

Abstract
Objectives
Plasmacytoid dendritic cells (pDCs), through the production of type 1 interferons (IFNs) and other cytokines, are major contributors to systemic lupus erythematosus (SLE) pathogenesis. IL‐3 promotes pDC survival, but its role in SLE is not well characterised. This study investigated serum IL‐3 and IFN levels, and a whole blood ‘IL‐3 gene signature’, in human SLE.
Methods
Serum cytokine levels were measured by ELISA in n = 42 SLE patients, and n = 44 healthy donors. IL‐3‐regulated genes were determined by RNASeq of healthy donor whole blood cells (WBCs) stimulated in vitro with IL‐3 for 6 or 24 h. Whole blood cell RNASeq analysis was undertaken in a separate cohort of n = 31 SLE patients, and n = 28 healthy donors.
Results
Serum IL‐3 levels correlated with IFNα (r = 0.612, 95% CI 0.455–0.733, P < 0.001) and type III IFN (r = 0.585, 95% CI 0.406–0.720, P < 0.0001). IL‐3 stimulation of WBC in vitro altered 794 genes (−1 ≥ logFC ≥ 1, FDR < 0.05), of which 35 overlapped with genes differentially expressed between SLE and healthy donors. These 35 genes were expressed in 27/31 SLE donors, revealing the presence of an ‘IL‐3 gene signature’. There was strong correlation between the IL‐3 signature and an IFN signature, as determined by hierarchical clustering of the 500 most variable genes in SLE donors (r = 0.939, 95% CI 0.898–0.964, P < 0.0001).
Conclusion
A dual IL‐3/IFN gene signature is a feature of SLE. An association between IL‐3 and IFN raises the possibility that dual blockade of IL‐3 and IFN may be especially useful for SLE patients with this dual cytokine gene signature.
Keywords: cytokines, gene signature, immunological disorders, interferons, interleukin-3, systemic lupus erythematosus
Through analysis of serum cytokine levels, and whole blood transcriptional profiling, a correlation between IL‐3 and interferon (IFN) (type I and type III) has been identified in systemic lupus erythematosus (SLE) patients. A correlation between both IFN‐α and type III IFN and IL‐3 in serum was seen, as was the presence of an ‘IL‐3 gene signature’ in a majority of SLE patients, which correlated strongly with an IFN gene signature.

Introduction
IL‐3 is a multi‐potent haematopoietic growth factor that contributes to the maturation and survival of a number of immune cell types. The role of IL‐3 in systemic lupus erythematosus (SLE) has been less studied compared to other cytokines, such as type I interferon (IFN) or B‐cell‐activating factor (BAFF), which are known to be mediators of pathogenesis. One previous study found elevated serum IL‐3 levels in SLE patients who were naïve to treatment with immunosuppressive drugs.1 Recent studies of murine lupus have more directly identified a role for IL‐3. In the MRL/lpr murine lupus model, IL‐3 administration exacerbated nephritis, and IL‐3 blockade alleviated disease and decreased autoantibody production.2 Plasma levels of IL‐3 increased during disease progression in these mice, and IL‐3 was produced by CD4+ T cells derived from the spleen and bone marrow, and by CD8+ T cells derived from the spleen. Recently, a therapeutic monoclonal antibody that neutralises IL‐3 and also depletes CD123+ cells was shown to decrease cell types (plasmacytoid dendritic cells [pDCs] and basophils) and cytokines implicated in the pathogenesis of SLE, including type I and III IFNs, in primary cells from SLE patients.3 These data raise the possibility of using IL‐3 blockade as a therapeutic strategy in SLE.
Here, we report an association between serum IL‐3 and IFN (IFNα and type III), and a dual IL‐3/IFN gene signature in whole blood from patients with SLE. This association suggests that SLE patients may benefit from therapeutic targeting of both IL‐3 and IFN.
