

Abstract
The parathyroid hormone receptor-1 (PTH1R) is a class B G protein–coupled receptor central to calcium homeostasis and a therapeutic target for osteoporosis and hypoparathyroidism. Here we report the cryo–electron microscopy structure of human PTH1R bound to a long-acting PTH analog and the stimulatory G protein. The bound peptide adopts an extended helix with its amino terminus inserted deeply into the receptor transmembrane domain (TMD), which leads to partial unwinding of the carboxyl terminus of transmembrane helix 6 and induces a sharp kink at the middle of this helix to allow the receptor to couple with G protein. In contrast to a single TMD structure state, the extracellular domain adopts multiple conformations. These results provide insights into the structural basis and dynamics of PTH binding and receptor activation.
Parathyroid hormone (PTH) and PTH-related peptide (PTHrP) are two endogenous ligands that play critical and distinct roles in skeletal development, calcium homeostasis, and bone turnover (1). Analogs of both PTH and PTHrP have been developed into therapeutic agents for osteoporosis, and PTH is used to treat hypoparathyroidism. In addition, tumor-produced PTHrP is a key factor driving cancer-associated hypercalcemia and cachexia (2), which is associated with weight loss disorder and is frequently the actual cause of death in cancer patients. Thus, modulating the PTH-PTHrP signaling axis is important for the development of treatments for a number of diseases, including osteoporosis and cancer.
The pleiotropic functions of PTH and PTHrP are mediated primarily through their binding and activation of the PTH type 1 receptor (PTH1R), a member of the class B G protein–coupled receptor (GPCR) subfamily, which also includes receptors for glucagon, glucagon-like peptides (GLPs), calcitonin, calcitonin gene–related peptide (CGRP), and other therapeutically important peptide hormones. The PTH1R couples primarily to stimulatory G protein (Gs), which is considered the major mediator of bone turnover and calcium homeostasis in response to PTH.
PTH1R contains two modular domains: a relatively large N-terminal extracellular domain (ECD) and a transmembrane domain (TMD). Activation of PTH1R is initiated by a rapid binding of the C-terminal region of peptide hormones to the receptor ECD, followed by a slow insertion of the N-terminal region of the peptide into the receptor’s TMD (3). Peptide hormone binding triggers the conformational changes in the TMD that mediate receptor activation. Previous crystal structures of the isolated PTH1R ECD bound to the C-terminal fragments of PTH and PTHrP have revealed that the peptides dock as amphipathic helices into a central groove formed by a three-layer α-β-βα fold in the ECD to resemble a “hot dog in a bun” configuration (4, 5). However, the structural basis by which the full-length PTH1R interacts with a functional peptide hormone and couples to downstream G protein remains un-known, and this is an obstacle to the development of clinically relevant PTH analogs and to understanding the fundamental mechanisms of GPCR signaling.
Recent technological advances in cryo–electron microscopy (cryo-EM) have revolutionized the progress in GPCR structural biology, resulting in a series of breakthrough structures, including several class A and class B GPCRs in complex with G proteins (6–13). In particular, cryo-EM structures of the glucagon-like peptide-1 receptor (GLP-1R) bound to GLP-1 (10) or a Gs-biased peptide Exendin-P5 (13) demonstrate that both peptides adopt a single continuous α helix, with their N termini pointing toward TM6, which induces a sharp kink at the middle of TM6 and an opening of the receptor’s cytoplasmic face to accommodate the α5 helix of the Ras-like domain of Gαs. The TM6 outward movement in Gs-bound GLP-1R has been observed in all other available class B GPCR–Gs complex structures. Nevertheless, the amino acid sequences of the class B GPCRs and their peptide ligands diverge considerably within the subfamily (3), such that the basis for ligand-binding specificity remains unknown.
We have reported previously that class B GPCRs vary widely in their requirement of the ECD for activation. For example, the ECD is not required for PTH1R activation when the peptide hormone is fused directly to the N-terminal end of its TMD (14, 15). In contrast, the ECD of GLP-1R is required for TMD activation even when the peptide ligand is covalently linked to the TMD or the receptor is activated by small molecules that interact with the intracellular side of TM6 (14, 15). To understand the specificity of peptide hormone recognition and receptor activation, we used single-particle cryo-EM to determine the structure of human PTH1R in complex with a long-acting PTH (LA-PTH) analog and Gs protein.
