

Summary
Valosin-containing protein (VCP)/p97 is an essential ATP-dependent protein unfoldase. Dominant mutations in p97 cause multisystem proteinopathy (MSP), a disease affecting the brain, muscle, and bone. Despite the identification of numerous pathways that are perturbed in MSP, the molecular-level defects of these p97 mutants are not completely understood. Here, we use biochemistry and cryo-electron microscopy to explore the effects of MSP mutations on the unfoldase activity of p97 in complex with its substrate adaptor NPLOC4·UFD1L (UN). We show that all seven analyzed MSP mutants unfold substrates faster. Mutant homo- and heterohexamers exhibit tighter UN binding and faster substrate processing. Our structural studies indicate that the increased UN affinity originates from a decoupling of p97’s nucleotide state and the positioning of its N-terminal domains. Together, our data support a gain-of-function model for p97-UN-dependent processes in MSP and underscore the importance of N-terminal domain movements for adaptor recruitment and substrate processing by p97.
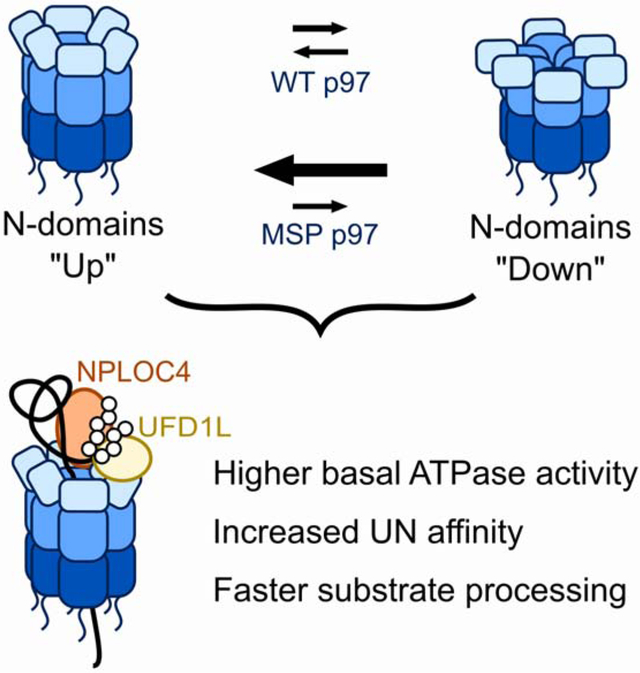
Graphical Abstract
eTOC Blurb
Mutations in p97 cause multisystem proteinopathy, but the underlying disease mechanisms are unknown. Blythe et al. demonstrate that these mutations cause conformational perturbations that increase substrate unfolding and cofactor binding, supporting a gain-of-function model for this disease and underscoring the importance of conformational changes for the regulation of p97 activity.
Introduction
Valosin-containing protein (VCP)/p97 is an essential AAA+ (ATPases Associated with various cellular Activities) ATPase involved in many cellular processes, such as ER-associated degradation (ERAD), aggregate clearance, and Golgi dynamics (van den Boom and Meyer, 2018). Each protomer within the barrel-shaped p97 homohexamer contains an N-terminal regulatory domain (NTD), two consecutive ATPase domains (D1 and D2), and an unstructured C-terminal tail (DeLaBarre and Brunger, 2003). The basic biochemical function of p97—unfolding proteins—is achieved by the ATP-powered threading of a substrate polypeptide through the central pore of the p97 hexamer (Blythe et al., 2017; Bodnar and Rapoport, 2017; Cooney et al., 2019; Twomey et al., 2019; Weith et al., 2018). This ATPase activity is coupled to large movements of the NTDs. While the NTDs favor a coplanar (‘down’) position with respect to the D1 ring when in the ADP-bound state, they switch to a coaxial (‘up’) position relative to the D1-D2 barrel when in the ATP-bound state (Banerjee et al., 2016; Tang et al., 2010). However, the function of these NTD movements during substrate processing remains unclear.
Substrates are delivered to p97 by a repertoire of adaptor proteins, many of which bind to the NTDs in mutually-exclusive ways (Hänzelmann and Schindelin, 2017). A common adaptor of p97 is the heterodimer UFD1ĹNPLOC4 (UN), which links p97 to numerous degradative processes (Meyer et al., 2000). Both UFD1L and NPLOC4 bind to p97 NTDs, either through a SHP motif (named from yeast Shp1) or a ubiquitin-like domain (UBD) (Hänzelmann and Schindelin, 2016; Isaacson et al., 2007; Le et al., 2016; Meyer et al., 2000). A recent structure of the fungal orthologues of p97·UN, Cdc48·Ufd1·Npl4, shows one UN adaptor positioned above the central pore of a Cdc48 hexamer and making additional contacts between its Mpr1–Pad1 N-terminal (MPN) domain and the D1 ATPase ring. The observed complex thus confirmed previous biochemical data suggesting a 1:1 stoichiometry of p97 hexamer and UN heterodimer (Bodnar et al., 2018; Hänzelmann and Schindelin, 2016). Similar to other p97 adaptors, UN recruits ubiquitinated substrates using ubiquitin-binding domains on both monomers (Meyer et al., 2000; Ye, 2003).