Results
Demographic and clinical characteristics
Systemic lupus erythematosus donors in the serum (Supplementary table 3) and transcriptional profiling (Supplementary table 4) cohorts were similar, with a female predominance and heterogeneous disease manifestations, mainly affecting the cutaneous, musculoskeletal and immunological systems. SLE patients were taking a range of immunomodulatory or immunosuppressive medications. Healthy donors were age and sex matched to each cohort, with donor characteristics presented in Supplementary table 5.
Correlation in peripheral blood between serum IL‐3 and type I and type III IFN levels, and between pDCs and basophils
Twenty‐five (of 42) SLE and 22 (of 44) healthy donors had detectable serum IL‐3 levels. Mean serum IL‐3 levels were not significantly higher in patients with SLE (272 ± 77 [SEM] pg mL−1, range 7.8–2000 pg mL−1) compared to healthy donors (400 ± 97 pg mL−1, range 7.8–2000 pg mL−1, P = 0.840; Figure 1a). There were no significant correlations between IL‐3 levels and higher disease activity (as defined by a SLEDAI ≥ 6, low complement or high anti‐dsDNA antibody levels, or raised inflammatory markers), or with disease manifestations or medication use in the SLE donors.
Figure 1.
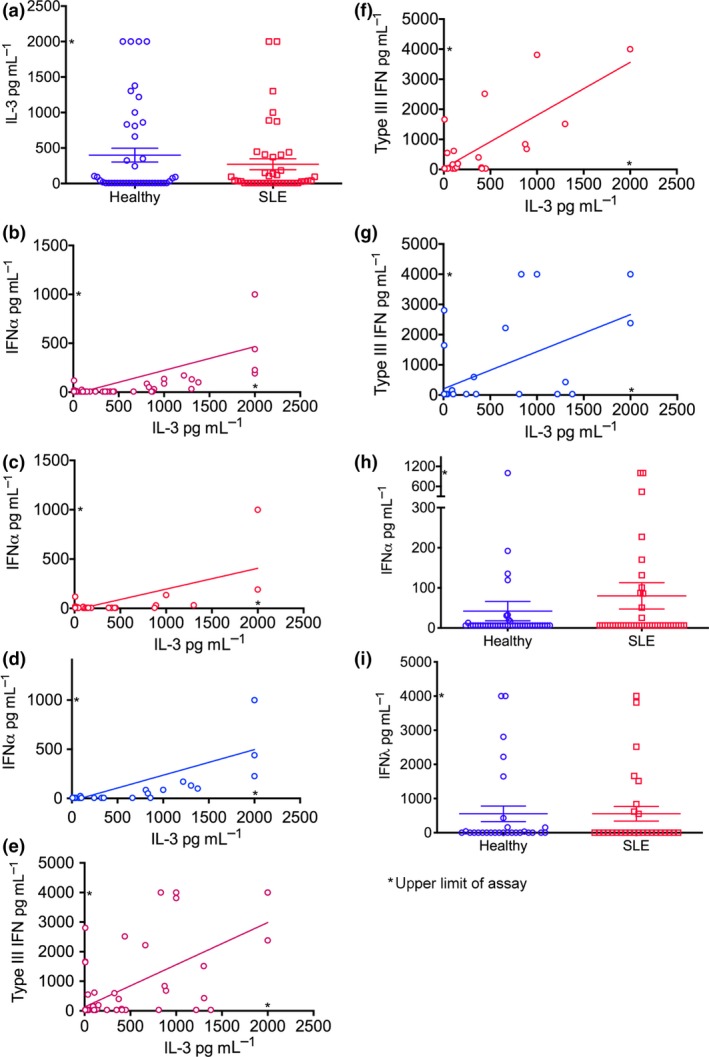
Serum IL‐3 levels correlate with IFNα and type III IFN in SLE patients and healthy donors. (a) Serum IL‐3 levels in SLE (n = 42) and healthy (n = 44) donors. Serum IL‐3 and IFNα levels in (b) SLE (n = 42) and healthy (n = 44), (c) SLE only and (d) healthy only, donors. Serum IL‐3 and type III IFN levels in (e) SLE (n = 36) and healthy (n = 37), (f) SLE only and (g) healthy only, donors. (h) Serum IFNα levels in SLE (n = 42) and healthy (n = 44) donors. (i) Serum IFNλ levels in SLE (n = 37) and healthy (n = 36) donors. Cytokine levels were determined by ELISA. Correlations were determined by the Spearman test. Data are depicted as mean ± SEM. n = 2 technical replicates.