Structure determination of the LA-PTH–PTH1R–Gs complex
To prepare a stable PTH1R–Gs complex for cryo-EM studies, we had to overcome several technical obstacles associated with low expression levels of the human PTH1R and the instability of the receptor–Gs complex. The expression level of PTH1R was increased by using a double tag of maltose binding protein at the receptor C terminus (fig. S1A). The stability of the receptor–Gs complex was achieved through coexpression of the receptor with a dominant negative form of the heterotrimeric Gs protein (13) and the use of the Gα- and Gβ-binding nanobody Nb35 (16) (fig. S1, B to D). The activation of the receptor was achieved, and the receptor–Gs complex was further stabilized by LA-PTH (17), a modified PTH/PTHrP chimera that activates the receptor with 10- to 100-fold higher potency than endogenous PTH or PTHrP, in both G protein–free and G protein–bound states (18, 19). The use of LA-PTH was particularly important for the stability of the receptor−Gs complex because it has a much longer residence time on the receptor compared with PTH (fig. S2A). This peptide induced sustained receptor signaling via adenosine 3′,5′-monophosphate (cAMP) (fig. S2B) derived from stable PTH1R–Gs complexes within endosomes (20). The PTH1R (residues 27 to 502) used in this study can be bound and activated by both PTH and LA-PTH almost equally as the full-length receptor (residues 27 to 593) (fig. S1, E to H).
A large dataset of 1.4 million particles was collected from vitrified samples of LA-PTH–bound receptor-Gs complex. After 2D averaging analysis and global 3D sorting on the whole complex, about 40% of the particle projections out of the semi-autopicked particle repertoire were subjected to the reconstruction and yielded a high-resolution cryo-EM density map at a nominal resolution of 3.0 Å. Further consecutive classification on the peptide-receptor region resulted in three distinct conformational states of the PTH1R–Gs signaling complex, designated state 1, state 2, and state 3, respectively (figs. S3 and S4). The interaction analysis is based on state 1 unless noted otherwise. The overall structure was built on the basis of the GLP-1R–Gs complex (13), and the ECD region was fitted into the map as a rigid body using the available PTHrP-PTH1R ECD crystal structure (4). Apart from the α-helical domain of Gαs, which is poorly resolved in most cryo-EM GPCR–G protein complex structures, the presence of the bound LA-PTH, the receptor, and Gs in the isolated complex was clearly visible in the EM map (Fig. 1A and fig. S5). Side chains of the majority of amino acid residues are well defined in all protein components. The final models contain 34 of the 36 LA-PTH residues, the Gαβγ subunits of Gs, and the PTH1R residues from V31ECD to F4838.67b (class B GPCR numbering in superscript) (21), the very last residue of helix 8, with a few missing residues in the extracellular loop 1 (ECL1; residues 251 to 272) and the intracellular loop 3 (ICL3; residues 394 to 398). Thus, the structure provides a detailed model of intermolecular interactions of the receptor with LA-PTH and Gs.
Fig. 1. Cryo-EM structure of LA-PTH–bound human PTH1R in complex with Gs.

(A) (Left) Cut-through view of cryo-EM density map that illustrates the LA-PTH–PTH1R–Gs complex and the disc-shaped micelle. The unsharpened cryo-EM density map at the 0.01 threshold shown as light gray surface indicates a micelle diameter of 12 nm. The colored cryo-EM density map is shown at 0.026 threshold. (Right) Cartoon representation of the LA-PTH–PTH1R–Gs complex is shown with annular lipids in purple stick representation. Green, PTH1R; orange, LA-PTH; gold, Gs Ras-like domain; light blue, Gβ; medium blue, Gγ; gray, Nb35. (B) Cryo-EM density of the ordered annular lipid layer around the receptor TMD shown in purple; numbers 1 through 7 represent TM1 to TM7; receptor ECD and G protein are omitted.
The EM maps also show that the lipid-modified moieties of Gαs and Gβγ were inserted into the disc-shaped detergent micelle (Fig. 1A and fig. S3), which has a diameter of 12 nm as indicated by the 2D class averages, mimicking well the morphology of the lipid bilayer. We also observed that the Gαs αN helix is almost parallel to the micelle (Fig. 1A). Unlike many GPCR cryo-EM structures, an ordered annular lipid belt wrapping the periphery of the receptor TMD is clearly discernible in the cryo-EM map, among which several cholesterol molecules were built (Fig. 1B). These structural lipids possibly constrain the mobility of the TMD and thus contribute to the stability of the receptor in its active state.