Mutations in p97 cause multisystem proteinopathy (MSP, also called IBMPFD), a fatal neurodegenerative disease (Watts et al., 2004). Symptoms of MSP manifest in middle age and vary in presentation, but can involve inclusion body myopathy, Paget’s disease of the bone, and frontotemporal dementia (Al-Obeidi et al., 2018; Nalbandian et al., 2011). Over 50 unique missense mutations in p97 have been identified, the majority of which lie at the interface between the NTD and D1 domains (Saracino et al., 2018). The mechanism by which these mutations cause disease remains controversial. Defects in numerous cellular pathways, such as endosomal trafficking, autophagy, mTOR regulation, and mitochondrial homeostasis have been reported in MSP mutants (Ching et al., 2013; Ju et al., 2009; Ritz et al., 2011; Zhang et al., 2017), yet the underlying functional changes in p97 and consequent cellular perturbations leading to disease pathology remain a mystery.
A key question regarding dominantly-inherited MSP is whether the disease is caused by a gain or loss of p97 function. This question is particularly relevant for therapy, since p97 inhibitors are being developed as cancer therapeutics (Anderson et al., 2015). Under basal conditions, i.e. in the absence of a protein substrate, MSP mutants exhibit higher ATPase activity in the D2 ring relative to wild-type p97 (Chou et al., 2014; Halawani et al., 2009; Niwa et al., 2012; Zhang et al., 2015), but whether this increased activity is productive or instead represents a decoupling of ATP hydrolysis from mechanical work is debated (Ju and Weihl, 2010; Tang and Xia, 2016). Structural studies suggest that MSP mutations cause a shift in the ‘up’/’down’ equilibrium of NTDs towards the ‘up’ conformation, regardless of the nucleotide state of p97 subunits (Huang et al., 2019; Niwa et al., 2012; Schuetz and Kay, 2016; Schütz et al., 2017; Tang and Xia, 2013; Tang et al., 2017). One likely consequence of this conformational preference is altered adaptor binding, and immunoprecipitations from MSP mutant cell lines have indeed shown differential adaptor associations (Fernández-Sáiz and Buchberger, 2010; Manno et al., 2010). However, the molecular mechanisms underlying a potentially modulated adaptor interaction and substrate-unfolding activity of p97 mutants remain elusive. Basic mechanistic studies of protein unfolding by MSP mutants have been stymied by the lack of an in vitro system, because native p97 substrates are not amenable to biochemistry. We previously overcame this barrier through the development of a GFP-based model substrate (Blythe et al., 2017), which allowed us to demonstrate that the MSP mutant p97-A232E unfolds substrate faster than wild type and that this difference could be normalized with low concentrations of the p97 inhibitor CB-5083 (Blythe et al., 2017).
In this study, we use model substrates in an in vitro reconstituted system to characterize the mechanistic details of MSP mutant p97 motors. We show that accelerated unfoldase activity and tighter UN binding are common features of MSP mutants. Importantly, these characteristics do not just apply to mutant homohexamers, but also heterohexamers that are formed from wild-type and mutant protomers and likely represent the relevant p97 species in heterozygote MSP patients. Our structural characterizations suggest that a preference for the ‘up’ conformation of NTDs in MSP mutant p97 is responsible for the higher UN affinity and the faster substrate unfolding we observe. These data support a gain-of-function model for UN-dependent substrate processing by p97 with MSP mutations.
Results
p97 MSP mutants show faster substrate processing
Our previous work employed a GFP-based model substrate to demonstrate p97 unfoldase activity (Blythe et al., 2017). To simplify this assay, we replaced GFP with mEos3.2 (Figure 1A), which has already been used to study p97/Cdc48 (Bodnar and Rapoport, 2017; Olszewski et al., 2019; Weith et al., 2018) and whose UV-induced backbone cleavage makes its unfolding irreversible (Wiedenmann et al., 2004; Zhang et al., 2012). As before, we modified a linear fusion of di-ubiquitin at the N-terminus of mEos3.2 with long K48-linked ubiquitin chains to allow recognition by UN and recruitment for p97·UN-mediated unfolding (Blythe et al., 2017).
Figure 1. MSP mutants have moderately accelerated unfoldase rates.