Serum IL‐3 levels positively correlated with serum IFNα in both SLE and healthy donors (r = 0.612, 95% CI 0.455–0.733, P < 0.0001; Figure 1b), although the correlation was weaker in patients with SLE (r = 0.376, 95% CI 0.072–0.616, P = 0.014; Figure 1c) compared to healthy (r = 0.784, 95% CI 0.629–0.879, P < 0.0001; Figure 1d) donors. An association was also found between IL‐3 and type III IFN levels (r = 0.585, 95% CI 0.406–0.720, P < 0.0001; Figure 1e), which was present in both SLE (r = 0.666, 95% CI 0.432–0.816, P < 0.0001; Figure 1f) and healthy (r = 0.515, 95% CI 0.213–0.724, P = 0.001; Figure 1g) donors. There was no significant difference in IFNα and type III IFN levels between SLE and healthy donors (Figure 1h and i, respectively).
A weaker correlation was seen between IL‐3 and IL‐4 levels (r = 0.279, 95% CI 0.038–0.489, P = 0.021; Supplementary figure 1a), and between IL‐3 and IL‐5 levels (r = 0.255, 95% CI 0.012–0.469, P = 0.035) in both SLE and healthy donors (Supplementary figure 1b). There were no statistically significant correlations between IL‐3 and BAFF, IFNγ, GM‐CSF, G‐CSF, Flt‐3l, IL‐2, IL‐6, IL‐13, IP‐10 or TNF in either group.
In SLE patients, there were moderate positive correlations between serum IL‐3 levels and pDCs (as % total WBC; r = 0.409, 95% CI 0.071–0.662, P = 0.016; Supplementary figure 2a), and basophils (as % total WBC, r = 0.372, 95% CI 0.029–0.637, P = 0.030; and as a count mL−1, r = 0.394, 95% CI 0.053–0.651, P = 0.021; Supplementary figure 2b and c). These correlations could reflect the reliance of pDCs and basophils on IL‐3 as a survival factor,5, 6 but similar significant positive correlations were not found in the healthy donor population (conversely, a negative correlation for pDCs as a count mL−1 was found; r = −0.347, 95% CI −0.616 to −0.005, P = 0.041). There were no significant correlations between IL‐3 levels and T‐cell numbers or percentages, in SLE or healthy donors.
IL‐3 gene signature differentiates SLE and healthy donors
To search for a potential ‘IL‐3 gene signature’, those genes altered in response to IL‐3 stimulation in vitro were determined by RNAseq analysis of WBCs from seven healthy donors stimulated with, or without, IL‐3, for 6 or 24 h. Using an FDR of < 0.05, and ≥ 2‐fold change in gene expression as thresholds, there were 794 differentially expressed genes (650 upregulated, 144 downregulated), inclusive of both time points (Figure 2a). Of these, 152 were differentially expressed at both time points, 592 at 24 h only, and 50 at 6 h only. The top 10 differentially expressed genes at 6 and 24 h are shown in Supplementary tables 6 and 7.
Figure 2.

IL‐3 gene signature differentiates SLE and healthy donors. (a) Numbers of differentially expressed genes between IL‐3 stimulated (for 6 or 24 h) and non‐stimulated, whole blood cells from healthy donors (n = 7), as analysed by RNAseq. Numbers enclosed within the circles represent genes that were differentially expressed (FDR < 0.05), with at least a 2‐fold change in gene expression. (b) Heat map showing expression of IL‐3‐regulated genes in SLE (n = 31) and healthy (n = 28) donors, as determined by RNAseq analysis of donor whole blood cells. (c) IL‐3 gene signature score, derived as the first principal component of the IL‐3 gene signature expression levels for each SLE and healthy donor individually. Data are shown as mean ± SEM, * P < 0.05. n = 2 technical replicates.