Molecular recognition of LA-PTH by PTH1R
The overall structure of the LA-PTH–bound PTH1R in complex with Gs is organized into three layers: the top layer is the receptor ECD, the middle layer is the receptor TMD embedded in a large disc-shaped micelle, and the bottom layer is the Gs protein (Fig. 1A). In the complex, LA-PTH adopts a single continuous helix that anchors the top ECD layer with the middle TMD layer, with the orientation of the LA-PTH helix and the ECD nearly perpendicular to the horizontal membrane (Figs. 1A and 2A). The structure of the receptor ECD and the C-terminal half of LA-PTH helix (hereafter referred to as LA-PTHC) closely resembles the crystal structure of a PTHrP fragment bound to the PTH1R ECD (4) (fig. S6), which is expected given that LA-PTH is a PTH/PTHrP chimera whose C-terminal fragment (residues 16 to 34) corresponds to the PTHrP sequence (fig. S2C). The N-terminal half of the LA-PTH helix (hereafter referred to as LA-PTHN; residues 1 to 16) adopts four helical turns and is inserted deeply into the TMD core. It is directly surrounded by TM1, TM2, TM3, TM5, TM6, and TM7. TM4 is the only transmembrane helix that does not contact the LA-PTH helix (Figs. 1B and 2). The EM map is very clear for the LA-PTH helix and the surrounding TM helices, allowing unambiguous assignment of most side chains of the bound LA-PTH and its binding pocket in the TMD core (figs. S3 and S5).
Fig. 2. Molecular recognition of LA-PTH by PTH1R.

(A) The binding mode of LA-PTH with PTH1R, showing that LA-PTHN (ribbon and stick representation, orange) penetrates into a pocket formed by all TM helices except TM4, and by ECL2 and ECL3 whereas its C-terminal half is recognized by the ECD (ribbon in transparent surface, green). Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr. (B and C) Detailed interactions of LA-PTH with the PTH1R TMD pocket with hydrogen bonds shown as dotted lines.
The binding of LA-PTH to PTH1R buries a total surface area of 3809 Å2, with the interaction of LA-PTHN with the TMD core contributing >60% of the buried surface, suggesting that interactions in this region provide the major binding energy for the formation of the complex. The stability of the interface between LA-PTHN and the TMD is well supported by an extensive network of complementary polar and nonpolar interactions between the bound peptide ligand and the TMD (table S1 and Fig. 2, B and C), with details described in the supplementary text.
Interactions between LA-PTH and PTH1R help rationalize numerous studies on receptor structure-activity relationship for PTH and PTH1R that have been reviewed in (1) and also point toward residues that are possible determinants for the mechanism of prolonged cAMP signaling. Originally designed to place the optimal signaling domain of PTH (residues 1 to 14) with the optimal binding domain of PTHrP (residues 15 to 36) (22), LA-PTH displays markedly enhanced affinity for the PTH1R when coupled to G proteins (17). This enhanced affinity is accompanied not only by prolonged endosomal cAMP production in cells but also by prolonged calcemic responses in mice and primates that are independent of the ligand half-life in the circulation (17). Specifically, LA-PTH has four major amino acid changes at positions 10, 11, 12, and 14 from PTH (fig. S2C). These changes make either additional hydrophobic contacts [Gly12→Ala (G12A) and H14W] or additional hydrogen bonds (N10Q and L11R) (table S1), thus increasing the binding affinity of LA-PTH to the receptor TMD and accounting for the long-acting property of LA-PTH.
The structure further unveils the complex network of interactions formed by the first four residues of LA-PTH. Consistent with a critical role for these residues in activating the receptor, an N-terminally truncated LA-PTH(5–36) peptide is a potent antagonist of PTH1R (fig. S7). Competition-binding isotherms indicated that this truncated peptide maintains most of the stabilizing interactions used by LA-PTH (fig. S7A) and is thus only missing the signaling interactions that permit Gs coupling and activation (fig. S7, B to E). Together with earlier studies on class B chimeric GPCRs and hybrid peptides (23–25), these results support the model that peptide ligands of class B GPCRs are composed of two independent domains, with the C-terminal residues contributing to peptide binding selectivity, whereas N-terminal residues switch on receptor activation.
