

Abstract
Background and Objectives
Suvorexant is an orexin receptor antagonist indicated for the treatment of insomnia, characterized by difficulties with sleep onset and/or sleep maintenance. As suvorexant is metabolized primarily by Cytochrome P450 3A (CYP3A), and its pharmacokinetics may be affected by CYP3A modulators, the effects of CYP3A inhibitors (ketoconazole or diltiazem) or an inducer (rifampin [rifampicin]) on the pharmacokinetics, safety, and tolerability of suvorexant were investigated.
Methods
In two Phase I, open-label, fixed-sequence trials (Studies P008 and P038), healthy subjects received a single oral dose of suvorexant followed by co-administration with multiple once-daily doses of strong/moderate CYP3A inhibitors (ketoconazole/diltiazem) or a strong CYP3A inducer (rifampin). Treatments were administered in the morning: suvorexant 4 mg with ketoconazole 400 mg (Study P008; N = 10), suvorexant 20 mg with diltiazem 240 mg (Study P038; N = 20), and suvorexant 40 mg with rifampin 600 mg (Study P038; N = 10). Area under the plasma concentration–time curve from time zero to infinity (AUC0–∞), maximum plasma concentration (Cmax), half-life (t½), and time to Cmax (tmax) were derived from plasma concentrations of suvorexant collected at prespecified time points up to 10 days following CYP3A inhibitor/inducer co-administration. Adverse events (AEs) were recorded.
Results
Co-administration with ketoconazole resulted in increased exposure to suvorexant [AUC0–∞: geometric mean ratio (GMR); 90% confidence interval (CI) 2.79 (2.35, 3.31)] while co-administration with diltiazem resulted in a lesser effect [GMR (90% CI): 2.05 (1.82, 2.30)]. Co-administration with rifampin led to a marked decrease (88%) in suvorexant exposure. Consistent with morning administration and known suvorexant pharmacology, somnolence was the most frequently reported AE.
Conclusions
These results are consistent with expectations that strong CYP3A inhibitors and inducers exert marked effects on suvorexant pharmacokinetics. In the context of a limited sample size, single suvorexant doses were generally well tolerated in healthy subjects when co-administered with/without a CYP3A inhibitor/inducer.
1. Introduction
Orexin antagonism has been extensively studied in the past decade as an alternative mechanism for the treatment of insomnia [1, 2]. Orexin A (OX-A) and orexin B (OX-B) are the key signaling neuropeptides in the orexinergic signaling pathway, which plays a central role in regulation of the transition between wake and sleep [3–5]. These neuropeptides exert their wake-promoting effects by activating their cognate orexin receptors (OX1R and OX2R) in the posterolateral hypothalamus and brain stem nuclei [2, 6, 7]. Suvorexant is an orally active, first-in-class orexin receptor antagonist indicated in the USA and other regions for the treatment of insomnia, characterized by difficulties with sleep onset and/or sleep maintenance [8, 9]. Data from clinical trials have demonstrated the efficacy of suvorexant in promoting sleep onset and maintenance with minimal residual next-day effects [10–16]. As the target patient population may include those with comorbidities and elderly patients, polypharmacy may be common in the treatment of insomnia and, as such, evaluation of the potential for drug–drug interactions (DDIs) is critical to inform both prescribing information and clinical practice.
Recent draft guidance from the US Food and Drug Administration (FDA) recommends that DDI management strategies should be developed when a clinically significant DDI for a drug is identified. These strategies may include contraindicating concomitant drug use, avoidance of concomitant drug use, temporary discontinuation of an interacting drug, dosage modifications of the new and concomitant drug, and specific monitoring strategies [17]. The overall objective of these DDI assessments is to provide an understanding of the mechanisms, magnitude, and potential consequences of any given DDI in order to optimize clinical response [17]. Cytochrome P450 3A (CYP3A) inhibition can increase exposure to gamma-aminobutyric acid (GABA)A-receptor hypnotics that are CYP3A substrates (e.g. zolpidem, eszopiclone) [18, 19]. It therefore follows that co-administration of CYP3A inhibitors and inducers with suvorexant, a CYP3A substrate [8], may alter suvorexant plasma concentrations, and subsequently, its therapeutic effect and safety profile. In vitro and in vivo characterization of suvorexant metabolism and disposition following single oral dose administration in humans has been reported else-where [20].
Based on the draft 2017 FDA DDI guidance, ketoconazole is designated as a clinical example of a strong CYP3A inhibitor [21] and is known to reduce metabolism of CYP3A substrates such as triazolam and alprazolam [22]. Although itraconazole or clarithromycin may reflect the preferred strong CYP3A index inhibitors for clinical investigation, at the time of suvorexant clinical development, ketoconazole was routinely utilized in clinical trials and its designation as a strong CYP3A inhibitor remains. As such, results from DDI trials with ketoconazole may be extrapolated to other examples of strong CYP3A inhibitors such as itraconazole [21]. Diltiazem, a calcium channel antagonist [23], has commonly been regarded as a moderate CYP3A inhibitor, such that the effects of diltiazem on CYP3A substrates may be extrapolated to the setting of co-administration with other moderate CYP3A inhibitors such as erythromycin [21]. Although the preferred categorization of diltiazem continues to evolve, the results reported herein are consistent with its designation and assessment as a moderate CYP3A inhibitor. Conversely, rifampin (rifampicin) is a strong inducer of CYP3A [17, 21], and is known to induce several-fold increases in hepatic and intestinal CYP3A protein expression [24]. Induction of CYP3A expression by rifampin can ultimately lead to > 90% decreases in plasma concentration of sensitive CYP3A substrates, and has been shown to reduce plasma concentration and effects of several GABAA-receptor hypnotics (e.g. triazolam, zolpidem) [25, 26].
Two Phase I, open-label, fixed-sequence clinical trials in healthy subjects evaluated the potential for CYP3A-mediated DDIs during the administration of suvorexant. Specifically, the pharmacokinetics of suvorexant were assessed following co-administration with ketoconazole, diltiazem, or rifampin. The safety and tolerability of suvorexant in the presence and absence of these inhibitors/inducers were also evaluated.
2. Methods
Two Phase I, open-label, fixed-sequence clinical studies [Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, Protocol MK-4305–008 (referred to as “P008”) and MK-4305–038 (“P038”)] were performed. Study P008 assessed the potential for a DDI arising from co-administration of suvorexant with ketoconazole, and Study P038 assessed co-administration of suvorexant with diltiazem or rifampin.
Study P008 was conducted at ProMedica Clinical Research Center, Inc., MA, USA, and Study P038 at Thomas Jefferson University Clinical Research Unit, PA, USA. Both studies were conducted in accordance with the principles of Good Clinical Practice and were approved by the appropriate institutional review boards and regulatory agencies. All subjects provided informed consent.
2.1. Study Populations and Procedures
Subjects in Study P008 were healthy men aged 18–45 years, with a body mass index (BMI) of ≤31 kg/m2 at pre-study screening. Study P038 subjects were healthy men and healthy non-pregnant women aged 18–50 years, with a BMI of ≥ 20 to ≤30 kg/m2 at pre-study screening. In both trials, subjects were non-smokers and in good overall mental and physical health based on vital signs, medical history, physical examination, laboratory safety tests, and had no abnormality on electrocardiograms (ECG) performed at screening. Those with a history of persistent difficulties in initiating or maintaining sleep for ≥ 3 months and those with obstructive sleep apnea, restless legs syndrome, narcolepsy, rapid eye movement, behavioral disorders (Study P038 only), or difficulty sleeping with sleep aids (Study P038 only) were excluded. In addition, subjects who could not refrain from prescription and/or non-prescription drugs and herbal remedies, and those who were frequent users of sedative hypnotics, including benzodiazepines, ‘soft’ GABAnergic agents, barbiturates, and/or other pharmaceutical sleep agents, were excluded. Subjects who had crossed three or more time zones in the 2 weeks prior to study initiation were also excluded from Study P008 only.
2.2. Treatments
Both studies consisted of two periods: in Period 1, subjects received a single oral dose of suvorexant (Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ, USA) only; in Period 2, they received a single oral dose of suvorexant co-administered with a CYP3A inhibitor or inducer (Fig. 1). All dosing occurred once daily in the morning. Selected CYP3A inhibitor and inducer doses were based on those known to be clinically relevant and/or able to maximize the inhibitory or induction potential described for CYP3A substrates [27–29].
Fig. 1.

Study design for a Study P008, b Study P038 diltiazem sub-study, and c Study P038 rifampin sub-study. D day, h hours
Suvorexant doses were guided in part by pharmacokinetic data from a rising single-dose clinical study in healthy males [30]. Assuming a maximum 10-fold increase in suvorexant area under the concentration–time curve from time of administration to infinity (AUC0–∞) and maximum observed concentration (Cmax) as a result of co-administration of ketoconazole, the suvorexant 4-mg dose selected for Study P008 would still have been associated with a lower exposure and similar Cmax compared with the well-tolerated 120-mg dose in the rising single-dose study. This approach was considered conservative, given that only a moderate increase in suvorexant exposure was expected in the presence of ketoconazole, based on published data for midazolam and triazolam [31, 32], which are extensively metabolized by CYP3A [33]. Two days of ketoconazole administration is sufficient to achieve maximal CYP3A inhibition prior to suvorexant dosing; however, due to the approximately 240-h pharmacokinetic sampling scheme required for suvorexant characterization, and the need to ensure continued ketoconazole-mediated CYP3A inhibition throughout the sample duration, ketoconazole was administered for an additional 9 days.
Co-administration of diltiazem was anticipated to have a lesser impact. Assuming a conservative maximum 5-fold increase in suvorexant AUC0–∞ and Cmax as a result of co-administration of diltiazem, the suvorexant 20-mg dose selected for Study P038 would have been associated with lower exposure (AUC0–∞ and Cmax), compared with the generally well-tolerated 240-mg dose in the rising single-dose study. Substantial CYP3A inhibition can be achieved with one day of diltiazem administration, but to ensure continued diltiazem-mediated CYP3A inhibition during the 120-h suvorexant pharmacokinetic sampling scheme, diltiazem was co-administered with suvorexant on Day 2 and for 4 subsequent days following suvorexant administration.
The selection of suvorexant 40 mg was largely informed by the maximum clinical dose that was investigated in parallel during Phase III clinical development, and owing to the pharmacokinetic properties across the clinically relevant dose range (10–40 mg), any effect of rifampin on suvorexant 40 mg could also support similar conclusions at the lower doses including those currently approved of 10–20 mg. In the current study, rifampin was administered for 13 days prior to administration of suvorexant to allow near-maximal levels of induction to be achieved, and then co-administered with a single suvorexant dose. Rifampin was further administered for an additional 3 days throughout the 96 h of suvorexant pharmacokinetic sampling to ensure continued CYP3A induction during this period for a total of 17 days.
2.3. Pharmacokinetic Evaluations
Venous blood samples were collected at prespecified time points for suvorexant plasma concentration analysis in both studies (Fig. 1; Table 1). Suvorexant pharmacokinetic parameters were evaluated and included: AUC0–∞, Cmax, apparent terminal elimination half-life (t½), and time to reach Cmax (tmax). These parameters were assessed following administration of suvorexant alone, and following co-administration with ketoconazole, diltiazem, or rifampin.
Table 1.
Blood sample collection times for suvorexant plasma concentration analysis
| Study | Drug regimen | Sampling times (h) |
|---|---|---|
| P008 | Suvorexant 4 mg | Pre-dose; post-dose: 0.5, 1, 2, 4, 6, 8, 12, 16, 24, 48, 72 |
| Suvorexant 4 mg + ketoconazole | Pre-dose (Day 2); post-dose: 0.5, 1, 2, 4, 6, 8, 12, 16, 24, 48, 72, 96, 120, 144, 168, 192, 216, 240 | |
| P038 | Suvorexant 20 mg | Pre-dose; post-dose: 0.5, 1, 2, 4, 6, 9, 12, 16, 24, 36, 48, 72, 96 |
| Suvorexant 20 mg + diltiazem sub-study | Pre-dose (Day 2); post-dose: 0.5, 1, 2, 4, 6, 9, 12, 16, 24, 36, 48, 72, 96, 120 | |
| Suvorexant 40 mg | Pre-dose; post-dose: 0.5, 1, 2, 4, 6, 9, 12, 16, 24, 36, 48, 72, 96 | |
| Suvorexant 40 mg + rifampin sub-study | Pre-dose (Day 14); post-dose: 0.5, 1, 2, 4, 6, 9, 12, 16, 24, 36, 48, 72, 96 |
h hours
2.4. Safety Assessments
Subjects were queried daily throughout the trials for the occurrence of adverse events (AEs). All AEs were evaluated with respect to seriousness, severity [mild (awareness of sign or symptom but easily tolerated), moderate (discomfort enough to cause interference with usual activity), severe (incapacitating with inability to work or do usual activity)], relation to trial drug, and action taken in response to their emergence. AEs considered events of clinical interest (ECIs) included: cataplexy, sleep paralysis (including sleep-onset paralysis), hypnagogic or hypnopompic hallucinations, suicidal ideation and/or behaviors, complex sleep-related behaviors, falls, and events associated with potential for abuse. Physical examinations were conducted at screening, pre-dose, and 24 h post-dose (ketoconazole only), and at the post-trial visit (14 days following the last dose of inhibitor/inducer). Vital signs measurements, in addition to ECG and laboratory safety analyses (hematology, serum chemistry, and urinalysis) were conducted at screening, pre-dose, and various time points between 1 to 8 and 24 h, and Day 3 (for Study P038 only) post-dose, and at the post-trial visit (14 days following the last dose of inhibitor/inducer).
2.5. Sample Analysis, Data Analysis, and Statistics
Plasma concentrations of suvorexant (molecular weight: 450.932 g/mol) were determined by liquid–liquid extraction followed by quantification using reversed-phase high-performance liquid chromatography–mass spectrometry [34]. The lower limit of quantification of the assay and its linear calibration range were 1 ng/mL and 1–1000 ng/mL, respectively. Suvorexant plasma concentrations, and their corresponding sampling times relative to the dosing time, were used to derive the pharmacokinetic parameters in each subject using the WinNonlin software (version 5.1.1/5.2.1; Pharsight Corporation, Mountain View, CA, USA). The apparent terminal rate constant (λ) was estimated by regression of the terminal log-linear portion of the plasma concentration–time profile. AUC from time of administration to the last time point of detectable plasma concentration (AUC0–last) was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations up to the last detectable plasma concentration. AUC0–∞ was estimated as the sum of AUC0–last and the extrapolated area given by the quotient of the last detectable concentration and λ. Cmax was obtained by inspection of the plasma concentration data. tmax was obtained by inspection of the plasma concentration data and, finally, the t½ was calculated as the quotient of ln(2) and λ.
Individual AUC0–∞ values were ln-transformed and evaluated with a mixed-effect analysis of variance (ANOVA) model, with treatment as a fixed effect and subject as a random effect. A two-sided 90% confidence interval (CI) for the true mean difference (suvorexant with CYP3A inhibitor or inducer—suvorexant alone) in ln-AUC was calculated using the mean square error from the ANOVA and referencing a t-distribution. These confidence limits were exponentiated to obtain a CI for the true geometric mean ratio (GMR) for AUC0–∞ (suvorexant with CYP3A inhibitor or inducer/suvorexant alone). Suvorexant Cmax was analyzed in a similar manner. No-effect boundaries of 0.5–2.0 were selected to inform sample size of the current studies and preliminary clinical development and trial conduct criteria. The totality of evidence, including clinical efficacy and safety data, and other approaches provide conclusive recommendations and interpretation of clinical relevance for any drug interactions and dosing guidelines.
AEs were tabulated for administration of suvorexant alone and in the presence of ketoconazole, diltiazem, or rifampin by frequency of occurrence. For vital signs, laboratory safety tests, and ECG monitoring, summary statistics of mean and standard error were generated for values and changes from baseline by part, treatment, and time point. Selected non-serious AEs (ECIs) were recorded and consisted of cataplexy, hypnagogic or hypnopompic hallucinations, sleep paralysis or sleep-onset paralysis, suicidal ideation and/or behaviors, complex sleep-related behaviors, falls, and selected events associated with potential for abuse.
Across both studies, sample sizes were selected based on study objectives and the pharmacokinetic variability of suvorexant established at the time of study conduct. Assuming a true GMR of 1.0, each of the sample sizes were expected to achieve a ≥ 80% probability that the 90% CI would fall between 0.5 and 2.0.
3. Results
3.1. Study Populations
Study P008 was conducted from November to December 2008, and Study P038 was conducted from August to October 2010, with treatment periods of approximately 4 and 8 weeks, respectively. Ten male subjects enrolled in and completed Study P008 (Table 2). A total of 30 subjects were enrolled in Study P038: 20 in the diltiazem sub-study, and 10 in the rifampin sub-study (Table 2). Seventeen and 10 subjects completed the diltiazem and rifampin sub-studies, respectively. In the diltiazem sub-study, one subject discontinued at the post-study visit having completed Periods 1 and 2.
Table 2.
Subject baseline characteristics and demographics in Studies P008 and P038
| Characteristic | Study P008 | Study P038 | |||||
|---|---|---|---|---|---|---|---|
| Ketoconazole study | Diltiazem sub-study | Rifampin sub-study | |||||
| Subjects (N = 10) | Male (n = 14) | Female (n = 6) | Total (N = 20) | Male (n = 8) | Female (n = 2) | Total (N = 10) | |
| Mean age, years (range) | 33 (22–45) | 42 (28–50) | 32 (23–41) | 39 (23–50) | 39 (28–45) | 29 (18–40) | 37 (18–45) |
| Mean height, cm (range) | 175 (161.0–185.0) | 178 (166.0–193.0) | 165 (151.0–174.0) | 174 (151.0–193.0) | 180 (171.0–188.0) | 165 (157.0–172.0) | 177 (157.0–188.0) |
| Mean weight, kg (range) | 81.9 (64.0–100.0) | 83.8 (74.1–94.4) | 71.2 (52.6–82.7) | 80.0 (52.6–94.4) | 85.6 (63.2–105.2) | 60.2 (59.7–60.6) | 80.5 (59.7–105.2) |
| Race, n (%) | |||||||
| White | 5 (50.0) | 3 (21.4) | 2 (33.3) | 5 (25.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Black | 4 (40.0) | 10 (71.4) | 4 (66.7) | 14 (70.0) | 8 (100.0) | 2 (100.0) | 10 (100.0) |
| Native American | 1 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Hispanic | 0 (0.0) | 1 (7.1) | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
All subjects were included in the pharmacokinetic analyses, with complete data available for all 10 (100%) subjects in Study P008, 18/20 [90% (including the subject who discontinued at the post-study visit)] in the P038 diltiazem sub-study, and all 10 (100%) in the P038 rifampin sub-study. Two subjects in Study P038 (both from the diltiazem sub-study) did not have a complete pharmacokinetic data set, as only data for Period 1 were available. Across both studies, all subjects (N = 40) were included in the safety evaluation.
3.2. Pharmacokinetic Evaluations
Arithmetic mean plasma concentration–time profiles for suvorexant alone and following co-administration with ketoconazole (400 mg), diltiazem (240 mg), or rifampin (600 mg) are provided in Fig. 2a–c, and the corresponding pharmacokinetic parameters are described in Table 3.
Fig. 2.
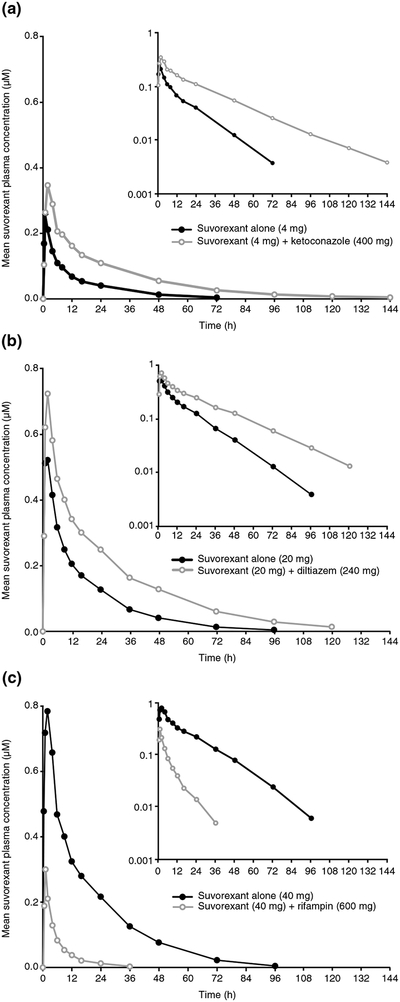
Arithmetic mean plasma concentration–time profile of suvorexant following administration of suvorexant alone or co-administered with a the strong CYP3A inhibitor ketoconazole (N = 10, Study P008), b the moderate CYP3A inhibitor diltiazem (suvorexant alone, N = 20; suvorexant plus diltiazem, N = 18; Study P038), or c the strong CYP3A inducer rifampin (N = 10, Study P038), in healthy subjects. Inset: semilog scale. CYP cytochrome P450, h hours
Table 3.
Summary of suvorexant pharmacokinetic parameters following administration of suvorexant alone or co-administered with the strong CYP3A inhibitor ketoconazole, moderate CYP3A inhibitor diltiazem, or the strong CYP3A inducer rifampin in healthy subjects
| Pharmacokinetic parameter | Study P008 | Study P038 | ||||
|---|---|---|---|---|---|---|
| Diltiazem sub-study | Rifampin sub-study | |||||
| Suvorexant 4 mg (N = 10) | Suvorexant 4 mg + ketoconazole (N = 10) | Suvorexant 20 mg (N = 20) | Suvorexant 20 mg + diltiazem (N = 18) | Suvorexant 40 mg (N = 10) | Suvorexant 40 mg + rifampin (N = 10)a | |
| GM AUC0–∞ (95% CI), μM·hb | 2.61 (1.84, 3.71) | 7.28 (5.13, 10.32) | 7.88 (6.57, 9.46) | 16.13 (13.38, 19.44) | 13.63 (10.53, 17.64) | 1.68 (1.30, 2.17) |
| GM Cmax (95% CI), μMb | 0.28 (0.22, 0.35) | 0.34 (0.27, 0.43) | 0.62 (0.54, 0.71) | 0.76 (0.66, 0.87) | 0.85 (0.70, 1.03) | 0.31 (0.25, 0.37) |
| Harmonic mean t½ (jack-knife SD), h | 11.2 (4.2) | 19.4 (6.9) | 12.4 (3.3) | 16.1 (5.3) | 12.9 (2.2) | 7.7 (3.4) |
| Median tmax (range), h | 1.0 (0.5–2.0) | 2.0 (1.0–4.0) | 1.5 (1.0–4.0) | 2.0 (1.0–4.0) | 2.0 (0.5–4.0) | 1.0 (0.5–2.0) |
AUC0–∞ area under the plasma concentration–time curve from time zero to infinity, CI confidence interval, Cmax maximum plasma concentration, CYP cytochrome P450, GM geometric mean, h hours, ln natural log, LSM least-squares mean, SD standard deviation, t½ half-life, tmax time to maximum plasma concentration
For one subject, the 36-h time point (Period 2) was excluded from the pharmacokinetic analysis due to an unusually high spike in plasma concentration relative to the 24-h time point data
Back-transformed LSM and CI from mixed-effects model performed on ln-transformed values
GMRs and their associated 90% CIs for the comparison of suvorexant in the presence and absence of inhibitors or an inducer of CYP3A are presented in Fig. 3. Co-administration of ketoconazole resulted in a moderate increase in suvorexant exposure (AUC0–∞) while co-administration of diltiazem resulted in a lesser effect. In contrast to exposure, marked differences in suvorexant Cmax were not observed following co-administration with strong or moderate CYP3A inhibitors. In contrast to co-administration of ketoconazole or diltiazem, co-administration of rifampin resulted in a decrease in suvorexant exposure (AUC0–∞, 88%) and suvorexant Cmax (64%).
Fig. 3.

GMRs (90% CI) for suvorexant co-administered with the strong CYP3A inhibitor ketoconazole, the moderate CYP3A inhibitor diltiazem, or the strong CYP3A inducer rifampin, versus suvorexant alone, for suvorexant AUC0–∞ (top) and Cmax (bottom). AUC0–∞ area under the concentration–time curve from time of administration to infinity, CI confidence interval, Cmax maximum observed concentration, CYP cytochrome P450, GMR least-squares geometric mean ratio
Compared with suvorexant administered alone (t½ range 11.2–12.9 h), suvorexant t½ was prolonged when co-administered with ketoconazole (19.4 h) or diltiazem (16.1 h; Table 3) and decreased with rifampin co-administration (7.70 h; Table 3).
3.3. Safety Assessments
The incidence of AEs is summarized in Table 4 for both trials. In Study P008, six (60%) subjects reported at least one AE with suvorexant administered alone and seven (70%) subjects reported at least one AE when suvorexant was co-administered with ketoconazole. In the P038 diltiazem sub-study, 20 (100%) subjects reported at least one AE with suvorexant administered alone and 18 (90%) with suvorexant co-administered with diltiazem (Table 4). All 10 (100%) subjects in the P038 rifampin sub-study reported at least one AE, with both suvorexant administered alone and when co-administered with rifampin (Table 4). Somnolence was the most frequently reported AE across all treatments for both studies, consistent with the reported pharmacology of suvorexant [8].
Table 4.
Summary of AEs
| AE | Study P008 | Study P038 | ||||
|---|---|---|---|---|---|---|
| Ketoconazole studya,b | Diltiazem sub-studya,c | Rifampin sub-studya,c | ||||
| Suvorexant 4 mg (N = 10) | Suvorexant 4 mg + ketoconazole (N = 10) | Suvorexant 20 mg (N = 20) | Suvorexant 20 mg + diltiazem (N = 20) | Suvorexant 40 mg (N = 10) | Suvorexant 40 mg + rifampin (N = 10) | |
| Subjects with ≥ 1 AE, n (%) | 6 (60) | 7 (70) | 20 (100) | 18 (90) | 10 (100) | 10 (100) |
| AEs reported by ≥ 2 subjects, any treatment group, n (%) | ||||||
| Somnolence | 6 (60) | 7 (70) | 19 (95) | 17 (85) | 10 (100) | 9 (90) |
| Headache | 0 (0) | 1 (10) | 0 (0) | 1 (5) | 1 (10) | 2 (20) |
| Vessel puncture site hematoma | 0 (0) | 0 (0) | 1 (5) | 1 (5) | 3 (30) | 0 (0) |
| Diarrhea | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) |
| Nausea | 0 (0) | 0 (0) | 2 (10) | 0 (0) | 0 (0) | 0 (0) |
| Abnormal dreams | 0 (0) | 0 (0) | 1 (5) | 1 (5) | 2 (20) | 3 (30) |
| ECI, n (%) | ||||||
| Euphoric mood | 0 (0) | 0 (0) | 0 (0) | 1 (5) | 1 (10) | 0 (0) |
| Hypnagogic hallucination | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Hypnopompic hallucination | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Sleep paralysis | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
AE adverse event, ECI event of clinical interest, MedDRA Medical Dictionary for Regulatory Activities
Although a subject may have had two or more AEs, the subject is counted only once in that category. The same subject may appear in different categories
MedDRA Version 12.0
MedDRA Version 13.1
All AEs were mild in intensity, except one moderate AE of headache reported in the P038 diltiazem sub-study, which resolved without intervention. All AEs in Study P008, and the majority of AEs in Study P038, were considered probably or possibly related to study treatment by the investigator. There were no serious AEs or deaths in either trial.
In Study P008, no AEs were considered to be ECIs. In Study P038, five AEs were considered ECIs (diltiazem sub-study, n = 1; rifampin sub-study, n = 4). These events were: euphoric mood (n = 1) with suvorexant 20 mg plus diltiazem, and euphoric mood (n = 1), sleep paralysis (n = 1), hypnagogic hallucination (n = 1), and hypnopompic hallucination (n = 1) with suvorexant 40 mg alone in the rifampin sub-study. These events were assessed as possibly related to study treatment and short-lived in duration, ranging from 1 min [hypnopompic hallucination (suvorexant 40 mg in the rifampin sub-study)] to 1 h [euphoric mood (suvorexant 20 mg plus diltiazem)].
Across both studies, no clinically meaningful changes in vital signs, laboratory safety tests, physical examination, or ECG monitoring were observed. Although some laboratory values fell outside the normal range, these variations were considered minor and, therefore, not clinically significant by the investigator and clinical monitor.
4. Discussion
Clinical pharmacokinetic DDI studies in healthy subjects were conducted with ketoconazole (strong CYP3A inhibitor), diltiazem (moderate CYP3A inhibitor), and rifampin (strong CYP3A inducer) to evaluate the potential for suvorexant to be subject to drug interactions mediated via inhibition or induction of CYP3A. Overall, CYP3A inhibition increased suvorexant exposure, while CYP3A induction decreased suvorexant exposure. Both studies used morning administration of suvorexant, and consistent with the known pharmacology of suvorexant, somnolence was the most widely reported AE [8].
Consistent with expectations of a CYP3A substrate, administration of suvorexant with strong or moderate CYP3A inhibitors resulted in increases in exposure (AUC0–∞). When suvorexant was co-administered with the strong CYP3A inhibitor ketoconazole or the moderate CYP3A inhibitor diltiazem, there were increases in suvorexant plasma concentrations compared with suvorexant alone (2.79- and 2.05-fold increases in AUC0–∞, respectively), with a prolongation in t½ (ketoconazole study, an increase of 8.2 h; diltiazem sub-study, an increase of 3.7 h). The observed increase in t½ with minimal increase in Cmax suggests that the observed increase in suvorexant AUC0–∞ following co-administration with ketoconazole may be due to an effect on systemic clearance, rather than on pre-systemic metabolism. While suvorexant is primarily cleared by CYP3A metabolism [8], the < 5-fold increase in AUC0–∞ following co-administration with ketoconazole was considered moderate and is likely due to the low clearance of suvorexant. When single doses of suvorexant 40 mg were co-administered with multiple doses of the strong CYP3A inducer rifampin, suvorexant exposure was reduced compared with when suvorexant was administered alone (approximately 88% reduction in suvorexant AUC0–∞ and 64% reduction in suvorexant Cmax). The larger impact of rifampin administration on suvorexant AUC0–∞ compared with Cmax indicates that the effects of this drug are mediated largely by changes in systemic clearance-related mechanisms. Consistent with previously reported pharmacokinetic parameters [35], suvorexant was readily absorbed, with a median tmax of 1–2 h in both trials summarized herein.
Although doses of suvorexant 20 mg were not specifically evaluated in the present trials, since the relative bioavailability between 10 and 20 mg tablets is equivalent, and suvorexant displays linear (time- and dose-independent) systemic pharmacokinetics (data on file), the anticipated suvorexant exposures in the presence of CYP3A inhibitors or inducers can be expected to be similar to those reported in the trials summarized herein. Further, given the collective pharmacokinetic data indicating the absence of a significant difference in elderly versus non-elderly healthy subjects and patients [8], the same assumptions may be applicable to elderly and non-elderly populations [12].
Notably, the observed increase in suvorexant exposure resulting from reduced CYP3A-mediated metabolism may be expected to lead to increased therapeutic effect and potential side effects, such as increases in next-day residual effects. However, in a randomized, double-blind, placebocontrolled, 4-period crossover polysomnography study in healthy subjects, no clinically meaningful effect on psycho-motor performance was observed following single doses of suvorexant 10 and 50 mg, whereas a statistically significant prolongation in reaction time (assessed using choice reaction time and simple reaction time) was observed with the 100 mg dose [14]. Suvorexant is approved at doses between 10 and 20 mg, and the need for dose adjustment or other clinical considerations in the setting of co-administration with strong or moderate CYP3A inhibitors is dependent on the approved dose in a particular geographical location. Local prescribing information should, therefore, be consulted for guidance. In contrast to co-administration with a CYP3A inhibitor, co-administration with a strong CYP3A inducer is likely to reduce suvorexant exposure and may, therefore, reduce efficacy [8]. In subsequent Phase II and III trials of the suvorexant development program, the concomitant use of mild-to-moderate CYP3A inhibitors (e.g. verapamil and diltiazem) and inducers (e.g. glucocorticoids) was permitted (data on file). Although only a limited number of subjects received these inhibitors (n = 15) and inducers (n = 25), an associated, significant effect on suvorexant exposure was not observed (data on file). This observation is consistent with expectations that co-administration of suvorexant with these mild-to-moderate inhibitors and inducers would exert much smaller effect sizes on suvorexant exposure than the strong inhibitors and inducers evaluated during Phase I development.
Consistent with morning administration and the known pharmacology of suvorexant [8], somnolence was the most frequently reported AE, with a greater incidence at higher doses of suvorexant.
Five AEs were considered ECIs and were reported following administration of suvorexant 40 mg alone or suvorexant 20 mg plus diltiazem in Study P038, including euphoric mood, hypnagogic and hypnopompic hallucinations, and sleep paralysis. Such sleep-related hallucinations have been previously reported in trials of zolpidem and zaleplon [18, 36].
The effects of CYP3A inhibition and induction are generalizable from single dose to chronic use; however, owing to the small sample size of the trials, the overall clinical response, efficacy, and safety should be informed by risk:benefit assessments.
5. Conclusions
CYP3A inhibition and induction altered the pharmacokinetic profile of single-dose suvorexant in healthy subjects. The relative magnitude of the impact was consistent with the effects expected of strong or moderate CYP3A modulators and continues to support the role of CYP3A in the biotransformation of suvorexant. In contrast, weak inhibitors/inducers will be expected to have smaller effect sizes and their impact is unlikely to be clinically relevant. Overall, it is recommended that clinicians refer to local labeling for specific guidance on the prescription of suvorexant in the presence of CYP3A inhibitors or inducers.
Key Points.
The pharmacokinetic interactions of inhibitors (ketoconazole or diltiazem) or an inducer (rifampin) of CYP3A on suvorexant are summarized.
Co-administration of ketoconazole or diltiazem with suvorexant resulted in increases in suvorexant AUC0–∞, and co-administration of rifampin with suvorexant resulted in a decrease in suvorexant AUC0–∞.
Single suvorexant doses were generally well tolerated in healthy subjects, both alone and when co-administered with a CYP3A inhibitor or inducer.
Acknowledgements
The authors would like to thank Xiaodong Li for serving as a statistician and Hong Sun for serving as a clinical monitor during these trials. Christopher Lines, PhD, and Tamara Cabalu, PhD, of Merck & Co., Inc., Kenilworth, NJ, USA provided comments and edits on a draft of the manuscript. Medical writing support, under the direction of the authors, was provided by Adele Blair, PhD, of CMC AFFINITY, a division of McCann Health Medical Communications Ltd., Glasgow, UK, in accordance with Good Publication Practice (GPP3) guidelines.
Funding These trials and medical writing support was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Footnotes
Data availability Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA’s data sharing policy, including restrictions, is available at http://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.
Conflict of interest REW, JBM, KLY, WL, DP, EM, and MC are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and may own stock and/or stock options. MPM-C was supported by National Institutes of Health Postdoctoral Training Grant No. T32GM008562. WKK has no disclosure to make.
Ethical approval All procedures performed in studies involving human subjects were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent Informed consent was obtained from all individual subjects included in the study.
References
- 1.Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roecker AJ, Coleman PJ. Orexin receptor antagonists: medicinal chemistry and therapeutic potential. Curr Top Med Chem. 2008;8(11):977–87. [DOI] [PubMed] [Google Scholar]
- 3.Coleman PJ, Gotter AL, Herring WJ, Winrow CJ, Renger JJ. The discovery of suvorexant, the first orexin receptor drug for insomnia. Annu Rev Pharmacol Toxicol. 2017;57:509–33. [DOI] [PubMed] [Google Scholar]
- 4.Gotter AL, Webber AL, Coleman PJ, Renger JJ, Winrow CJ. International Union of Basic and Clinical Pharmacology. LXXXVI. Orexin receptor function, nomenclature and pharmacology. Pharmacol Rev. 2012;64(3):389–420. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida Y, Fujiki N, Nakajima T, Ripley B, Matsumura H, Yoneda H, et al. Fluctuation of extracellular hypocretin-1 (orexin A) levels in the rat in relation to the light-dark cycle and sleep-wake activities. Eur J Neurosci. 2001;14(7):1075–81. [DOI] [PubMed] [Google Scholar]
- 6.Scammell TE, Winrow CJ. Orexin receptors: pharmacology and therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2011;51:243–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573–85. [DOI] [PubMed] [Google Scholar]
- 8.Sharp Merck & Corp Dohme. Belsomra (suvorexant) Package Insert. 2014. http://www.merck.com/product/usa/pi_circulars/b/belsomra/belsomra_pi.pdf. Accessed 26 June 2018. [Google Scholar]
- 9.Kawabe K, Horiuchi F, Ochi M, Nishimoto K, Ueno SI, Oka Y. Suvorexant for the treatment of insomnia in adolescents. J Child Adolesc Psychopharmacol. 2017;27:792–5. [DOI] [PubMed] [Google Scholar]
- 10.Herring WJ, Snyder E, Budd K, Hutzelmann J, Snavely D, Liu K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265–74. [DOI] [PubMed] [Google Scholar]
- 11.Herring WJ, Connor KM, Ivgy-May N, Snyder E, Liu K, Snavely DB, et al. Suvorexant in patients with insomnia: results from two 3-month randomized controlled clinical trials. Biol Psychiatry. 2016;79(2):136–48. [DOI] [PubMed] [Google Scholar]
- 12.Herring WJ, Connor KM, Snyder E, Snavely DB, Zhang Y, Hutzelmann J, et al. Suvorexant in elderly patients with insomnia: pooled analyses of data from Phase III randomized controlled clinical trials. Am J Geriatr Psychiatry. 2017;25(7):791–802. [DOI] [PubMed] [Google Scholar]
- 13.Michelson D, Snyder E, Paradis E, Chengan-Liu M, Snavely DB, Hutzelmann J, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a Phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461–71. [DOI] [PubMed] [Google Scholar]
- 14.Sun H, Kennedy WP, Wilbraham D, Lewis N, Calder N, Li X, et al. Effects of suvorexant, an orexin receptor antagonist, on sleep parameters as measured by polysomnography in healthy men. Sleep. 2013;36(2):259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vermeeren A, Vuurman E, Bautmans A, Li X, Vets E, Lewis N, et al. Suvorexant, a dual orexin receptor antagonist, does not impair next day driving performance in healthy elderly subjects. Sleep. 2012;35(Abstract Supplement):pA226 (Abstract 0670). [Google Scholar]
- 16.Vermeeren A, Vuurman E, Van Oers A, Van Leeuwen C, Jongen S, Bautmans A, et al. Effects of suvorexant, an orexin receptor antagonist on next day driving performance in healthy non-elderly subjects. Neuropsychopharmacology. 2012;38(Suppl 1):S320–1. [Google Scholar]
- 17.U.S. Food and Drug Administration. Clinical Drug Interaction Studies — Study Design, Data Analysis, and Clinical Implications Guidance for Industry DRAFT GUIDANCE. 2017. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. Accessed 28 June 2018. [Google Scholar]
- 18.RxList. Ambien (zolpidem) Package Insert. 2004. https://www.rxlist.com/ambien-drug/patient-images-side-effects.htm. Accessed 29 June 2018. [Google Scholar]
- 19.Sunovion Pharmaceuticals Inc. Lunesta [package insert]. 2014. http://www.lunesta.com/PostedApprovedLabelingText.pdf. Accessed 29 June 2018. [Google Scholar]
- 20.Cui D, Cabalu T, Yee KL, Small J, Li X, Liu B, et al. In vitro and in vivo characterisation of the metabolism and disposition of suvorexant in humans. Xenobiotica. 2016;46(10):882–95. [DOI] [PubMed] [Google Scholar]
- 21.U.S. Food and Drug Administration. Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. 2018. https://www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm. Accessed 3 July 2018. [Google Scholar]
- 22.Greenblatt DJ, Wright CE, von Moltke LL, Harmatz JS, Ehrenberg BL, Harrel LM, et al. Ketoconazole inhibition of triazolam and alprazolam clearance: differential kinetic and dynamic consequences. Clin Pharmacol Ther. 1998;64(3):237–47. [DOI] [PubMed] [Google Scholar]
- 23.Sutton D, Butler AM, Nadin L, Murray M. Role of CYP3A4 in human hepatic diltiazem N-demethylation: inhibition of CYP3A4 activity by oxidized diltiazem metabolites. J Pharmacol Exp Ther. 1997;282(1):294–300. [PubMed] [Google Scholar]
- 24.Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT. Pharmacokinetic interactions with rifampicin : clinical relevance. Clin Pharmacokinet. 2003;42(9):819–50. [DOI] [PubMed] [Google Scholar]
- 25.Villikka K, Kivisto KT, Backman JT, Olkkola KT, Neuvonen PJ. Triazolam is ineffective in patients taking rifampin. Clin Pharmacol Ther. 1997;61(1):8–14. [DOI] [PubMed] [Google Scholar]
- 26.Villikka K, Kivisto KT, Luurila H, Neuvonen PJ. Rifampin reduces plasma concentrations and effects of zolpidem. Clin Pharmacol Ther. 1997;62(6):629–34. [DOI] [PubMed] [Google Scholar]
- 27.Backman JT, Olkkola KT, Neuvonen PJ. Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin Pharmacol Ther. 1996;59(1):7–13. [DOI] [PubMed] [Google Scholar]
- 28.Stoch SA, Friedman E, Maes A, Yee K, Xu Y, Larson P, et al. Effect of different durations of ketoconazole dosing on the single-dose pharmacokinetics of midazolam: shortening the paradigm. J Clin Pharmacol. 2009;49(4):398–406. [DOI] [PubMed] [Google Scholar]
- 29.Friedman EJ, Fraser IP, Wang Y-H, Bergman AJ, Li C-C, Larson PJ, et al. Effect of different durations and formulations of diltiazem on the single-dose pharmacokinetics of midazolam: how long do we go? J Clin Pharmacol. 2011;51(11):1561–70. [DOI] [PubMed] [Google Scholar]
- 30.Sun H, Kennedy WD, Lewis N, Laethem T, Tee K, Li X, et al. The single dose pharmacokinetic (PK) and pharmacodynamic (PD) profiles of suvorexant (MK-4305), a dual orexin receptor antagonist, in healthy male subjects. Sleep Biol Rhythms. 2011;9(4):332 (Abstract). [Google Scholar]
- 31.Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55(5):481–5. [DOI] [PubMed] [Google Scholar]
- 32.Varhe A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patients receiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;56(6 Pt 1):601–7. [DOI] [PubMed] [Google Scholar]
- 33.Kronbach T, Mathys D, Umeno M, Gonzalez FJ, Meyer UA. Oxidation of midazolam and triazolam by human liver cytochrom P450IIIA4. Mol Pharmacol. 1989;36:89–96. [PubMed] [Google Scholar]
- 34.Breidinger SA, Simpson RC, Mangin E, Woolf EJ. Determination of suvorexant in human plasma using 96-well liquid-liquid extraction and HPLC with tandem mass spectrometric detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;1002:254–9. [DOI] [PubMed] [Google Scholar]
- 35.Yee KL, McCrea J, Panebianco D, Liu W, Lewis N, Cabalu T, et al. Safety, tolerability, and pharmacokinetics of suvorexant: a randomized rising-dose trial in healthy men. Clin Drug Invest. 2018;38(7):631–8. [DOI] [PubMed] [Google Scholar]
- 36.Drugs.com. Sonata (zaleplon) Package Insert. 2004. https://www.drugs.com/sonata.html. Accessed 3 July 2018. [Google Scholar]