Thirty‐five of these genes were also found to be differentially expressed (with a fold change of ≥ 2) between SLE and healthy donor whole blood cells (WBCs) by RNAseq analysis (Supplementary table 8). These 35 genes were more highly expressed in all but four of the SLE donors, but in only six of the healthy donors (Figure 2b), suggesting the presence of an ‘IL‐3 gene signature’ in most SLE donors. Additionally, the mean IL‐3 gene signature score, derived as the first principal component of the genes in the signature (shown in green in Figure 2b), was significantly higher in SLE patients than in healthy donors (Figure 2c).
Correlation of IL‐3 gene signature with IFN gene signature
Given the association of IL‐3 and IFN levels in the serum, a possible association at the transcriptional level in whole blood cells was explored. An ‘IFN gene signature’ (EPST11, ISG15, HERC5, IFI44, OAS3, OAS1, LY6E, CMPK2, RSAD2, IFI44L, IFIT1, IFIT3, USP18, SIGLEC1, IFI27, OTOF)7, 8, 9, 10 that differentiated most SLE from most healthy donors was determined by hierarchical clustering and pathway analysis of the 500 most variably expressed genes in SLE donors (Figure 3a), that is those genes that were most differentially expressed in SLE patients compared to healthy donors and were known interferon‐inducible genes per the Interferome database.7
Figure 3.

Correlation between IL‐3 and IFN gene signature scores in SLE and healthy donors. (a) Heat map showing expression of IFN‐regulated genes (EPST11, ISG15, HERC5, IFI44, OAS3, OAS1, LY6E, CMPK2, RSAD2, IFI44L, IFIT1, IFIT3, USP18, SIGLEC1, IFI27, OTOF) in SLE (n = 31) and healthy (n = 28) donors, as determined by RNAseq analysis of donor whole blood cells, and hierarchical clustering of the 500 most variably expressed genes (based on standard deviation) in SLE donors (FDR < 0.05). (b) IFN and IL‐3 gene signature scores in SLE (n = 31) and healthy (n = 28) donors, as determined by RNAseq analysis of donor whole blood cells. IL‐3 and IFN gene signature scores were determined by taking the first principal component of the panel of genes comprising each gene signature. Correlation was determined by the Spearman test, P < 0.05. n = 2 technical replicates.
Single gene scores for both the IL‐3 and IFN gene signatures were determined for each SLE patient and healthy donor, as the first principal component11 from each gene signature. A strong correlation between IL‐3 and IFN gene signature scores was found (r = 0.939, 95% CI 0.898–0.964, P < 0.0001; Figure 3b).
Discussion
We have shown a correlation between IL‐3 and IFN, in two separate cohorts of SLE patients and healthy donors, using serum cytokine analysis and transcriptional profiling of WBCs. This correlation has not previously been described in SLE, or in other diseases, and was evident even though mean IL‐3 levels in the serum were not elevated in SLE patients compared to healthy donors.