Conformational dynamics of the PTH1R–Gs complex
The relatively weak density for the ECD in the initial EM map suggests that multiple conformations are present simultaneously in the active PTH1R–Gs complex. To address the dynamic agonist recognition by the PTH1R, we employed extensive particle classifications that yielded three distinct subclasses and revealed the position of the ECD in three different conformational states relative to the TMD core, with nominal global resolutions of 3.0, 3.5, and 4.0 Å, respectively. In contrast, a single homogeneous conformation of the receptor TMD and TMD-bound LA-PTHN was evident throughout all three major conformational states (Fig. 3 and figs. S4 and S8).
Fig. 3. Conformational dynamics of the LA-PTH–PTH1R complex.

(A) Three conformational states of the LA-PTH–PTH1R complex. (B and C) The relative movement of the Ca positions of LA-PTH (B) and the PTH1R ECD and TMD in four independent MD simulations (C). The u and r in parentheses indicate that the Gs-binding interface is unrestrained or restrained, respectively, during simulation. RMSF, root mean square fluctuation; RMSD, root mean square deviation.
The first two conformational states are very similar, with the ECD and LA-PTHC rotated about 15° against each other (Fig. 3 and fig. S8A). In the third conformation, the LA-PTHC is bent, resulting in a loss of the interaction of LA-PTHC with the ECD and, thereby, a largely unresolved ECD density in the EM map compared with the other two conformational states (Fig. 3 and fig. S8B). However, the distinctive conformation of LA-PTHN among all three conformational states implies that stable interactions between LA-PTHN and the TMD core likely provide the driving energy that induces receptor activation, an observation consistent with previous studies (14). Molecular dynamics (MD) simulation results corroborate the EM structures, revealing that the LA-PTHN and the TMD core are in a single dominant conformational state in the simulation, whereas the ECD can twist around the LA-PTH helix (Fig. 3 and movie S1). Flexibility of the ECD might be required for initial binding of the peptide to the receptor, which is the first step in the two-step/two-domain model of ligand binding and receptor activation (26, 27). The third conformational state also implies that the separation of the LA-PTHC from ECD is likely the first step of the receptor–ligand dissociation process.
Basis of peptide hormone specificity
Peptide ligands of class B GPCRs vary greatly in their amino acid sequences (Fig. 4), which contribute to the specificity of ligand recognition by each receptor. Comparison of the LA-PTH–PTH1R–Gs complex with three other distinct class B GPCR–Gs complexes shows the underlying structural features that determine receptor–ligand specificity. The major difference among receptors lies in the position and orientation of their ECDs (Fig. 4A), which hold the corresponding peptide ligands in different conformations and orientations. The specificity of interaction between the ECD and the C-terminal half of the peptide ligand resembles what has been observed in the previously reported crystal structures of ECD-peptide complexes (4, 5) (fig. S6). Here we focus on differences in the N-terminal halves of the peptide helices in the TMD cores. Figure 4B illustrates shapes and sizes of the TMD pockets and the N-terminal halves of their corresponding peptide ligands. The TMD pockets of class B GPCRs typically range in size from 3300 to 3700 Å3, and only 32 to 61% of the pocket space is occupied by a peptide ligand. Despite a common structural packing of TM helices that construct the pocket, the pocket shape is unique for each receptor, owing to low conservation of the side chains lining it (fig. S9); thus, each pocket is capable of accommodating specific residues of a particular peptide ligand. In addition, different positions of the ECD among PTH1R, GLP-1R, and CGRP receptors help tweak the peptide helices differently into the TMD pocket (Fig. 4B), there-by contributing additional specificity to ligand binding.
Fig. 4. Basis of peptide hormone specificity.
