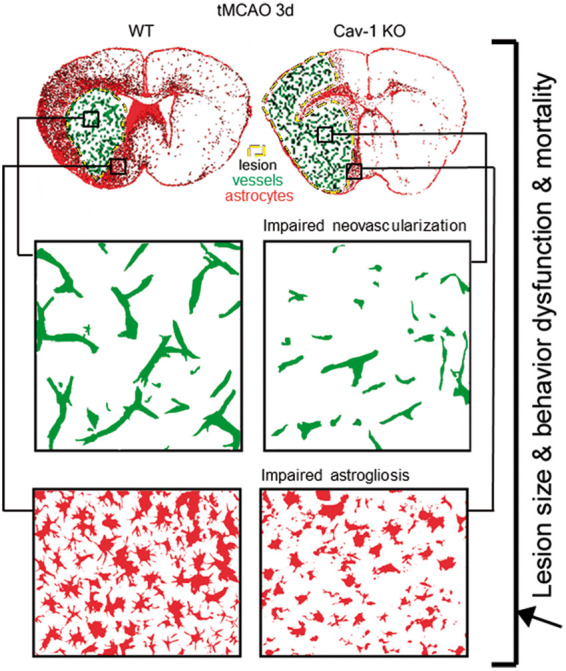
Summary figure of the differences observed between WT and Cav-1 KO mice. Increased lesion sizes associated with behavioral dysfunction such as sensorimotor deficits were observed in parallel to impaired neovascularization in the core of the lesion and altered astrogliosis, more specifically changes in astrocytic morphology preventing proper scar formation in the peri-lesion.
Presence of Cav-1 benefits recovery post-stroke
Several studies have shown changes in Cav-1 expression in different preclinical models of brain disorders with a dual interpretation of its role.7,18 This is true for stroke research where preclinical studies in a variety of animal models report deleterious or beneficial roles of Cav-1 after stroke. Here we chose the filament tMCAO model as the middle cerebral artery is the most commonly affected blood vessel in human ischemic stroke29 and this model has been used in many studies addressing pathophysiological processes or neuroprotective agents.22 It induces ischemia in the striatum and cortex with a quantifiable neurological deficit and models ischemia with reperfusion as seen in most stroke patients. We identified 35 min occlusion as a good compromise between variability of lesion size and severity in the C57Bl6/J WT mice, as recently published.30 In our model, Cav-1 KOs displayed larger lesion sizes at time of sacrifice compared to WTs, suggesting a beneficial role of Cav-1 in tMCAO. We observed a difference between groups as early as 6 h after injury, with a more prominent loss of neurons in the absence of Cav-1.
The major preclinical outcome measures for stroke pharmacotherapy studies are final lesion size and sensorimotor neurological deficits. The latter are more clinically relevant and also more challenging to accurately assess as deficits can rapidly resolve in rodent models.29 A broad variety of tests have been described to assess post-stroke deficits.25 To our knowledge, we are the first to assess and describe sensorimotor deficits in Cav-1 KO mice following stroke. We chose three tests to discriminate between general neurological deficits (neuroscore), locomotor activity (rotarod) and fine movements (adhesive removal). In the Cav-1 KO group, at each time point, the neuroscore was worse, the time to fall from the rotarod shorter and the time to contact and remove the adhesive longer than in the WT group. Overall, this highlights greater sensorimotor deficits for the Cav-1 KO group in agreement with larger lesion volumes. Others have reported neurological abnormalities including clasping, abnormal spinning, muscle weakness, reduced activity and gait abnormalities in Cav-1 KO mice.31,32 However, we did not observe this and found no significant differences in the three behavioral tests used in this study between WT and Cav-1 KO mice after sham injuries (Supplementary Figure 3).
Our results are in agreement with a previous study19 where Cav-1 KO mice had increased lesion volumes at an early time point in a model of permanent distal MCAO (MCA sutured locally after a craniotomy). In the cited work, the authors suggested an important role of increased Cav-1 at 48 h, and at one and two weeks for tissue protection, with a significant decrease in apoptosis, a significant increase in neovascularization as well as increased numbers of new endothelial cells in the lesion core. However, in a rat model of 2 h tMCAO, Gu et al.17 obtained opposite results showing decreased Cav-1 expression at 24, 48 and 72 h in the ipsilateral hemisphere. In the same study, a decrease in Cav-1 expression was associated with an increase in metalloproteinase activity at 24 h after a 15-min tMCAO in Cav-1 KO mice.17 The discrepancy between our results and Gu et al.17 may be explained by differences in the experimental protocols, including MCA occlusion time (35 min versus 15 min in mice), different stroke models in different species (rat or mouse), as well as analysis of homogenates of the entire ischemic hemisphere (here) versus homogenates of the ischemic core only in Gu et al. Our behavioral outcomes after stroke support the idea that endogenous Cav-1 plays a beneficial role in limiting the extension of the lesion after 35 min tMCAO in mice.
Benefit from post-stroke neovascularization
Studies showed Cav-1 expression in various cell types of the NVU, and in a juvenile rat TBI model, we previously showed an increase in Cav-1 levels in endothelial cells and astrocytes.12 We explored the localization of Cav-1 protein here at 3 dpi by immunofluorescence in WT animals. Cav-1 was present in CD31-positive endothelial cells. This result is in agreement with previous work, as endothelial cells are one of the cell populations that express the highest level of caveolae and Cav-1 and caveolae are thought to constitute up to 30% of the total endothelial cell surface in capillaries.7 Interestingly, the ischemic hemisphere appeared to have increased levels of Cav-1 after stroke compared to contralateral hemisphere. This was also seen in previous studies showing increased Cav-1 expression in the ischemic hemisphere after photothrombotic ischemia in mice28 and tMCAO in rats.19 Here we confirm it in a stroke model with reperfusion. Previous studies extensively investigated the correlation between Cav-1 expression and protection against BBB disruption and suggested a protective role for Cav-1 against BBB breakdown during focal cerebral ischemia.28 The deficiency in neovascularization in Cav-1 KO mice has already been assessed in a permanent cerebral ischemia model of distal ligation of the MCA19 and in a hind limb ischemia model.33 In the work by Jasmin et al., neovascularization was demonstrated by PCNA (proliferating cell nuclear antigen)-positive laminin-labelled blood vessels. Here we use a different MCAO model and marker of cell proliferation, Ki67, and confirm these results. We observed endothelial cell proliferation in the ischemic lesion in both groups (Figure 4) in agreement with the previous studies.7 However, Cav-1 KO mice exhibited significantly less neovascularization with fewer Ki67-positive endothelial cells compared to WT and decreased vessel density. This suggests a lower number of new endothelial cells in the ischemic lesion when Cav-1 is absent. Neovascularization is a dynamic process of endothelial cell proliferation, migration and differentiation and a well-established event occurring in the ischemic lesion.7 It is essential for brain recovery as it stimulates blood flow, collateralization and neuroplasticity.33 The impaired neovascularization observed might contribute to the increased ischemic injury observed in Cav-1 KO mice. Mechanistically, Cav-1 could act as a “differentiation sensor” to modulate regulators of neovascularization, such as vascular endothelial growth factor (VEGF), produced by astrocytes or eNOS to coordinate proliferation and differentiation of endothelial cells.7
Benefit from post-stroke astrogliosis
We identified other Ki67-positive proliferating cells as reactive astrocytes and microglia (Supplementary Figure 5). Some studies showed Cav-1 in astrocyte cultures in vitro10,11 and in an in vitro model of ischemia reperfusion (oxygen glucose deprivation/reoxygenation: OGD-R).42 To our knowledge, apart from our previous study showing localization of Cav-1 protein in rat astrocytes after juvenile TBI in vivo,12 we show for the first time, Cav-1 localization in adult mice astrocytes positive for GS and GFAP after ischemic stroke (Figure 1(e) to (h)). However, the emerging view is that astrocytes constitute a heterogeneous population8 and the role of Cav-1 in astrocytes is unknown in both normal and pathological brain tissue. Astrogliosis is defined as the response of astrocytes to brain stress, damage and disease with changes in gene expression, cellular structure and functions.8 Marked diffuse reactive astrogliosis is generally found in areas surrounding severe focal lesions forming a glial scar,34 thought to be part of the first phase after focal injury such as ischemia, comprising cell death and inflammation35 that may have beneficial effects on brain injury outcome.36 The role of Cav-1 in astrocytes after ischemia was not yet investigated. We show morphological differences in reactive astrocytes in the absence of Cav-1; specifically, the ramification pattern is different with a significant decrease in the total length of segments and maximum branch length in Cav-1 KO mice. The low level of ramification and process complexity of Cav-1 KO astrocytes could prevent scar formation and may explain the lower visually observed labeling intensity. We quantified the number of astrocytes at 3 dpi in the images used for the morphology analysis (Figure 5(d)) and found higher numbers of GFAP-positive astrocytes in the Cav-1 KO compared to the WT group. Both groups show Ki67-positive astrocytes after tMCAO.
We observed better recovery of WT mice at 3 dpi in behavioral tests, while Cav-1 KO animals had modified reactive astrocytes and absence of a glial scar in the ischemic peri-lesion. One other study also found less astroglial differentiation in the dentate gyrus of adult Cav-1 KO mice37 indicating that genetic ablation of Cav-1 may directly inhibit the formation of new reactive astrocytes and be detrimental to ischemic recovery. While traditionally viewed as a barrier to axon regeneration, beneficial functions of the glial scar have recently been identified.38 An emerging concept is that the astrocytic scar could aid central nervous system regeneration. In a model of spinal cord injury (SCI), RNA sequencing revealed that astrocytes and non-astrocyte cells in the lesion express multiple axon-growth-supporting molecules. In contrast, preventing astrocytic scar formation significantly reduced this stimulated axon regrowth.39 The glial scar has been widely studied in SCI but more work is needed in TBI and ischemic stroke.
Although several studies investigated Cav-1 signaling pathways of astrocytes in vitro,40–42 the mechanism by which Cav-1 regulates astrogliosis is still unclear. In an OGD-R in vitro model, a recent study suggested that basic fibroblast growth factor (bFGF) might protect astrocytes from injury by up regulating the Cav-1/VEGF signaling pathway42 indicating in addition to its roles in neovascularization and neurogenesis, this pathway may be involved in astrogliosis, astroglial scar formation and neuroinflammation after cerebral ischemia.42
Limitations of the present study include providing a mechanism of how Cav-1 might promote revascularization and astroglial scar formation after stroke. Currently only the Cav-1 KO mouse is available and to prove a primary role of Cav-1, deletion of Cav-1 using a tamoxifen-inducible conditional deletion at a delayed time point after infarct resolution would be an interesting approach. Likewise, endothelial/astrocyte-specific overexpression of Cav-1 to rescue revascularization and astrogliosis will be topics for future research.
Summary
Altogether, characterization of Cav-1 KO mice brings insight into the role of caveolin-1 in brain injuries. Here, we investigated endothelial cells and astrocytes of the NVU, for a novel perspective on approaches against ischemia injury. We saw better survival and recovery after stroke if Cav-1 was present. These results point towards a protective role of endogenous Cav-1 in the NVU in the first days after stroke. Our hypothesis is that via its signaling functions, Cav-1 and caveolae formation could play a role in neovascularization and astrogliosis after ischemic injury. A key protein in ‘wound-healing’, mimicking caveolin functions could be a novel neuroprotective strategy.
Supplemental Material
Supplemental material for Involvement of caveolin-1 in neurovascular unit remodeling after stroke: Effects on neovascularization and astrogliosis by Camille Blochet, Lara Buscemi, Tifenn Clément, Sabrina Gehri, Jérôme Badaut and Lorenz Hirt in Journal of Cerebral Blood Flow & Metabolism
Acknowledgements
The authors thank Leonardo Restivo for help with behavioral analysis using the NeuroBAU platform of the Department of Fundamental Neurosciences. The authors acknowledge the Cellular Imaging Facility (CIF) for imaging support. The authors thank Melanie Price for help with carefully reading and editing the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by the Swiss National Science Foundation 31003A_163465 / 1 (LH, JB), Eranet Neuron TRAINS (JB); TRAIL- Laboratory of Excellence TRAIL (ANR-10-LABX-57 to JB).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article
Authors’ contributions
CB, LB, JB and LH contributed to study conception. CB acquired the data and CB, LB, SG and TC analyzed the data. CB, JB, LB and LH drafted the manuscript and figures. JB and LH contributed equally to this work and share last authorship. All authors read and approved the final version of the manuscript.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics – 2016 update: a report from the American Heart Association. Circulation 2016; 133: e38–360. [DOI] [PubMed] [Google Scholar]
- 2.Meadows KL. Experimental models of focal and multifocal cerebral ischemia: a review. Rev Neurosci 2018; 29: 661–674. [DOI] [PubMed] [Google Scholar]
- 3.Goyal M, Menon BK, van Zwam WH, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet 2016; 387: 1723–1731. [DOI] [PubMed] [Google Scholar]
- 4.Neuhaus AA, Couch Y, Hadley G, et al. Neuroprotection in stroke: the importance of collaboration and reproducibility. Brain 2017; 140: 2079–2092. [DOI] [PubMed] [Google Scholar]
- 5.Zhang JH, Badaut J, Tang J, et al. The vascular neural network – a new paradigm in stroke pathophysiology. Nat Rev Neurol 2012; 8: 711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Persidsky Y, Ramirez SH, Haorah J, et al. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol 2006; 1: 223–236. [DOI] [PubMed] [Google Scholar]
- 7.Xu L, Guo R, Xie Y, et al. Caveolae: molecular insights and therapeutic targets for stroke. Expert Opin Ther Targets 2015; 19: 633–650. [DOI] [PubMed] [Google Scholar]
- 8.Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol 2014; 7: a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hnasko R, Lianti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv 2003; 3: 20. [DOI] [PubMed] [Google Scholar]
- 10.Ikezu T, Ueda H, Trapp BD, et al. Affinity-purification and characterization of caveolins from the brain: differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain Res 1998; 804: 177–192. [DOI] [PubMed] [Google Scholar]
- 11.Cameron PL, Ruffin JW, Bollag R, et al. Identification of caveolin and caveolin-related proteins in the brain. J Neurosci 1997; 17: 9520–9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badaut J, Ajao DO, Sorensen DW, et al. Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: signs of blood-brain barrier healing? Neuroscience 2015; 285: 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stern CM, Mermelstein PG. Caveolin regulation of neuronal intracellular signaling. Cell Mol Life Sci 2010; 67: 3785–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Razani BW, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev 2002; 54: 37. [DOI] [PubMed] [Google Scholar]
- 15.Okamoto T, Schlegel A, Scherer PE, et al. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem 1998; 273: 5419–5422. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Cardena G, Martasek P, Masters BS, et al. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J Biol Chem 1997; 272: 25437–25440. [DOI] [PubMed] [Google Scholar]
- 17.Gu Y, Zheng G, Xu M, et al. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem 2012; 120: 147–156. [DOI] [PubMed] [Google Scholar]
- 18.Gu Y, Dee CM, Shen J. Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front Biosci 2011; 3: 1216–1231. [DOI] [PubMed] [Google Scholar]
- 19.Jasmin JF, Malhotra S, Singh Dhallu M, et al. Caveolin-1 deficiency increases cerebral ischemic injury. Circ Res 2007; 100: 721–729. [DOI] [PubMed] [Google Scholar]
- 20.Knowland D, Arac A, Sekiguchi KJ, et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014; 82: 603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science 1997; 278: 2075–2080. [DOI] [PubMed] [Google Scholar]
- 22.Fluri F, Schuhmann MK, Kleinschnitz C. Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther 2015; 9: 3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bederson JB, Pitts LH, Tsuji M, et al. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 1986; 17: 472–476. [DOI] [PubMed] [Google Scholar]
- 24.Hirt L, Badaut J, Thevenet J, et al. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke 2004; 35: 1738–1743. [DOI] [PubMed] [Google Scholar]
- 25.Balkaya M, Krober JM, Rex A, et al. Assessing post-stroke behavior in mouse models of focal ischemia. J Cereb Blood Flow Metab 2013; 33: 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison H, Young K, Qureshi M, et al. Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci Rep 2017; 7: 13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Castro Ribeiro M, Badaut J, Price M, et al. Thrombin in ischemic neuronal death. Exp Neurol 2006; 198: 199–203. [DOI] [PubMed] [Google Scholar]
- 28.Choi KH, Kim HS, Park MS, et al. Regulation of caveolin-1 expression determines early brain edema after experimental focal cerebral ischemia. Stroke 2016; 47: 1336–1343. [DOI] [PubMed] [Google Scholar]
- 29.Macrae IM. Preclinical stroke research – advantages and disadvantages of the most common rodent models of focal ischaemia. Br J Pharmacol 2011; 164: 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benakis C, Brea D, Caballero S, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat Med 2016; 22: 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trushina E, Du Charme J, Parisi J, et al. Neurological abnormalities in caveolin-1 knock out mice. Behav Brain Res 2006; 172: 24–32. [DOI] [PubMed] [Google Scholar]
- 32.Gioiosa L, Raggi C, Ricceri L, et al. Altered emotionality, spatial memory and cholinergic function in caveolin-1 knock-out mice. Behav Brain Res 2008; 188: 255–262. [DOI] [PubMed] [Google Scholar]
- 33.Zhang ZG, Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol 2009; 8: 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol 2010; 119: 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014; 81: 229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci 2015; 16: 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Lau WM, So KF, et al. Caveolin-1 promote astroglial differentiation of neural stem/progenitor cells through modulating Notch1/NICD and Hes1 expressions. Biochem Biophys Res Commun 2011; 407: 517–524. [DOI] [PubMed] [Google Scholar]
- 38.Adams KL, Gallo V. The diversity and disparity of the glial scar. Nat Neurosci 2018; 21: 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson MA, Burda JE, Ren Y, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016; 532: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jo A, Park H, Lee SH, et al. SHP-2 binds to caveolin-1 and regulates Src activity via competitive inhibition of CSK in response to H2O2 in astrocytes. PLoS One 2014; 9: e91582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu L, Wang L, Wen Z, et al. Caveolin-1 is a checkpoint regulator in hypoxia-induced astrocyte apoptosis via Ras/Raf/ERK pathway. Am J Physiol Cell Physiol 2016; 310: C903–910. [DOI] [PubMed] [Google Scholar]
- 42.Liu M, Wu Y, Liu Y, et al. Basic Fibroblast growth factor protects astrocytes against ischemia/reperfusion injury by upregulating the caveolin-1/VEGF signaling pathway. J Mol Neurosci 2018; 64: 211–223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material for Involvement of caveolin-1 in neurovascular unit remodeling after stroke: Effects on neovascularization and astrogliosis by Camille Blochet, Lara Buscemi, Tifenn Clément, Sabrina Gehri, Jérôme Badaut and Lorenz Hirt in Journal of Cerebral Blood Flow & Metabolism