The correlation between IL‐3 and IFN (IFNα and type III) may be partly explained by the survival effect of IL‐3 on pDCs,5 which produce both these types of IFN; a moderate correlation between serum IL‐3 levels and the number of pDCs in peripheral blood was also observed in this study. Recently, IL‐3 was found to enhance IFNα production by pDCs from healthy donors stimulated with RNA‐containing immune complexes.12 Whether IL‐3 increased IFNα production through enhancing pDC survival, or via another mechanism, was not elucidated in that study, but the upregulation of known IFN‐inducible genes in response to IL‐3 may also contribute to our findings. Of interest in that study, GM‐CSF also potently increased pDC‐derived IFNα production,12 independent of pDC proliferation. Supernatant from T cells activated with CD3/CD28 was also able to stimulate IFNα production from pDCs, in both SLE and healthy donors. Depletion of GM‐CSF from the activated T‐cell supernatants reduced the stimulatory effect of the T‐cell supernatants on pDC‐derived IFNα production, as did blockade of the GM‐CSF receptor and shared β subunit of the GM‐CSF and IL‐3 receptors. In contrast, depletion of IL‐3 or blockade of the IL‐3 receptor alone did not, indicating that T‐cell‐derived GM‐CSF may play an important role in this interaction. However, in our study, a correlation between serum GM‐CSF and IFNα was not observed. A weak correlation of IL‐3 with IL‐4, and with IL‐5, was evident in our study, as was a positive correlation between IL‐3 and basophil numbers. A potential explanation could be the survival effect of IL‐3 on basophils, which have been postulated to contribute to SLE by elaborating Th2 cytokines (such as IL‐4 and IL‐5) when activated by IgE‐containing immune complexes.13
Although type I IFNs are well‐known pathogenic cytokines in SLE, data suggesting dysregulation of type III IFNs in SLE have only emerged recently. There are four subtypes of type III IFN – IFNλ1 (IL‐29), IFNλ2 (IL‐28A) and IFNλ3 (IL‐28B) and IFNλ4.14 In our study, we have measured the combined levels of IFNλ1‐3, but not IFNλ4. It is possible that delineating the different subtypes of type III IFN may reveal correlations with disease parameters, as each subtype may play a different role in disease. For example, serum IFNλ1 levels have previously been associated with the presence of anti‐dsDNA antibodies, glomerulonephritis and arthritis,15 and elevated IFNλ2 mRNA transcripts have been found in activated CD4+ T cells from lupus patients compared with healthy controls.16 Another potential limitation of this study is the possible influence of background therapies on cytokine levels and gene expression in the SLE patients. Although we could not detect any correlation between IL‐3 and particular medications, the relatively small numbers of patients on individual immunosuppressants made assessment for this potential confounding factor difficult.
The presence of an IFN gene signature in SLE is well known and may predict more responsiveness to anti‐IFN therapies.17 In this study, we have found an IL‐3 gene signature occurring concurrently with an IFN gene signature in the majority of SLE patients, which may be partly due to the presence of a large number of IFN‐inducible genes in response to IL‐3 stimulation. The findings from this study raise the possibility that those with a dual IFN/IL‐3 gene signature may derive additional therapeutic benefit from blockade of both IL‐3 and IFN.
Methods
Serum cytokine analysis, and quantification of peripheral blood cell types, was performed in SLE patients from The Royal Melbourne Hospital, and on healthy donors from The Walter and Eliza Hall Institute's Volunteer Blood Donor Registry.
RNAseq analysis of IL‐3 upregulated gene expression was performed on whole blood cells collected from healthy donors in The Walter and Eliza Hall Institute's Volunteer Blood Donor Registry.
Differential gene expression between SLE donors from The Monash Lupus Clinic and healthy donors from The Skin and Cancer Foundation was determined by RNAseq analysis of donor whole blood cells.
The study was conducted with approval from the Human Research Ethics Committees of each institution, and all donors gave written consent prior to study participation. Donors were ≥ 18 years old, and SLE donors fulfilled the ACR (American College of Rheumatology)4 classification criteria for SLE.
Experimental methods and bioinformatic analysis are described in further detail in the Supplementary methods. Data from RNASeq analyses have been deposited online in the Gene Expression Omnibus (accession numbers GSE110041 and GSE112087).
Conflict of interest
SO, IPW, AH and EM have received research funding from CSL Limited. KM, MN, GV, EM, ADN and NJW are employees of CSL Limited, and KM, GV, EM, ADN and NJW hold stock in CSL Limited.
Funding
This work was supported by the Reid Charitable Trusts, NHMRC Australia (IPW Clinical Practitioner Fellowship (1023407) and NHMRC Program Grant (1016647), SO Postgraduate Scholarship (1039026)), operational infrastructure grants through the Australian Government Institute for Research and Innovation in Social Services and the Victorian State Government, Arthritis Australia (South Australia Lupus, Scleroderma and Sjogren's Support Group Grant), CSL Limited and Janssen Research and Development, LLC.