(A) Schematic for the substrate design and unfoldase assay. A linear fusion of di-ubiquitin on the N-terminus of mEos3.2 is irradiated with UV light to induce backbone cleavage and a shift in fluorescence emission from green to red. K48-linked ubiquitin chains of variable length are then enzymatically attached to the linearly fused ubiquitins. Unfolding of the substrate by p97·UFD1L·NPLOC4 (p97·UN) can be observed by the loss of red fluorescence, as the two polypeptides of split mEos3.2 cannot re-associate once separated. (B) Example time courses and single-exponential fits for substrate unfolding by wild-type p97·UN and two MSP mutants, showing a loss of mEos3.2 fluorescence over time. Experiments were performed under single-turnover conditions with saturating p97·UN over substrate. (C) Relative rates of substrate unfolding for seven MSP mutants of p97, normalized to the rate of wild type. Technical replicates shown. See also Figure S1.
We showed earlier that the MSP mutant p97-A232E processes substrate at approximately 140% compared to the rate of wild-type p97 under single-turnover conditions (Blythe et al., 2017). To determine whether faster unfoldase activity was a consistent characteristic of MSP mutants, we used our mEos3.2 substrate to assess the unfoldase activity of six additional mutants. All tested mutants showed accelerated substrate unfolding under single-turnover conditions, with rates ranging from ~ 115 to 160 % of the wild-type rate (Figure 1B–C, Figure S1A–C). Albeit moderate, this unfolding acceleration for MSP mutants was consistently observed and highly reproducible in biological and technical replicates. To rule out that the differences in activity relative to wild-type p97 were caused by heterologous expression, we also analyzed the unfoldase activities of p97 variants expressed in human cells. Compared to wild-type E. coli-expressed protein, wild-type p97 expressed in human cells showed faster unfolding and was able to process substrate even in the absence of exogenously added UN, which was likely a consequence of residual contamination with co-purified adaptors (Figure S1D). Importantly, however, p97-A232E isolated from human cells was still a faster unfoldase than human wild-type p97 (Figure S1E), confirming our results with the variants expressed in E. coli. Given that human p97 is decorated with post-translational modifications (Ewens et al., 2010; Hänzelmann and Schindelin, 2017) and may co-purify with endogenous adaptors when expressed in mammalian cells, we chose to continue our characterization of MSP mutations with E. coli-expressed proteins.
Our initial experiments used homohexameric wild-type or mutant p97. However, patients are heterozygous for MSP mutations (Watts et al., 2004) and thus likely have mixed p97 hexamers. To mimic this scenario, we co-expressed untagged wild-type and C-terminally His-tagged p97-A232E-mutant protomers in E. coli and used stepwise elution from a nickel affinity column to isolate three fractions of p97 hexamers with increasing ratios of p97-A232E:wild-type protomers (Figure 2A). These results confirmed that mutant and wild-type p97 protomers can co-assemble. The unfoldase rates of these mixed hexamers lied between those of wild-type and p97-A232E homohexamers and increased with the number of mutant protomers (Figure 2B–C). Our results suggest that the presence of MSP-mutant protomers imparts an improved ability for the hexamer to process substrate.
Figure 2. Unfoldase rates correlate with the number of mutant subunits in mixed p97 hexamers.

(A) Purification scheme for mixed hexamers. Wild-type and His6-tagged A232E-mutant p97 were co-expressed in E. coli, three fractions of mixed hexamers with increasing numbers of p97-A232E protomers were purified by stepwise elution from a Ni-NTA column (middle), and their relative composition was analyzed by SDS PAGE and Coomassie staining (right). (B) Example traces for the fluorescence decrease their single-exponential fits during single-turnover substrate unfolding by mixed p97 hexamers. (C) Relative rates for substrate unfolding by mixed hexamers and p97-A232E homo-hexamers compared to wild-type homo-hexamers. Technical replicates shown.
MSP mutant p97-A232E does not alter p97·UN complex structure
To understand the basis for this increased activity, we analyzed whether the disease mutation affected the overall structure and conformation of UN-bound p97. The complex was formed by mixing the p97-A232E hexamer with excess UN adaptor (1:1.5) in the presence of ATP and vitrified for single particle cryogenic electron microscopy (cryo-EM) analysis. The final reconstruction was refined to 4.26 Å (Figure S2). Similar to previous p97 structures (Banerjee et al., 2016; Bodnar et al., 2018), the p97-A232E hexamer was in a symmetric conformation, with the NPLOC4 portion of the UN heterodimer resolved in a position atop the central channel (Figure 3A). The NTDs were observed in an ‘up’ conformation, with extra density for the UBX of NPLOC4 on one of the NTDs (Figure 3A), which is consistent with a recent cryo-EM structure of UN-bound Cdc48 (Bodnar et al., 2018). The previously published structures of p97-ATPγS (PDB:5ftn, (Banerjee et al., 2016)) and the Cdc48·UN complex (PDB: 6chs, (Bodnar et al., 2018)) both dock well into our map (Figure 3B–C), showing that the A232E point mutation did not cause any major changes in the conformation of the p97 hexamer or the position and orientation of the bound UN cofactor.
Figure 3. Cryo-EM structure of the p97-A232E·UN complex.