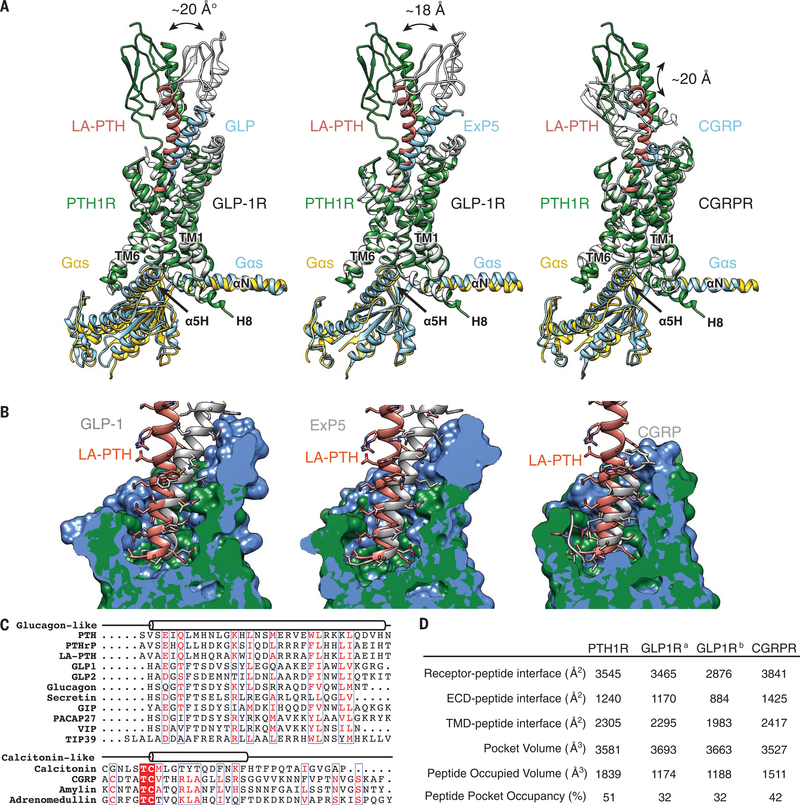
(A) Pairwise comparison of the LA-PTH bound to PTH1R with GLP-1, Exendin-P5 (ExP5), and CGRP in complex with their corresponding receptors, showing the relative positions of peptide ligands and their receptor ECDs. The RAMP1 component was omitted from the CGRPR figure for clarity. (B) Pairwise comparison of the PTH1R structure with GLP-1R bound to GLP-1, ExP5, and CGRPR, showing the pocket shapes accommodating respective peptide ligands. (C) Sequence alignment of class B GPCR peptide ligands. (D) Pocket size and occupancy of peptide ligands in class B GPCR structures. GLP-1Ra is a GLP-1–bound GLP-1R structure, and GLP-1Rb is an ExP5–bound GLP-1R structure.
The ECD of class B GPCRs displays a broad spectrum of relative stability, ranging from the poorly resolved calcitonin receptor (CTR) ECD to the resolved CTR ECD to the CGRP receptor (CGRPR) ECD whose mobility is restricted by interactions with RAMP1 (11, 12). Of the class B GPCR complex structures resolved to date, the orientation of the ECDs of PTH1R and GLP-1R is partially limited by the extended peptide α helix that extends from the ECD to the receptor TMD. One notable difference between the active-state structures of PTH1R and GLP-1R is in the ECL1 region, which is unstructured in PTH1R but has a helical extension in GLP-1R (left and middle panels in Fig. 4A) that forms several additional direct contacts with the C-terminal portion of the bound GLP-1 helix. The ECL1 in both rat and human GLP-1R is in a position close enough to form van der Waals interactions with the first two helical turns of the N-terminal helix of the ECD, whereas there is no interaction between PTH1R ECD and its TMD. The differences mentioned above likely contribute to the greater mobility of the PTH1R ECD compared with that of GLP-1R, resulting in the unwinding of LA-PTH from the active state of the PTH1R complex. In contrast, the occupancy of the LA-PTHN ligand in the TMD pocket is much higher than that of GLP-1 (63 versus 38%), suggesting substantially tighter binding and thus providing the potential structural basis for the high potency of LA-PTH that sustains cAMP responses after receptor internalization to endosomes. PTH(1–14) peptide analogs that contain the same or a very similar set of six substitutions that are incorporated into the N-terminal portion of LA-PTH exhibit much greater potency on a PTH1R TMD construct than the unmodified PTH(1–14) peptide (28). Together, these observations provide a rationale for the fact that ECD is required for GLP-1R activation but dispensable for PTH1R activation (14, 26).
Toward a common mechanism of class B GPCR activation and Gs coupling
Despite the different conformations and orientations of LA-PTH, GLP-1, and CGRP, the first residue at the N-terminal helical end of each peptide is located at the same relative position near TM6 in the TMD pocket of the cognate receptor (Fig. 4, A and B). Both LA-PTH and GLP-1 have one additional N-terminal residue before the N-terminal helical end. CGRP has five additional N-terminal residues, which structurally turn away from the bottom of the agonist-binding pocket (12) (Fig. 4C). These extra residues also point toward TM6, pushing the C-terminal half of TM6 outward and partially unwinding this portion of the TM6 helix. There is also a similar outward movement of this region in TM7 (Figs. 4A and 5A). A near-80° sharp kink is seen between P4156.47b and G4186.50b in the middle of TM6, which is stabilized by H4206.52b, Q4517.49b, and N3745.50b that form capping interactions with the three backbone carbonyls from the broken helix residues 415 to 417 at the kink (Fig. 5B). N3745.50b, P4156.47b, G4186.50b, H4206.52b, and Q4517.49b (NPGHQ motif) are conserved across the class B GPCRs, with the only exception being H7.49b (fig. S9). Thus, despite different shapes and sizes of the TM pockets and the different peptide sequences, class B GPCRs share a common mechanism of ligand-mediated receptor activation through key conserved residues that stabilize the sharp kink at the middle of TM6.
Fig. 5. Common mechanism of class B GPCR activation and Gs coupling.
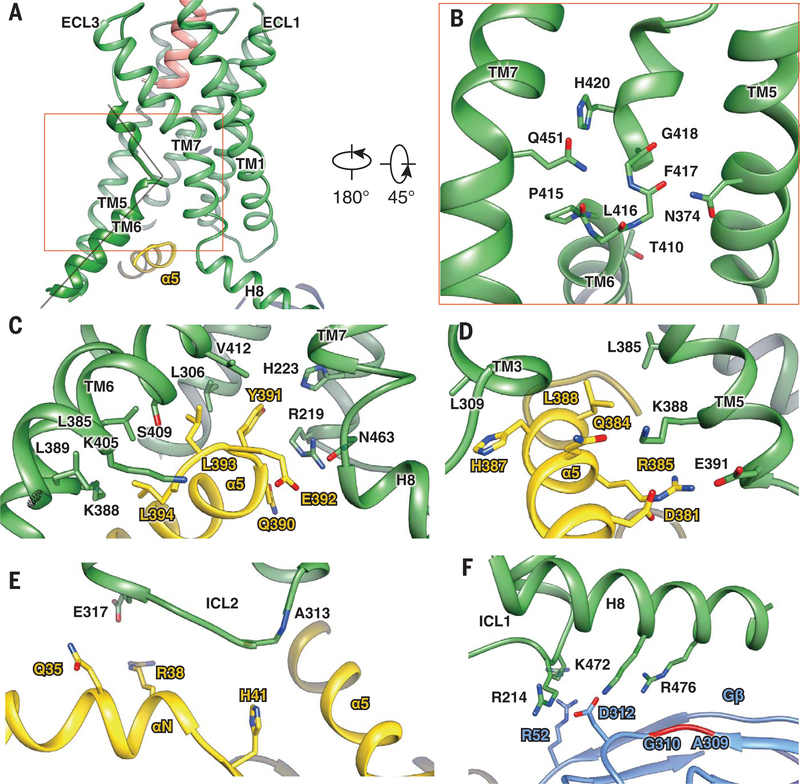
(A) The relative position of the LA-PTH N terminus (orange) toward the sharp kink of TM6 (green). (B) The kink and helix capping at TM6 are stabilized by conserved interactions with residues from TM5 (N3745.50b) and TM7 (Q4517.49b). (C to F) The PTH1R–Gs interface, including Gαs α5-PTH1R helix 8 interface (C); Gαs α5-PTH1R TM3/TM5 interface (D); PTH1R ICL2-Gαs αN helix interface (E); and helix 8–Gβ interface (F). PTH1R, green; Gαs, yellow; and Gβ, cyan.
This sharp kink results in a pronounced outward movement of the cytoplasmic end of the TM6 helix. This outward movement is also accompanied by a 90° rotation of the helical wheel at the T4106.42b position (fig. S10), which normally forms a conserved polar core with residues in TM2, TM3, and TM7 in the inactive GLP-1R structure (fig. S9). Rearrangement of this polar core has been proposed as a conserved mechanism for constitutively active class B GPCR mutants (29, 30), and it is consistent with the constitutively active PTH1R mutant H223R2.50b, which causes Jansen-type metaphyseal chondrodysplasia (31). Thus, the structure and sequence analyses indicate that the key residues that keep the receptors in an inactive state and the key residues that are involved in receptor activation are highly conserved among class B GPCRs.
The outward movement of TM6 leads to a large opening of the cytoplasmic cavity for coupling Gs. Compared with typical class A GPCRs, which undergo rapid deactivation (32, 33), PTH1R can remain activated long after peptide dissociation (34), a phenomenon that was considered as a possible “molecular memory” of the active PTH1R state. Given the large degree of conformational changes between the active and inactive states of TM6, and prolonged signaling of PTH1R, we reason that the transition between these two states is separated by a high-energy barrier, which may explain slow kinetics of receptor activation and deactivation. A similar phenomenon of molecular memory and mechanism remains to be seen for other class B GPCRs.
The overall assembly of the receptor–Gs complex is remarkably similar among class B GPCR structures solved to date (10–13). This outward movement of TM6 and the resulting cavity formed on the cytoplasmic surface were not present in the recent crystal structure of an engineered PTH1R in complex with a PTH peptide, likely because of the TMD mutations that were introduced for thermal stability, as these mutations prevented activation and signaling (35). In the current structure, the main PTH1R–Gs complex is anchored by the α5 helix of Gαs, which fits snugly into the cytoplasmic cavity of the TMD (Fig. 5, C and D). Additional contacts are observed between the extended helix 8 of the receptor and the Gβ subunit (Figs. 1 and 5F). The PTH1R residues that interface with the Gs protein are highly conserved across class B GPCRs (fig. S9). Structure comparison of PTH1R–Gs complexes with other class B GPCR–Gs complexes reveals substantial similarity in the G protein–binding interface, consistent with a common mechanism of Gs protein engagement (36). The only major difference is in the ICL2 conformation, in which PTH1R and GLP-1R are most similar to each other but different from CTR and CGRPR. In this conformation, the CGRPR F246 from ICL2 and corresponding F253 residue in CTR make much closer contacts with the αN helix and α5 helix of the Gαs than the corresponding residue in PTH1R and GLP-1R.
Outlook
We used cryo-EM to solve the near-atomic resolution structure of the LA-PTH–bound PTH1R in complex with Gs. This structure provides a comprehensive understanding of how PTH1R interacts with an agonist peptide ligand and couples to Gs and further reveals the prolonged active state of PTH1R that sustains endosomal cAMP signaling. Structural comparison with other classB GPCRs provides the basis of peptide binding specificity as well as the common mechanism of ligand-induced receptor activation and coupling to downstream Gs protein. Given the relatively conserved complex assembly and key structural components, the principles derived from these structural observations should be applicable to the entire family of class B GPCRs. Moreover, PTH is a classic endocrine hormone identified more than 80 years ago, with a rich history of physiological and pharmacological studies. The structure reported in this paper provides a foundation to systematically rationalize the extensive body of biochemical and mutational data developed for the PTH1R and from which to move toward new treatments of bone and mineral diseases, such as osteoporosis and cancer cachexia. In addition, the extensive ordered lipids around the PTH1R TMD provide a structural template for studying membrane protein–lipid interactions.
Supplementary Material
ACKNOWLEDGMENTS
The cryo-EM data were collected at the Center of Cryo-Electron Microscopy, Zhejiang University, with the assistance of S. Chang and X. Zhang.
Funding: This work was partially supported by National Key Basic Research Program of China (2018YFC1003600 to Y.Z.); Zhejiang Province Science Fund for Distinguished Young Scholars (LR19H310001 to Y.Z.); Shanghai Science and Technology Development Fund (18ZR1447800 to L.-H.Z. and 16ZR1407100 to A.D.); the Young Innovator Association of CAS (2018325 to L.-H.Z.); SA-SIBS Scholarship Program (L.-H.Z. and D.Y.); the Fudan-SIMM Joint Research Fund (FU-SIMM-20174003 to L.-H.Z. and M.-W.W.); National Key R&D Program of China grant (2018YFA0507000 to M.-W.W.); National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program,” China (2018ZX09711002-002-005 to D.Y. and 2018ZX09735-001 to M.-W.W.); the National Natural Science Foundation of China (31300245 to L.-H.Z.; 81573479 and 81773792 to D.Y.); Ministry of Science and Technology (China) grants 2012ZX09301001, 2012CB910403, 2013CB910600, XDB08020303, and 2013ZX09507001 (to H.E.X.); Novo Nordisk-CAS Research Fund (NNCAS-2017-1-CC to D.Y.); the Jay and Betty Van Andel Foundation (H.E.X.); National Institutes of Health grants (GM127710 to H.E.X.; R01-DK102495, R01-DK111427, and R01-DK116780 to J.-P.V.; DK11794 to T.J.G.; and T32-GM008424 to A.D.W.); and the Cotswold Foundation Fellowship Awards (to F.G. J.-A. and A.D.W.).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: Density maps and structure coordinates have been deposited to the Electron Microscopy Database and the Protein Data Bank with accession numbers EMD-0410 and PDB ID 6NBF for conformation state 1, EMD-0411 and PDB ID 6NBH for conformation state 2, and EMD-0412 and PDB ID 6NBI for conformation state 3.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Gardella TJ, Vilardaga JP, Pharmacol. Rev 67, 310–337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kir S. et al. , Nature 513, 100–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pal K, Melcher K, Xu HE, Acta Pharmacol. Sin 33, 300–311 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pioszak AA, Parker NR, Gardella TJ, Xu HE, J. Biol. Chem 284, 28382–28391 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pioszak AA, Xu HE, Proc. Natl. Acad. Sci. U.S.A 105, 5034–5039 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang Y. et al. , Nature 558, 553–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koehl A. et al. , Nature 558, 547–552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Draper-Joyce CJ et al. , Nature 558, 559–563 (2018). [DOI] [PubMed] [Google Scholar]
- 9.García-Nafría J, Nehmé R, Edwards PC, Tate CG, Nature 558, 620–623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y. et al. , Nature 546, 248–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang YL et al. , Nature 546, 118–123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang YL et al. , Nature 561, 492–497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang YL et al. , Nature 555, 121–125 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Zhao LH et al. , J. Biol. Chem 291, 15119–15130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin Y. et al. , Cell Discov. 2, 16042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rasmussen SG et al. , Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimizu M. et al. , J. Bone Miner. Res 31, 1405–1412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hattersley G, Dean T, Corbin BA, Bahar H, Gardella TJ, Endocrinology 157, 141–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda A. et al. , Proc. Natl. Acad. Sci. U.S.A 110, 5864–5869 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vilardaga JP, Jean-Alphonse FG, Gardella TJ, Nat. Chem. Biol 10, 700–706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM, Proc. Natl. Acad. Sci. U.S.A 110, 5211–5216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardella TJ et al. , J. Biol. Chem 270, 6584–6588 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Bergwitz C. et al. , J. Biol. Chem 271, 26469–26472 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Vilardaga JP, Lin I, Nissenson RA, Mol. Endocrinol 15, 1186–1199 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Gourlet P. et al. , Eur. J. Biochem 239, 349–355 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Luck MD, Carter PH, Gardella TJ, Mol. Endocrinol 13, 670–680 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Dean T, Khatri A, Potetinova Z, Willick GE, Gardella TJ, J. Biol. Chem 281, 32485–32495 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Shimizu N, Guo J, Gardella TJ, J. Biol. Chem 276, 49003–49012 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Yin Y. et al. , J. Biol. Chem 292, 9865–9881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou XE, Melcher K, Xu HE, Protein Sci. 28, 487–501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schipani E, Kruse K, Jüppner H, Science 268, 98–100 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Dror RO et al. , Proc. Natl. Acad. Sci. U.S.A 108, 18684–18689 (2011). [Google Scholar]
- 33.Kohlhoff KJ et al. , Nat. Chem 6, 15–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferrandon S. et al. , Nat. Chem. Biol 5, 734–742 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehrenmann J. et al. , Nat. Struct. Mol. Biol 25, 1086–1092 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Glukhova A. et al. , ACS Pharmacol. Transl. Sci 1, 73–83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.