Supporting information
Acknowledgments
The authors thank the donors for participation in the study. The authors also thank Jenni Harris, Lina Laskos, Naomi Sprigg and Cathy Quillici at The Walter and Eliza Hall Institute's Volunteer Blood Donor Registry and Susan Tadros, Clare O'Neill and our clinical colleagues in the rheumatology, renal and dermatology units at The Royal Melbourne Hospital for their assistance with donor recruitment.
References
- 1. Fishman P, Kamashta M, Ehrenfeld M et al Interleukin‐3 immunoassay in systemic lupus erythematosus: preliminary data. Int Arch Allergy Immunol 1993; 100: 215–218. [DOI] [PubMed] [Google Scholar]
- 2. Renner K, Hermann FJ, Schmidbauer K et al IL‐3 contributes to development of lupus nephritis in MRL/lpr mice. Kidney Int 2015; 88: 1088–1098. [DOI] [PubMed] [Google Scholar]
- 3. Oon S, Huynh H, Tai TY et al A cytotoxic anti‐IL‐3Rα antibody targets key cells and cytokines implicated in systemic lupus erythematosus. JCI Insight 2016; 1: e86131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997; 40: 1725. [DOI] [PubMed] [Google Scholar]
- 5. Grouard G, Rissoan M, Filgueira L et al The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)‐3 and CD40‐ligand. J Exp Med 1997; 185: 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Voehringer D. Basophil modulation by cytokine instruction. Eur J Immunol 2012; 42: 2544–2550. [DOI] [PubMed] [Google Scholar]
- 7. Samarajiwa S, Forster S, Auchetti K et al INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res 2009; 37: D85–D857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cervantes‐Garcia K, Husi H. Integrative analysis of multiple sclerosis using a systems biology approach. Sci Rep 2018; 8: 5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kennedy WP, Maciuca R, Wolslegel K et al Association of the interferon signature metric with serological disease manifestations but not global activity scores in multiple cohorts of patients with SLE. Lupus Sci Med 2015; 2: e000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lisney A, Szelinski F, Reiter K et al High maternal expression of SIGLEC1 on monocytes as a surrogate marker of a type 1 interferon signature is a risk factor for the development of autoimmune congenital heart block. Ann Rheumatic Dis 2017; 76: 1476–1480. [DOI] [PubMed] [Google Scholar]
- 11. Langfelder P, Horvath S. Eigengene networks for studying the relationships between co‐expression modules. BMC Syst Biol 2007; 1: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leonard D, Eloranta ML, Hagberg N et al Activated T cells enhance interferon‐-α production by plasmacytoid dendritic cells stimulated with RNA‐containing immune complexes. Ann Rheum Dis 2016; 75: 1728–1734. [DOI] [PubMed] [Google Scholar]
- 13. Charles N, Hardwick D, Daugas E et al Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat Med 2010; 16: 701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amezcua‐Guerra LM, Ferrusquía‐Toriz D, Castillo‐Martinez D et al Limited effectiveness for the therapeutic blockade of interferon α in systemic lupus erythematosus: a possible role for type III interferons. Rheumatology 2015; 54: 203–205. [DOI] [PubMed] [Google Scholar]
- 15. Wu Q, Yang Q, Lourenco E et al Interferon‐lambda1 induces peripheral blood mononuclear cell‐derived chemokines secretion in patients with systemic lupus erythematosus: its correlation with disease activity. Arthritis Res Ther 2011; 13: R88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin SC, Kuo CC, Tsao JT et al Profiling the expression of interleukin (IL)‐28 and IL‐28 receptor α in systemic lupus erythematosus patients. Eur J Clin Invest 2012; 42: 61–69. [DOI] [PubMed] [Google Scholar]
- 17. Furie R, Khamashta M, Merrill JT et al Anifrolumab, an anti‐interferon‐α receptor monoclonal antibody, in moderate to severe systemic lupus erythematosus. Arthritis Rheumatol 2017; 69: 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials