

Abstract
Background
Single-cell RNA-sequencing (scRNA-seq) is a transformative technology, allowing global transcriptomes of individual cells to be profiled with high accuracy. An essential task in scRNA-seq data analysis is the identification of cell types from complex samples or tissues profiled in an experiment. To this end, clustering has become a key computational technique for grouping cells based on their transcriptome profiles, enabling subsequent cell type identification from each cluster of cells. Due to the high feature-dimensionality of the transcriptome (i.e. the large number of measured genes in each cell) and because only a small fraction of genes are cell type-specific and therefore informative for generating cell type-specific clusters, clustering directly on the original feature/gene dimension may lead to uninformative clusters and hinder correct cell type identification.
Results
Here, we propose an autoencoder-based cluster ensemble framework in which we first take random subspace projections from the data, then compress each random projection to a low-dimensional space using an autoencoder artificial neural network, and finally apply ensemble clustering across all encoded datasets to generate clusters of cells. We employ four evaluation metrics to benchmark clustering performance and our experiments demonstrate that the proposed autoencoder-based cluster ensemble can lead to substantially improved cell type-specific clusters when applied with both the standard k-means clustering algorithm and a state-of-the-art kernel-based clustering algorithm (SIMLR) designed specifically for scRNA-seq data. Compared to directly using these clustering algorithms on the original datasets, the performance improvement in some cases is up to 100%, depending on the evaluation metric used.
Conclusions
Our results suggest that the proposed framework can facilitate more accurate cell type identification as well as other downstream analyses. The code for creating the proposed autoencoder-based cluster ensemble framework is freely available from https://github.com/gedcom/scCCESS
Keywords: Autoencoder, Cluster ensemble, Single cells, scRNA-seq, Single-cell transcriptome, Cell type identification
Background
Transcriptome profiling by single-cell RNA-sequencing (scRNA-seq) is a fast-emerging technology for studying complex tissues and biological systems at the single-cell level [1]. Identification of cell types present in a biological sample or system is a vital part of scRNA-seq data analysis workflow [2]. The key computational technique for unbiased cell type identification from scRNA-seq data is unsupervised clustering [3]. Typically, this is achieved by using a clustering algorithm to partition cells in a scRNA-seq dataset into distinct groups and subsequently annotating each group to a type of cell based on cell type marker genes and/or other biological knowledge of cell type characteristics [4].
Due to the critical role played by cell type identification for downstream analyses, significant effort has been devoted to tailoring standard clustering algorithms or developing new ones for scRNA-seq data clustering and cell type identification [5]. These include standard k-means clustering, hierarchical clustering, and variants that are specifically designed for scRNA-seq data (i.e. RaceID/RaceID2 [6], CIDR [7]) as well as more advanced methods that utilise likelihood-based mixture modelling (countClust) [8], density-based spatial clustering [9] and kernel-based single-cell clustering (SIMLR) [10]. Several studies have compared and summarised various clustering algorithms used for scRNA-seq data analysis [11–13].
One of the key challenges in scRNA-seq data clustering is handling specific characteristics of the data including high feature-dimensionality and high feature-redundancy. This is because typically a large number of genes are profiled in the experiment but only a small proportion of them are cell type-specific and therefore informative for cell type identification. Hence, clustering directly on the original high-dimensional feature space may result in suboptimal partitioning of the cells due to low signal-to-noise ratio. To reduce the high feature-dimensionality in scRNA-seq data for visualisation and downstream analyses, various dimension reduction techniques, including traditional approaches as well as newly developed ones, have been applied to scRNA-seq data. These include generic methods such as principal component analysis (PCA), independent component analysis (ICA), non-negative matrix factorization [14], and t-distributed stochastic neighbour embedding (tSNE) [15], as well as other methods developed for scRNA-seq data, such as zero inflated factor analysis (ZIFA) [16]. Recently, deep learning techniques such as scvis, a deep generative model [17], and a scNN [18], a neural network model, were developed specifically for scRNA-seq data dimension-reduction. While these new developments are primarily focused on scRNA-seq data visualisation, they represent the first applications of deep learning techniques for scRNA-seq data analysis.
Ensemble learning is an established field in machine learning and has a wide application in bioinformatics [19]. Ensemble clustering via random initialisation is a popular ensemble learning method for clustering [20]. While this approach was found to improve stability of the k-means clustering algorithm, it appeared to have a less consistent effect on clustering accuracy [21]. Ensemble clustering via random projection is an alternative ensemble learning method for clustering. This approach was applied to DNA microarray data analysis and resulted in improved clustering accuracy [22]. Weighted ensemble clustering combines multiple clustering outputs based on their respective quality [23]. Recently, cluster ensembles have been generated by combining outputs from different upstream processing and similarity metrics [24] or different clustering algorithms for cell type identification from scRNA-seq data [25, 26]. While these heuristic methods were found to be effective for improving clustering accuracy in cell type identification, they are ad-hoc in nature and may not fully explore characteristics and biological signals in scRNA-seq data when clustering.
To extract biological signal from scRNA-seq data while at the same time addressing the issues of high feature-dimensionality and high feature-redundancy, here we propose an autoencoder-based cluster ensemble framework for the robust clustering of cells for cell type identification from scRNA-seq data. The proposed framework first randomly projects the original scRNA-seq datasets into sub-spaces to create ‘diversity’ [27] and then trains autoencoder networks to compress each such random projection to a low-dimensional feature space. Subsequently, clusterings are generated on all encoded datasets and consolidated into a final ensemble output.
The proposed framework of ensemble clustering via autoencoder-based dimension-reduction and its application to scRNA-seq is a principled approach and the first of its kind. We demonstrate that (1) the autoencoder-reduced ensemble clustering of scRNA-seq data significantly improves clustering accuracy of cell types, whereas simple ensemble clustering without autoencoder-based dimension reduction showed no clear improvement; (2) improvement of clustering accuracy in general increases with the ensemble size; and (3) the proposed framework can improve cell type-specific clustering when applied using either the standard k-means clustering algorithm or a state-of-the-art kernel-based clustering algorithm (SIMLR) [10] specifically designed for scRNA-seq data analysis. This demonstrates that the proposed framework can be coupled with different clustering algorithms to facilitate accurate cell type identification and other downstream analyses.
Results
Hyperparameter optimisation for autoencoders
We undertook a grid search to optimise three hyperparameters including random projection size, encoded feature space size and autoencoder learning rate during backpropagation; this was performed across four datasets (Table 2) using the ARI, NMI, FM and Jaccard index metrics discussed above. Together, the four metrics across four datasets made a total of sixteen dimensions across which to optimise. We used Pareto analysis [28] to select an appropriate combination of parameters from across all four optimisation datasets without giving priority to any single dataset or metric. A Pareto rank of 1 indicates an optimal clustering results on a selection of optimisation datasets using a combination of hyperparameters.
Table 2.
Summary of the experimental scRNA-seq datasets used for hyperparameter optimisation and method evaluation
| Repository | Source | # cell | # class | Ref. | Protocol | Purpose |
|---|---|---|---|---|---|---|
| GSE60361 | Mouse cortex | 3005 | 7 | [33] | SMARTer | Optimisation |
| GSE45719 | Mouse embryogenesis | 300 | 8 | [34] | SMART-seq2 | Optimisation |
| GSE67835 | Adult and fetal human brain | 466 | 8 | [35] | SMARTer | Optimisation |
| E_MTAB_3929 | Human embryogenesis | 1529 | 5 | [36] | SMART-seq2 | Optimisation |
| GSE84371 | Mouse neurons | 1402 | 8 | [37] | Smart-seq2 | Evaluation |
| GSE82187 | Mouse striatum | 705 | 10 | [38] | SMARTer & Smart-seq2 | Evaluation |
| Broad portal | Human archived brain | 14963 | 19 | [39] | Drop-seq | Evaluation |
| Broad portal | Mouse archived brain | 13313 | 26 | [39] | Drop-seq | Evaluation |
As the Pareto front becomes larger and more ambiguous as more datasets are included, we tested the robustness of each parameter set by obtaining the Pareto front for all possible combinations of 1, 2, 3 or all 4 datasets and counting the number of such combinations for which the given parameter set appears in the Pareto front (Fig. 1). We determined that the most robust high-accuracy results were obtained by selecting 2048 genes during random projection; producing an encoded feature space of 16 dimensions; and training the autoencoder using a learning rate of 0.001. All evaluation benchmarks were undertaken using this parameter combination and a hidden layer width of 128.
Fig. 1.

Hyperparameter optimisation for autoencoders using Pareto analysis. Left panel: PCA visualisation of the four evaluation metrics (i.e. ARI, NMI, FM and Jaccard) on each of the four optimisation datasets. Each point corresponds to a single combination of hyperparameter values including random projection size, encoded feature space size, and autoencoder learning rate during backpropagation; each combination/point is colour-coded by the number of times it was assigned Pareto rank 1 (i.e. the combination that gives best clustering performance) across all possible combinations of the four optimisation datasets. Right panel: Autoencoder architecture as determined by the hyperparameter optimisation procedure
Ensemble of k-means clustering
We first asked if the ensemble of autoencoder-based clustering can improve upon the performance of a single clustering run on a single encoded dataset. To test this, we first used a standard k-means clustering algorithm (“Clustering algorithms” section) to create base clustering results and tested the performance of different ensemble sizes based on ARI, NMI, FM and Jaccard. Note that we repeatedly ran the entire procedure multiple times to account for the variability in the clustering results. We found that in general the overall ensemble clustering performance improves as the number of base clustering runs increases (Fig. 2, light blue boxes) according to all four evaluation metrics and in all four datasets used for evaluation. These results demonstrate that the ensemble of autoencoder-based clustering framework can indeed improve cell type identification for the k-means clustering algorithm.
Fig. 2.

Ensemble of k-means clustering results on the four scRNA-seq datasets. Red boxes represent ensemble of k-means clustering on the raw input expression matrix without using the autoencoder framework. Light blue boxes represent autoencoder-based k-means cluster ensemble
We wondered if such an improvement from ensemble clustering is independent of the random projection and autoencoder framework. Hence, we compared the performance of k-means clustering on the raw input expression matrix without using the autoencoder framework (that is, without applying the random projection and autoencoder steps) with different ensemble sizes. We found that the improvement in clustering performance was diminished in most cases (Fig. 2, red boxes), suggesting that the improved clustering performance is due to the random projection and autoencoder steps implemented in the proposed framework in addition to the ensemble step. Notably, the autoencoder framework also enhanced the data signal-to-noise ratio in most cases as can been seem from the improved performance of autoencoder-based k-means cluster compared to direct k-means clustering on the raw input at the ensemble size of 1.
Another interesting observation is that the variance of the clustering outputs in general decreased with the increasing number of base clusterings (Fig. 2). While the ensemble of k-means clustering without random projection and autoencoder does not improve cell type identification accuracy, it does reduce the clustering variability, and therefore resulted in more stable and reproducible clustering outputs compared to a single run of k-means clustering. These results are consistent with previous findings [21]. In comparison, autoencoder-based ensemble clustering led to both more accurate cell type identification and a reduction of variability, both of which are desirable characteristics for scRNA-seq data analysis.
Autoencoder-based SIMLR ensemble
While the autoencoder-based cluster ensemble framework is able to improve the performance of a standard k-means clustering algorithm in both accuracy and reproducibility of cell type clustering, we wondered if such an ensemble framework could also improve the performance of the latest clustering algorithm. To this end, we applied the proposed framework to a state-of-the-art kernel-based clustering algorithm, SIMLR, designed specifically for cell type identification on scRNA-seq data. Because the computational complexity of SIMLR grows exponentially with the number of cells in a dataset, we focused our evaluation on the two smaller datasets (i.e. GSE84371 and GSE82187). Similar to k-means clustering, we found in these cases that the performance of the autoencoder-based SIMLR ensemble improved with increased ensemble size (Fig. 3). Clustering variability also generally decreased with larger ensemble sizes. These results demonstrate that the proposed autoencoder-based cluster ensemble framework can also lead to more accurate cell type identification and clustering reproducibility from scRNA-seq data for SIMLR.
Fig. 3.

Evaluation of autoencoder-based SIMLR ensemble. Ensemble sizes range from 1 to 100 were tested using four evaluation metrics in two scRNA-seq datasets
While cell type clustering accuracy improves with the larger ensemble sizes for both k-means clustering algorithm and SIMLR (Figs. 2 and 3), we observed that this improvement plateaus at an ensemble size of 50 (Fig. 3). We therefore recommend an ensemble size of 50 as a good trade-off between clustering output quality and computational time. Note that the computational complexity of the proposed cluster ensemble framework increases linearly with respect to the ensemble size.
Performance comparison of autoencoder-based cluster ensemble
Typically, a single run of a clustering algorithm is used to identify cell types from a given scRNA-seq dataset. An interesting question is how much improvement the proposed autoencoder-based cluster ensemble offers compared to the common clustering procedure where a clustering algorithm is directly applied to raw gene expression data (that is, without random projection and autoencoder steps). To address this, we next quantified cell type clustering accuracy from the direct application of k-means and SIMLR clustering to the raw gene expression input and compared these with the autoencoder-based cluster ensemble of k-means and SIMLR, respectively. Note that an ensemble size of 50 was used for the cluster ensemble. k-means clustering and the random projection step in ensemble clustering are non-deterministic; while SIMLR contains technically non-deterministic steps (including a k-means step), we found that repeated runs on the same raw dataset with different random seeds produced identical clustering partitions. Consequently, SIMLR may be thought of as functionally deterministic. To account for variability in the clustering results for stochastic methods, we repeated clustering ten times and calculated the standard deviation across multiple runs. Table 1 summaries these results.
Table 1.
Comparison of direct application of k-means and SIMLR clustering on raw gene expression data with autoencoder-based k-means and SIMLR ensemble
| Raw | Autoencoder | |||||||
|---|---|---|---|---|---|---|---|---|
| ARI | NMI | FM | Jaccard | ARI | NMI | FM | Jaccard | |
| k-means | ||||||||
| Mouse neurons | 0.22 ±0.02 | 0.36 ±0.03 | 0.39 ±0.02 | 0.22 ±0.01 | 0.38 ±0.02 | 0.56 ±0.01 | 0.53 ±0.02 | 0.34 ±0.02 |
| Mouse striatum | 0.36 ±0.07 | 0.69 ±0.06 | 0.51 ±0.06 | 0.31 ±0.05 | 0.45 ±0.01 | 0.75 ±0.01 | 0.58 ±0.01 | 0.37 ±0.01 |
| Human archived brain | 0.29 ±0.01 | 0.49 ±0.01 | 0.35 ±0.01 | 0.21 ±0.01 | 0.37 ±0.01 | 0.56 ±0 | 0.43 ±0.01 | 0.27 ±0.01 |
| Mouse archived brain | 0.32 ±0.01 | 0.49 ±0 | 0.35 ±0.01 | 0.21 ±0.01 | 0.43 ±0.01 | 0.58 ±0 | 0.46 ±0.01 | 0.3 ±0.01 |
| SIMLR | ||||||||
| Mouse neurons | 0.44 ±0 | 0.65 ±0 | 0.58 ±0 | 0.39 ±0 | 0.71 ±0 | 0.7 ±0 | 0.81 ±0 | 0.67 ±0 |
| Mouse striatum | 0.55 ±0 | 0.81 ±0 | 0.67 ±0 | 0.34 ±0 | 0.8 ±0.02 | 0.87 ±0.01 | 0.85 ±0.02 | 0.74 ±0.03 |
Cell type identification accuracy were quantified by the four evaluation metrics
Specifically, the autoencoder-based k-means ensemble improved cell type clustering for an average of about 30% in the four evaluation datasets according to all four evaluation metrics (Table 1). Clustering variability was also typically smaller using the autoencoder-based k-means ensemble. Perhaps more strikingly, the cell type clustering accuracy as measured by ARI and Jaccard metrics for the autoencoder-based SIMLR ensemble improved about 50% to 100% compared to using SIMLR alone on the raw expression matrix for the mouse neurons and striatum datasets. Moreover, we found that the cell type clustering accuracy of SIMLR is substantially better than the standard k-means clustering algorithm, suggesting SIMLR is indeed an effective clustering algorithm for scRNA-seq data analysis. Therefore, the further gain in clustering accuracy by coupling SIMLR with the proposed autoencoder-based cluster ensemble is of practical importance and will add to the state-of-the-art methods for scRNA-seq data analysis.
Comparison of autoencoder-based cluster ensemble with PCA-based clustering
We next compared the performance of autoencoder-based cluster ensemble with PCA-based clustering. PCA is a commonly used dimension reduction method and has been widely used for reducing the high-dimensionality of scRNA-seq data prior to clustering cell types. By benchmarking the performance of cell type clustering across the evaluation datasets, we found that in almost all cases autoencoder-based clustering ensemble outperformed PCA-based approach for both k-means clustering and SIMLR according to all four evaluation metrics (Fig. 4). We confirmed the statistical significance of these performance improvements using the Wilcoxon Rank Sum test. These results further demonstrate the utility of the autoencoder-based cluster ensemble for more accurate clustering of cell types in scRNA-seq datasets.
Fig. 4.

Comparison of autoencoder-based clustering framework with PCA-based dimension reduction and clustering using the four evaluation metrics. Statistical significance (p<0.001; denoted by ⋆) of either autoencoder with k-means clustering against the rest for human and mouse archived brain datasets, or autoencoder with SIMLR clustering against the rest for mouse neurons and striatum datasets were performed using Wilcoxon Rank Sum test (two-sided)
Discussions
There may be further opportunities to build on the proposed method:
Firstly, we performed hyperparameter optimization over four datasets searching for the most robust configuration. While our chosen configuration was the most consistently accurate over all possible combinations of these four datasets, we saw that it was not among the most accurate configurations for two of the optimization datasets individually. Additionally, there is no guarantee that this combination falls into a global or near-global optimum across scRNA-seq datasets in general, or that such an optimum exists. Devising a way to produce parameter configurations based on individual dataset characteristics without ground-truth labels may be an avenue for further exploration.
Secondly, the current iteration of our proposed method uses random subspace projection to reduce the dimension of datasets prior to autoencoder training. An additional direction for future research may include exploring other methods of basic dimension reduction, such as weighted gene selection based on variability or other metrics; this may be more useful in capturing cell type-specific characteristics by retaining genes containing more biological signal related to cell type.
Lastly, while the clustering algorithms k-means and SIMLR were used as independent components in our current proposed framework, an interesting direction for future work might be the development of an artificial neural network architecture and training method which performs simultaneous dimension reduction and clustering. An integrated approach such as this may facilitate the exploration of clustering output in the reduced feature space.
Conclusions
High throughput scRNA-seq technology is transforming biological and medical research by allowing the global transcriptome profiles of individual cells from heterogeneous samples and tissues to be quantified with high precision. Cell type identification has become essential in scRNA-seq data analysis, and clustering has been the key computational approach used for this task. In this study, we have proposed an autoencoder-based ensemble clustering approach by incorporating several state-of-the-art techniques in a computational framework.
We evaluated the proposed clustering framework on its impact on the level and robustness of cell type identification accuracy using a collection of scRNA-seq datasets with pre-defined cell type annotations. Based on previously defined gold standards for each scRNA-seq dataset, we demonstrate that the proposed framework is highly effective for cell type identification. The application of the proposed framework to both a standard k-means clustering algorithm and a state-of-the-art kernel-based clustering algorithms, SIMLR, illustrates its generalisability and applicability to other clustering algorithms. We therefore envision the proposed framework being flexibly adopted into the common workflow for scRNA-seq data analysis.
Methods
The autoencoder-based cluster ensemble framework is summarised in Fig. 5a. The proposed framework accepts scRNA-seq data in the form of an N×M expression matrix (denoted as X) where N is the number of cells and M is the number of genes.
Fig. 5.
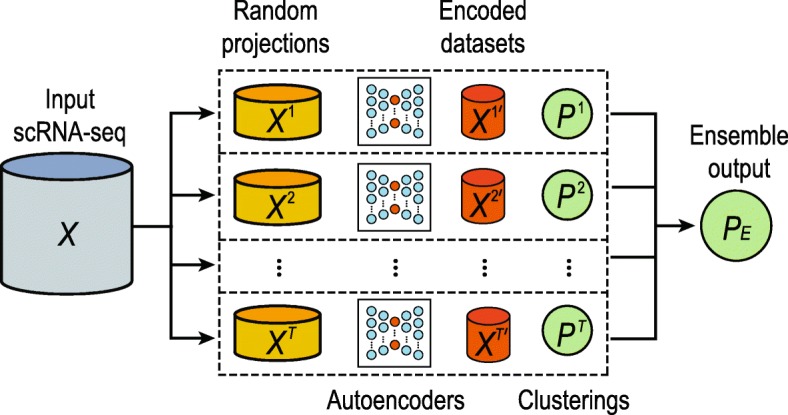
A schematic illustration of the proposed autoencoder-based cluster ensemble framework. The first step is the sampling of multiple random projections from the original input scRNA-seq data set. A separate autoencoder artificial neural network is trained on each of these random projections and used to encode the data to a smaller-dimensional space. Subsequently, clustering of each encoded dataset is conducted using an arbitrary clustering method; the final clustering output is produced by integrating individual clustering results using a fixed-point algorithm [31]
Dimension reduction by autoencoders
Genes are randomly selected from the input dataset to produce a set of “random projection” datasets Xt (t=1,...,T), each with a dimension of N×M′. The purpose of this step is to create ‘diversity’ [27] in subsequent encodings and individual clusterings of these datasets to achieve a more robust consensus in the resulting ensemble. Following the random projection step, each matrix Xt is then used to train a fully-connected autoencoder neural network. An autoencoder is an artificial neural network consisting of two sub-networks: an encoder and a decoder, intersecting at a ‘bottleneck’ layer of a smaller size than the original input. The network is trained to reconstruct the original input with minimum error, forcing the network to learn to encode the information contained within the smaller latent space of the output of the bottleneck layer[29].
In the autoencoders used with our framework, the encoder accepts samples of cell data from X as input. The encoder contains a single hidden layer and an output layer which produces reduced-dimension encodings of the aforementioned samples. The decoder subnetwork accepts these encoded samples as input, passing these through a single hidden layer and an output layer which produces reconstructions of the original samples. In both subnetworks, the activation function of the hidden layer is a ‘Leaky ReLu’ [30]; linear activation is applied to all other layers.
Each autoencoder is trained by minimising reconstruction error using the mean squared loss function:
where Xt is the input expression matrix from the tth random projection and is the autoencoder’s reconstruction of Xt.
Following training, each matrix Xt is fed through its respective autoencoder and a low-dimension encoded dataset is extracted from the encoder output. Training and hyperparameter optimisation of autoencoders are discussed in Hyperparameter optimisation for autoencoders section.
Clustering algorithms
To perform clustering on dimension-reduced datasets generated from autoencoders, we utilised both a standard k-means clustering algorithm with Lloyd’s implementation [32] and a kernel-based clustering algorithm (SIMLR) specifically designed for scRNA-seq data analysis [10].
Given an initial set of random centres m1,...,mK, and a distance matrix D (typically computed from Euclidean space), the algorithm first finds the closest cluster centres for each of all cells based on their expression profiles :
and then updates the cluster centres:
The output is the assignment of each cell based on its expression profile x to a cluster k∈1,...,K.
SIMLR calculates the distance matrix for cells using multiple kernels as follows:
where wl is the weight of a Gaussian kernel function for a pair of cells defined as follows:
where εij is the variance and |xi−xj|2 is the squared distance between cell i and j, calculated from their expression profiles xi and xj. To test the proposed framework, we utilised SIMLR (Version 1.8.0) implemented in Bioconductor (Release 3.7).
The number of clusters to be created was set according to the number of pre-defined cell types/classes in each scRNA-seq data for both the k-means clustering and SIMLR (see “Evaluation metrics” section). After obtaining individual clustering outputs (denoted as Pt) from either k-means clustering or SIMLR, a fixed-point algorithm for obtaining hard least squares Euclidean consensus partitions was applied to compute the consensus PE of individual partitions [31]:
| 1 |
in which wb is the weight associated with individual clustering output and is set to 1 in our case, and D(Pt,PE)2 is the squared Euclidean function for computing the distance of an individual partition with the consensus partition.
Together, the proposed autoencoder-based cluster ensemble framework can be summarised in pseudocode as below.

Data description and evaluation
This section summarises the scRNA-seq datasets and performance assessment metrics utilised for method evaluation.
scRNA-seq datasets
A collection of eight publicly available scRNA-seq datasets (Table 2) were utilized in this study. These datasets were downloaded from the NCBI GEO repository, the EMBL-EBI ArrayExpress repository, or the Broad Institute Single-Cell database portal. The log2-transformed transcripts per million (TPM) or counts per million (CPM) values (as determined by the original publication for a given dataset) were used to quantify full length gene expression for datasets generated by SMARTer or SMART-seq2 protocols. UMI-filtered counts were used to quantify gene expression for the Drop-seq datasets. All datasets have undergone cell-type identification using biological knowledge from their respective original publications which we retain for evaluation purposes. For each dataset, genes detected in less than 20% of cells were removed. This step trims the number of genes and allows only those that are expressed in at least a subset of cells to be considered for subsequent analyses. Four datasets were used to optimise autoencoder hyperparameters. We present evaluation benchmarking results for four additional datasets.
Evaluation metrics
A common approach to assess the performance of clustering methods for cell type identification in scRNA-seq data analysis is to compare the concordance of the clustering outputs of cells with a ‘gold standard’. As mentioned above, such a gold standard may be obtained from orthogonal information such as cell type marker genes and/or other biological knowledge of cell type characteristics. In this study, cell type annotations from their original publications are used as ‘gold standards’.
For each dataset, the number of clusters for both k-means clustering and SIMLR was set as the number of pre-defined classes based on its original publication and the concordance between the clustering outputs and the ‘gold standard’ were measured using different metrics. Here we employed a panel of four evaluation metrics including Adjusted Rand index (ARI), normalized mutual information (NMI), Fowlkes-Mallows index (FM), and Jaccard index [40] (Fig. 6).
Fig. 6.
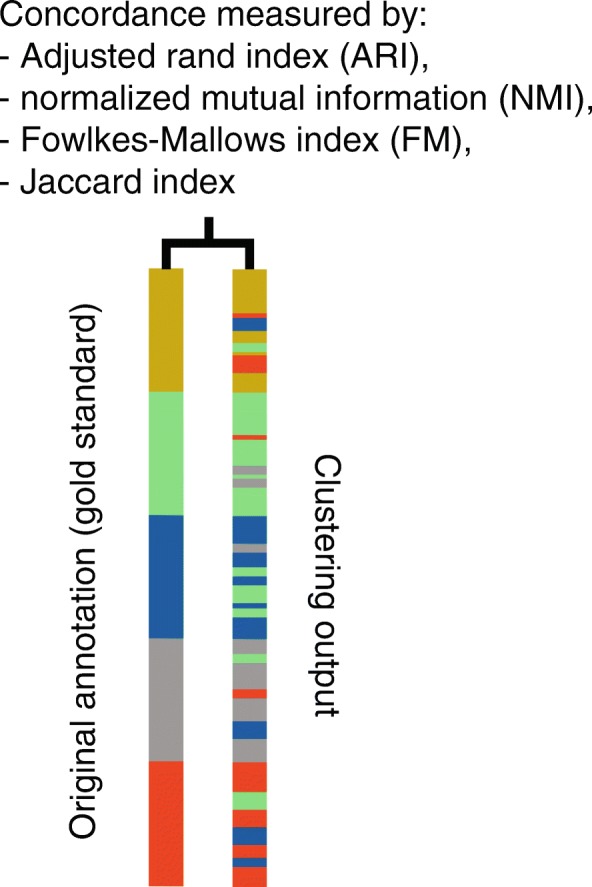
A schematic showing the quantification of concordance of the clustering output with the original ’gold standard’ annotation using a panel of evaluation metrics
Let G, P be the cell partitions based on the gold standard and the clustering output respectively. We define a, the number of pairs of cells assigned to the same group in both partitions; b, the number of pairs of cells assigned to the same cell type in the first partition but to different cell types in the second partition; c, the number of pairs of cells assigned to different cell types in the first partition but to the same cell type in the second partition; and d, the number of pairs of cells assigned to different cell types in both partitions. ARI, FM, and Jaccard index can then be calculated as follows:
Let G={u1,u2,...,uk} and P={v1,v2,...,vk} denote the gold standard and the clustering partition across K classes, respectively. NMI is defined as follows:
where I(G,P) is the mutual information of G and P, defined as
and H(G) and H(P) are the entropy of partitions G and P calculated as
where N is the total number of cells.
Acknowledgements
The authors thank their colleagues at the School of Mathematics and Statistics; and School of Life and Environmental Sciences for informative discussion and valuable feedback.
About this supplement
This article has been published as part of BMC Bioinformatics, Volume 20 Supplement 19, 2019: 18th International Conference on Bioinformatics. The full contents of the supplement are available at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-20-supplement-19.
Abbreviations
- ARI
Adjusted rand index
- CPM
Counts per million
- FM
Fowlkes-Mallows index
- PCA
principal component analysis
- scRNA-seq
Single-cell RNA-seq
- SIMLR
kernel-based clustering algorithm
- tSNE
t-distributed stochastic neighbour embedding
- TPM
Transcripts per million
Authors’ contributions
PY and TAG conceived the study. TAG led the experimental design and data analyses. TK contributed to the data curation and analyses; LN contributed to the algorithm design with input from DT; PY and TAG interpreted the experimental results with input from JGB and JYHY; PY and TAG wrote the manuscript with input from all authors. All authors reviewed and approved the final version of the manuscript.
Funding
Publication of this supplement was funded by the Australian Research Council Discovery Early Career Researcher Award (DE170100759) and a National Health and Medical Research Council (NHMRC) Investigator Grant (1173469) to P.Y., the Australian Research Council Discovery Projects (DP170100654) to JYHY and PY, the National Health and Medical Research Council (NHMRC)/Career Development Fellowship (1105271) to JYHY, the Australian Government Research Training Program Scholarship to TAG and the Judith and David Coffey Life Lab Gift scholarship to TAG.
Availability of data and materials
The datasets generated and/or analysed during the current study are available from the GEO, EBI, and Broad repositories. Details of datasets are listed in Table 1.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ziegenhain C, Vieth B, Parekh S, et al. Comparative analysis of single-cell rna sequencing methods. Mol Cell. 2017;65(4):631–43. doi: 10.1016/j.molcel.2017.01.023. [DOI] [PubMed] [Google Scholar]
- 2.Trapnell C. Defining cell types and states with single-cell genomics. Genome Res. 2015;25(10):1491–8. doi: 10.1101/gr.190595.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bacher R, Kendziorski C. Design and computational analysis of single-cell rna-sequencing experiments. Genome Biol. 2016;17(1):63. doi: 10.1186/s13059-016-0927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell rna sequencing. Mol Cell. 2015;58(4):610–20. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Kiselev VY, Andrews TS, Hemberg M. Challenges in unsupervised clustering of single-cell rna-seq data. Nat Rev Genet. 2019;20:273–282. doi: 10.1038/s41576-018-0088-9. [DOI] [PubMed] [Google Scholar]
- 6.Grün D, Lyubimova A, Kester L, et al. Single-cell messenger rna sequencing reveals rare intestinal cell types. Nature. 2015;525(7568):251. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- 7.Lin P, Troup M, Ho JW. Cidr: Ultrafast and accurate clustering through imputation for single-cell rna-seq data. Genome Biol. 2017;18(1):59. doi: 10.1186/s13059-017-1188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dey KK, Hsiao CJ, Stephens M. Visualizing the structure of rna-seq expression data using grade of membership models. PLoS Genet. 2017;13(3):1006599. doi: 10.1371/journal.pgen.1006599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macosko EZ, Basu A, Satija R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–14. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang B, Zhu J, Pierson E, Ramazzotti D, Batzoglou S. Visualization and analysis of single-cell rna-seq data by kernel-based similarity learning. Nat Methods. 2017;14(4):414. doi: 10.1038/nmeth.4207. [DOI] [PubMed] [Google Scholar]
- 11.Freytag S, Tian L, Lönnstedt I, Ng M, Bahlo M. Comparison of clustering tools in R for medium-sized 10 × Genomics single-cell RNA-sequencing data. F1000Research. 2018;7:1297. doi: 10.12688/f1000research.15809.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duò A, Robinson MD, Soneson C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Research. 2018;7:1141. doi: 10.12688/f1000research.15666.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim T, Chen IR, Lin Y, Wang AY-Y, Yang JYH, Yang P. Impact of similarity metrics on single-cell rna-seq data clustering. Brief Bioinforma. 2018. 10.1093/bib/bby076. [DOI] [PubMed]
- 14.Shao C, Höfer T. Robust classification of single-cell transcriptome data by nonnegative matrix factorization. Bioinformatics. 2017;33(2):235–42. doi: 10.1093/bioinformatics/btw607. [DOI] [PubMed] [Google Scholar]
- 15.Maaten Lvd, Hinton G. Visualizing data using t-sne. J Mach Learn Res. 2008;9(Nov):2579–605. [Google Scholar]
- 16.Pierson E, Yau C. Zifa: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 2015;16(1):241. doi: 10.1186/s13059-015-0805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding J, Condon A, Shah SP. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat Commun. 2018;9(1):2002. doi: 10.1038/s41467-018-04368-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin C, Jain S, Kim H, Bar-Joseph Z. Using neural networks for reducing the dimensions of single-cell rna-seq data. Nucleic Acids Res. 2017;45(17):156. doi: 10.1093/nar/gkx681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang P, Hwa Yang Y, B Zhou B, Y Zomaya A. A review of ensemble methods in bioinformatics. Curr Bioinforma. 2010;5(4):296–308. doi: 10.2174/157489310794072508. [DOI] [Google Scholar]
- 20.Vega-Pons S, Ruiz-Shulcloper J. Int J Pattern Recogn Artif Intell. 2011; 25(03):337–72.
- 21.Kuncheva LI, Vetrov DP. Evaluation of stability of k-means cluster ensembles with respect to random initialization. IEEE Trans Patt Anal Mach Intell. 2006;28(11):1798–808. doi: 10.1109/TPAMI.2006.226. [DOI] [PubMed] [Google Scholar]
- 22.Avogadri R, Valentini G. Fuzzy ensemble clustering based on random projections for dna microarray data analysis. Artif Intell Med. 2009;45(2-3):173–83. doi: 10.1016/j.artmed.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 23.Ren Y, Domeniconi C, Zhang G, Yu G. Weighted-object ensemble clustering. In: Data Mining (ICDM), 2013 IEEE 13th International Conference On. IEEE: 2013. p. 627–36.
- 24.Kiselev VY, Kirschner K, Schaub M, et al. Sc3: consensus clustering of single-cell rna-seq data. Nat Methods. 2017;14(5):483. doi: 10.1038/nmeth.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Huh R, Culpepper HW, Lin Y, Love MI, Li Y. Safe-clustering: Single-cell aggregated (from ensemble) clustering for single-cell rna-seq data. Bioinformatics. 2018;35(8):1269–77. doi: 10.1093/bioinformatics/bty793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Risso D, Purvis L, Fletcher RB, et al. clusterexperiment and RSEC: A bioconductor package and framework for clustering of single-cell and other large gene expression datasets. PLoS Comput Biol. 2018;14(9):1006378. doi: 10.1371/journal.pcbi.1006378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuncheva LI, Hadjitodorov ST. Using diversity in cluster ensembles. In: 2004 IEEE International Conference On Systems, Man and Cybernetics. IEEE: 2004. p. 1214–9. 10.1109/icsmc.2004.1399790.
- 28.Ngatchou P, Zarei A, El-Sharkawi A. Intelligent Systems Application to Power Systems, 2005. Proceedings of the 13th International Conference On. Arlington: IEEE; 2005. Pareto multi objective optimization. [Google Scholar]
- 29.Hinton GE, Salakhutdinov RR. Reducing the dimensionality of data with neural networks. Science. 2006;313(5786):504–7. doi: 10.1126/science.1127647. [DOI] [PubMed] [Google Scholar]
- 30.Maas AL, Hannun AY, Ng AY. Rectifier nonlinearities improve neural network acoustic models. In: in ICML Workshop on Deep Learning for Audio, Speech and Language Processing. Atlanta: 2013.
- 31.Hornik K. A clue for cluster ensembles. J Stat Softw. 2005;14(12):1–25. doi: 10.18637/jss.v014.i12. [DOI] [Google Scholar]
- 32.Lloyd S. Least squares quantization in pcm. IEEE Trans Inf Theory. 1982;28(2):129–37. doi: 10.1109/TIT.1982.1056489. [DOI] [Google Scholar]
- 33.Zeisel A, Muñoz-Manchado AB, Codeluppi S, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell rna-seq. Science. 2015;347(6226):1138–42. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 34.Deng Q, Ramsköld D, Reinius B, Sandberg R. Single-cell rna-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343(6167):193–6. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- 35.Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, Gephart MGH, Barres BA, Quake SR. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci. 2015;112(23):7285–90. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petropoulos S, Edsgärd D, Reinius B, Deng Q, Panula SP, Codeluppi S, Reyes AP, Linnarsson S, Sandberg R, Lanner F. Single-cell rna-seq reveals lineage and x chromosome dynamics in human preimplantation embryos. Cell. 2016;165(4):1012–26. doi: 10.1016/j.cell.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Habib N, Li Y, Heidenreich M, et al. Div-seq: Single-nucleus rna-seq reveals dynamics of rare adult newborn neurons. Science. 2016;353(6302):925–8. doi: 10.1126/science.aad7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gokce O, Stanley GM, Treutlein B, et al. Cellular taxonomy of the mouse striatum as revealed by single-cell rna-seq. Cell Rep. 2016;16(4):1126–37. doi: 10.1016/j.celrep.2016.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Habib N, Avraham-Davidi I, Basu A, et al. Massively parallel single-nucleus rna-seq with dronc-seq. Nat Methods. 2017;14(10):955. doi: 10.1038/nmeth.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wagner S, Wagner D. Comparing Clusterings: an Overview: Universität Karlsruhe, Fakultät für Informatik Karlsruhe; 2007.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the GEO, EBI, and Broad repositories. Details of datasets are listed in Table 1.