(A) Side (left) and top (right) views of the final sharpened density map for the p97-A232E·UN complex. Subunits and domains are colored: NPLOC4 (orange), NTDs (light blue), D1 (sky blue), D2 (navy blue). (B) Atomic model for the Cdc48·Npl4 complex (PDB:6chs, (Bodnar et al., 2018)) docked into the map for p97-A232E·UN. (C) Atomic model for ATPγS-bound wild-type p97 (PDB:5ftn, (Banerjee et al., 2016)) docked into the p97-A232E·UN complex map. See also Figure S2.
p97’s unfoldase rate is not correlated with its ATPase rate
MSP mutations are known to increase the basal ATPase rate of p97 (Chou et al., 2014; Halawani et al., 2009; Niwa et al., 2012; Zhang et al., 2015), and, hence, one possible explanation for faster substrate unfolding is a faster consumption of ATP. Since substrate processing stimulates p97’s ATP hydrolysis (Blythe et al., 2017; Bodnar and Rapoport, 2017), we compared the working ATPase rate of MSP mutants after a 15-minute incubation with excess substrate and UN adaptor. At that point, most p97 hexamers are expected to have stalled on a substrate’s ubiquitin chain. Previous studies indicated that p97 in the absence of deubiquitinases fails to thread the long ubiquitin modifications present on model substrates, leading to just a single turnover of protein unfolding (Bodnar and Rapoport, 2017). Yet, despite this stall in translocation, p97 continues to hydrolyze ATP, allowing a comparison of working ATPase rates for these substrate-engaged motors. For wild-type p97, the ATP-hydrolysis activity increased from a basal rate of 0.27 ± 0.02 ATP per second per hexamer to 6.8 ± 0.1 ATP per second per hexamer in the presence of excess UN and substrate. Under these conditions, mutants showed only a 1.5-fold higher ATPase rate than wild type, much less than the 5- to 10-fold difference observed for the basal ATPase activity in the absence of substrate (Figure 4A, C). Furthermore, all three wild-type:p97-A232E mixed hexamer fractions showed essentially identical working ATPase rates compared to wild-type p97, despite also exhibiting 5- to 10-fold higher basal rates (Figure 4B, D). For all tested p97 variants, the working ATPase rates were not well correlated with the unfoldase rates (Figure 4E), implying that faster ATP hydrolysis is not responsible for accelerated substrate unfolding by MSP mutants.
Figure 4. ATPase rates do not correlate with unfoldase rates.

Normalized basal ATPase rates (A, B) and working ATPase rates in the presence of substrate (C, D) for wild-type p97 and MSP-mutant homo-hexamers (A, D), as well as mixed hexamers containing wild-type and p97-A232E mutant protomers (B, D). Rates were normalized to wild-type p97, which together with p97-A232E homo-hexamers is shown in all panels for comparison. (E) A plot of unfoldase rate versus working ATPase rate shows no clear correlation. All rates were normalized to the unfoldase and ATPase rates of wild-type p97. Shown are technical replicates (N ≥ 3) and their S.D..
MSP mutants have increased affinity for UN
Faster substrate processing could be explained by faster delivery to p97 and initiation of unfolding, which we explored by developing a FRET-based assay for the dynamics of UN binding to p97. Structural data suggest that the N-termini of p97 and bound NPLOC4 are close to each other (Isaacson et al., 2007). We therefore designed an NPLOC4 construct susceptible to N-terminal labeling with a TRITC donor fluorophore (Theile et al., 2013) to complement a previously published p97 construct with an N-terminal Cy5 acceptor dye (Xue et al., 2016). Incubation of UTRITCN with Cy5p97 led to a decrease in TRITC fluorescence, indicative of FRET (Figure 5A). Importantly, the loss of donor fluorescence was rescued by the addition of unlabeled UN, providing evidence that this FRET assay is specific for the formation of the p97·UN complex (Figure 5A).
Figure 5. FRET-based assay for UN binding to p97.

(A) Fluorescence spectra, showing that the emission of a TRITC donor dye attached to the N-terminus of NPLOC4 in the UTRITCN heterodimer (red trace) is quenched due to FRET upon addition of excess p97 with a Cy5 acceptor dye attached to an N-terminal ybbR tag (purple trace). FRET is prevented and donor fluorescence is restored upon addition of excess unlabeled UN (black trace). (B) FRET-based competition binding assay, monitoring the recovery of TRITC donor fluorescence upon mixing various concentrations of unlabeled p97 with a complex formed from UTRITCN and Cy5-labeled wild-type, p97-A232E mutant, or mixed p97 hexamers in ATP. Ki values were calculated from curve fits of IC50. Shown are technical replicates (N = 3) and their S.D.. See also Table 1 and Figure S3.
By employing this FRET-based assay, we were able to measure the equilibrium (Figure S3A) and kinetic (Figure S3B–D) constants for UN binding to p97 in various nucleotide states. For wild-type p97, the equilibrium dissociation constant was 2-fold lower in ATP (302 nM) than ADP (636 nM), and further reduced in the presence of the non-hydrolyzable ATP analog ATPγS (177 nM; Table 1). These KD values are comparable to those measured by SPR (100–400 nM) (Chia et al., 2012), but ~3–10-fold lower than the KD measured by ITC in the absence of nucleotide (1.7 μM) (Hänzelmann et al., 2011). Similar to our data, SPR studies also report an increase in UN affinity when p97 is in an ATP state (Chia et al., 2012).
Table 1.
Kinetic and equilibrium binding constants for the interaction of UTRITCN with Cy5p97 and its mutants (technical replicates N ≥ 3 ± S.D.).
| KD (nM) | kon (×106 M−1s−1) | koff (s−1) | Ki (nM) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ATP | ADP | ATPYS | ATP | ADP | ATPYS | ATP | ADP | ATPYS | ATP | |
| WT | 302 ± 51 | 636 ± 96 | 177 ± 32 | 1.6 ± 0.3* | n.d. | 1.9 ± 0.3* | 0.48 ± 0.01 | n.d. | 0.33 ± 0.01 | 110 ± 20 |
| R155H | 6.3 ± 0.1* | n.d. | n.d. | 53 ± 1 | n.d. | n.d. | 0.332 ± 0.001 | n.d. | n.d. | n.d. |
| A232E | 4.6 ± 0.2* | 50 ± 11 | 4.6 ± 0.2* | 54 ± 1 | 3.3 ± 0.7* | 61 ± 1 | 0.25 ± 0.01 | 0.165 ± 0.003 | 0.28 ± 0.01 | 3.8 ± 0.6 |
| T262A | 3.5 ± 0.1* | n.d. | n.d. | 60 ± 1 | n.d. | n.d. | 0.210 ± 0.001 | n.d. | n.d. | n.d. |
| Mixed 2 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 17 ± 2 |
Strikingly, the p97-A232E mutant bound UN much more tightly, with a 12-fold higher affinity relative to wild-type p97 in the ADP-bound state, and a 65-fold higher affinity in the presence of ATP (Table 1). Like wild type, the mutant thus showed weaker binding with ADP versus ATP or ATPγS (50 nM versus 4.6 nM), yet the difference was > 10-fold larger and indicative of the p97-A232E mutant being particularly well suited for UN binding when in the ATP bound state. The increase in affinity is solely a consequence of a much faster on-rate, while the off-rate remains unchanged (Table 1). The p97-R155H and p97-T262A mutants in the presence of ATP have UN affinities very similar to that of p97-A232E, suggesting that this phenomenon of tight UN binding is a common feature of MSP mutants. Indeed, previous work demonstrated increased co-immunoprecipitation of UN with MSP mutants (Fernández-Sáiz and Buchberger, 2010; Manno et al., 2010). Other adaptors, such as p47, bind MSP-mutant p97 with higher affinity as well (Bulfer et al., 2016; Rao et al., 2017; Tang et al., 2017); however, in those cases, tighter binding was observed only in the ADP state, whereas we measured tighter binding of UN in any nucleotide state.
To probe the adaptor affinity of mixed hexamers, we used a competition assay (Figure 5B, Figure S3E), in which the decrease of FRET, i.e. the increase in donor fluorescence upon binding of unlabeled p97, was measured upon incubating UTRITCN and Cy5p97-A232E with increasing concentrations of unlabeled mixed hexamers. UN-binding affinities were determined as Ki values from IC50 curves (Table 1), showing a nice agreement with the directly measured KD values when titrating in wild-type and p97-A232E homo-hexamers, and intermediate affinities for mixed hexamers. For mixed hexamers with different numbers of MSP-mutant subunits, we observed similarly increased UN affinities (Figure S3F), indicating that only a subset of mutant protomers may be required for tighter binding of this cofactor. Importantly, no exchange of protomers between p97 hexamers occurred on the time scale of these competition-binding experiment (Figure S3G).
P97-A232E mutant prefers ‘up’ NTD conformation
To explore the structural basis for the enhanced UN-binding affinity of MSP mutants, we visualized p97 in different nucleotide states by cryo-EM. Previous p97/Cdc48 structural studies have established that the NTDs vary in their conformation and position relative to the D1 ATPase ring depending on the nucleotide state, moving between an ‘up’ conformation in ATP or ATPγS and a ‘down’ conformation in ADP (Banerjee et al., 2016; Bodnar et al., 2018; Huang et al., 2019; Schuetz and Kay, 2016; Schütz et al., 2017; Tang et al., 2010). Since UN binding was modulated by nucleotide, we were interested in exploring how MSP mutations affected the overall equilibrium of NTDs. We therefore collected single-particle cryo-EM data of wild-type and A232E mutant p97 in the presence of ATP or ADP, and analyzed the positions of the NTDs in 2D class averages (Figure S4). We assigned ‘up’ or ‘down’ states to class averages with visible NTDs, and classified averages with unresolved, dynamic NTDs as ‘mobile’ (Figure S4). Although this technique is limited in visualizing and properly assigning all NTD conformations, especially when differential states are occupied within the same hexamer, we observed clear trends in NTD conformations depending on the p97 variant and the bound nucleotide (Figure 6A).
Figure 6. MSP mutations alter NTD conformational dynamics.

(A) Reference-free 2D-class averages from single-particle cryo-EM datasets for p97 and p97-A232E in the presence of ATP or ADP were compared to back projections of the maps for p97 in ADP (EMD-3297) and p97 in ATPγS (EMD-3299), and NTD conformations were assigned to ‘up’ and ‘down’, or ‘mobile’ when NTDs were not resolved. Shown on the left are representative class averages for the ‘mobile’ conformation (top with grey outline, taken from p97-A232E in ATP), the ‘up’ conformation (middle with blue outline, taken from p97 in ATP), and the ‘down’ conformation (bottom with orange outline, taken from p97 in ATP). The proportion of particles in ‘down,’ ‘up,’ and ‘mobile’ conformations are plotted on the right as a percentage of total particles classified. NTDs in the ‘mobile’ conformation either originate from averaging mixed ‘up’ and ‘down’ states in more dynamic parts of the hexamer or from actual mobility after dislodging from the defined ‘down’ state, and are therefore in the bar graph grouped together with the ‘up’ state. See also Figure S4. (B) Model for the increased unfoldase activity of MSP mutant p97. While the NTDs in wild-type hexamers are moving between ‘up’ and ‘down’ conformations dependent on the nucleotide state, the NTDs of MSP-mutant hexamers remain in an ‘up’ conformation, allowing both, faster UN binding and faster substrate processing.
In the presence of ATP, with subunits actively hydrolyzing and transitioning through the various stages of the ATPase cycle, the locations of NTDs for wild-type p97 were highly variable, occupying both the ‘up’ and ‘down’ states with approximately equal distribution. In contrast, all visible NTDs in the p97-A232E mutant under the same conditions were in the ‘up’ position (Figure 6A). Thus, although both motors hydrolyze ATP and sample the different nucleotide states, only the NTDs of wild-type p97 seem to display the associated conformational changes and switch between ‘up’ and ‘down’ positions. Consistently, we found that in the presence of excess ADP, all resolved NTDs of wild-type p97 were in the ‘down’ conformation, while the p97-A232E mutant still showed some particles with their NTDs in the ‘up’ state (Figure 6A). Notably, the dataset for ADP-bound p97-A232E had the highest percentage of ‘mobile’ NTDs, indicating that the ADP-induced ‘down’ state is destabilized in this MSP mutant. There is also a fraction of wild-type p97 particles with ‘mobile’ NTDs, which may account for the UN-binding competency of p97-ADP (Table 1). The observed general patterns demonstrate that the conformational equilibrium of the p97-A232E mutant is shifted toward the ‘up’ state and decoupled from the nucleotide state, which is consistent with previous studies on MSP disease mutations (Huang et al., 2019; Niwa et al., 2012; Schuetz and Kay, 2016; Schütz et al., 2017; Tang and Xia, 2013; Tang et al., 2010, 2017). Our findings for the ADP-bound hexamers agree with the ‘up’/’down’ equilibrium assessments of p97 truncations (p97-ND1) by NMR (Schuetz and Kay, 2016; Schütz et al., 2017) and expand upon this analysis by showing that the conformational differences persist also in full-length p97 during active ATP hydrolysis. Our p97-A232E·UN structure and the previous Cdc48·UN structure (Bodnar et al., 2018) suggest that UN binds to p97 when NTDs are in the ‘up’ conformation, which explains why MSP mutants with their NTD conformational equilibrium shifted toward the ‘up’ state show much higher UN affinity in all nucleotide states. This model for UN preferentially binding and then stabilizing NTDs in the ‘up’ conformation is also consistent with our kinetic measurements, where MSP mutants showed faster on-rates, but no effect on the off-rate.
Discussion
Here, we demonstrate that moderately accelerated unfoldase activity is a defining feature among p97 hexamers with MSP mutations. We show for the first time that mixed hexamers with wild-type and mutant subunits are fully functional and display faster substrate unfolding as well. These data contradict a previously proposed dominant-negative model for MSP mutations (Ju and Weihl, 2010; Tang and Xia, 2016), hypothesizing that mutant p97 protomers poison otherwise wild-type hexamers and impair their ability to unfold protein substrates. Since MSP patients are heterozygote and likely contain mixed p97 hexamers (Weihl et al., 2006; Manno et al., 2010; Arhzaouy et al., 2012; Kimura et al., 2013), our findings are of considerable significance for the development of future therapies based on small-molecule inhibitors or modulators of p97 activity.
To understand the basis for faster substrate processing, we probed two steps in p97-mediated unfolding: 1) UN-cofactor binding, which affects substrate recruitment and subsequent engagement, and 2) ATP hydrolysis during substrate processing, which reports on the speed of the working motor. Using a FRET-based assay for p97·UN complex formation, we discovered a 30- to 60-fold increase in UN affinity for mutant p97 homohexamers, and a 5- to 15-fold increase for heterohexamers containing wild-type and mutant subunits. Interestingly, this increased affinity of the mutant is due to a faster on rate, whereas the off rate and therefore the residence time of UN on p97 remains unchanged. The faster binding is consistent with our observation that MSP mutations preferentially induce the UN-binding-competent ‘up’ state of NTDs. In the presence of ATP, the p97-A232E mutant had all visible NTDs in the ‘up’ conformation, whereas wild-type p97 showed an approximately equal ‘up’/’down’ distribution. The shift of the conformational equilibrium itself cannot explain the mutant’s 60-fold increase in UN affinity, but it is likely the nucleotide-dependent dynamic switching of NTDs that limits the UN-binding kinetics for wild-type p97 and whose elimination increases the on-rate for MSP mutants.
Nevertheless, this faster UN binding is apparently not responsible for the observed faster substrate processing by MSP mutants. Substrate unfolding under single-turnover conditions showed no dependence on the concentrations of UN or p97 (Figure S1A), indicating that initial UN·substrate binding to the motor is not the rate-determining step. Furthermore, despite their differences in substrate-unfolding rates, wild-type and mutant p97 exhibit similar working ATPase rates, speaking against ATP hydrolysis being rate-limiting. We therefore propose that uncoupling the NTDs from the nucleotide state in MSP-mutant subunits facilitates yet another step in substrate processing that follows initial UN binding, for instance the correct docking of UN on top of the D1 ATPase ring for substrate engagement by the AAA+ motor (Bodnar et al., 2018) or the initiation of translocation itself. Despite their similarities in UN binding, different MSP mutations may differentially affect this rate-limiting step, which would explain the diversity in unfolding-rate acceleration we observed. Further work will be required to delineate the roles of NTD movements during substrate engagement, unfolding, and translocation, and to explore how individual MSP mutations perturb these steps that are not captured in this study.
Our biochemical and structural data support a gain-of-function model for the molecular cause of MSP pathology. Faster substrate processing or tighter interactions with UN under cellular conditions where cofactor binding is rate-limiting could enhance p97·UN-dependent processes and lead to the inappropriate extraction, unfolding, or degradation of protein targets. This hypothesis is supported by studies on an MSP mouse model that demonstrated increased turnover of IκB-α and consequently improper activation of NF-κB signaling (Custer et al., 2010). Since IκB-α degradation is p97·UN dependent, the hyperstimulated NF-κB phenotype can be explained by the increased activity of mutant p97·UN (Dai et al., 1998; Li et al., 2014; Nalbandian et al., 2011). Similarly, increased processing of TDP-43 and mitofusin have been demonstrated in MSP mutant Drosophila (Ritson et al., 2010; Zhang et al., 2017). Another consequence of p97·UN hyperactivity could be the formation of detrimental aggregates, if the 26S proteasome, responsible for ubiquitin-dependent protein degradation downstream of p97, is unable to keep pace with the increased rate of p97-mediated substrate unfolding. Tissues affected by MSP show aggregates that are decorated with ubiquitin modifications (Nalbandian et al., 2011), which would be consistent with an overburdened proteasome.
The p97 system is extremely complex, and we cannot discount other explanations for MSP pathology that are consistent with our biochemical studies. An alternative hypothesis for disease is that improved UN affinity leads to an adaptor imbalance that generates cellular perturbations. Based on published values (Gillen and Forbush, 1999; Reitsma et al., 2017; Xue et al., 2016) and our affinity measurements, we can estimate intracellular concentrations of approximately 50 nM free UN, 480 nM free p97, and 80 nM p97-UN complex in wild-type cells. In contrast, MSP-mutant cells are expected to have essentially all of their UN adaptor bound to p97, leading to intracellular concentrations of 140 nM p97·UN complex and 430 nM free p97. If free UN is needed for initial substrate binding in the cellular context, as has been suggested for mitophagy (Kimura et al., 2013), the lack of this free UN pool in mutant cells may impair p97·UN-dependent functions. Alternatively, with less free p97 available to bind other adaptors, this imbalance in UN interactions may cause a loss of function in other p97-dependent pathways. We cannot rule out that the gain of p97·UN function we observed for MSP mutants in vitro is physiologically irrelevant, and a potential gain or loss of function with another p97 adaptor is responsible for pathology. In particular, UBXD1 has a decreased affinity for MSP-mutant p97 due to its preference for binding the NTDs in the ‘down’ conformation (Ritz et al., 2011; Schuetz and Kay, 2016; Schütz et al., 2017), and MSP mutations have been associated with failures in lysosomal degradation, which is mediated by UBXD1 (Ju et al., 2009; Kirchner et al., 2013; Papadopoulos et al., 2017; Ritz et al., 2011). Thus, a perhaps more realistic scenario is that the MSP pathology is the combined outcome of a complex series of molecular perturbations to different pathways. Regardless, the dominant inheritance of MSP and the observed lack of a poison-subunit effect in substrate unfolding are most consistent with an increase, not decrease, in p97’s biochemical function.
If MSP is indeed caused by a gain of function in p97·UN-dependent pathways, a p97 inhibitor could be an effective treatment for this disease. Though an inhibitor would not rectify the causal defect—aberrant NTD conformations—it could minimize the harmful downstream effects. Such an approach was also demonstrated to be viable in Drosophila, where physiological and morphological defects in muscle tissue caused by the expression of mutant p97 were reversed through treatment with low doses of p97 inhibitor (Zhang et al., 2017). Currently, there is no treatment for MSP, but our biochemical work suggests an exciting potential avenue for therapeutic intervention. More work is needed to identify the molecular basis for MSP pathology and explore whether p97 inhibitors may be effective treatments.
STAR Methods
Lead contact and materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andreas Martin (a.martin@berkeley.edu).
Experimental model and subject details
Bacterial strains
Proteins were expressed in E. coli strains BL21(DE3) and Rosetta(DE3)pLysS as indicated.
Cell lines
Proteins were expressed in Expi293 suspension cells (Thermo Fisher). Cells were grown in Expi293 Expression Medium (Thermo Fisher) at 37 °C with 8% CO2.
Method detail
Protein purification and labeling
Expression, purification, and labeling of wild-type UN, p97-His, and Cy5p97-His were carried out as previously described (Blythe et al., 2017; Xue et al., 2016). MSP mutations were introduced into these constructs using site-directed mutagenesis, and mutant proteins were expressed and purified similar to wild-type.
For FRET studies, a sortase recognition motif was added to the N-terminus of NPLOC4 (Hanzelmann and Schindelin, 2011) to form MGGG-NPLOC4. To prevent labeling of His-Ufd1 (Fernández-Sáiz and Buchberger, 2010), site directed mutagenesis was used to remove residue G2. This modified UN was expressed and purified as previously described (Blythe et al., 2017) with one modification. Prior to gel filtration, 30 μM UN was labeled using 500 μM peptide (TRITCWSHPQFEKLPETGG, GenScript) and 5 μM sortase (Theile et al., 2013) in labeling buffer (25 mM HEPES pH 7.4, 150 mM NaCl, 10 mM CaCl2, 1 mM DTT) at 25 °C for 2 hours. The reaction was diluted in s trep buffer (100 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA) and incubated with 2 mL of Strep-Tactin resin (IBA Lifesciences) for 10 minutes at 4 °C with agit ation. The resin was washed with 5 column volumes of strep buffer, and the protein was eluted with strep buffer plus 50 mM biotin.
A plasmid for co-expression of p97 and p97-A232E-His was constructed via Gibson cloning by inserting sequences of p97-His and p97-A232E-His into a pCOLA-Duet backbone. Protein was expressed in Rosetta(DE3)pLysS overnight at 16 °C with 0.4 mM IPTG. Cell pellet was resuspen ded in nickel binding buffer (50 mM Tris pH 7.4, 500 mM KCl, 5 mM MgCl2, 20 mM imidazole, 5% glycerol, 2 mM b-mercaptoethanol) with protease inhibitors and sonicated. Clarified lysate was bound to a HisTrapHP (GE Healthcare), and proteins were eluted in a stepwise gradient of 12% (wash), 20% (“Mixed 1”), 35% (“Mixed 2”), and 55% (“Mixed 3”) nickel binding buffer with 300 mM imidazole. Proteins were further purified on a Superose 6 column (GE Healthcare) in storage buffer (20 mM HEPES pH 7.4, 250 mM KCl, 1 mM MgCl2, 5% glycerol, 0.5 mM TCEP), concentrated, and flash frozen.
Plasmids encoding p97-FLAG and p97-A232E-FLAG for mammalian expression were made by inserting p97 sequences into a pcDNA3.1 backbone. Expi293 cells were transfected with 1.1 mg DNA per 1 L of cells using polyethyleneimine “Max” (MW 40,000) (Polysciences) at a ratio of 1:2.7 DNA:PEI. After 72 hours, cells were harvested at 3000×g for 15 minutes and washed once with 30 mL cold PBS. Cell pellet was resuspended in lysis buffer (50 mM Tris pH 7.4, 150 mM KCl, 5 mM MgCl2, 5% glycerol, 0.5% Triton X-100) with protease inhibitors (Roche) and incubated with rotation at 4 °C for 30 minutes. Clarified lysate was incubated with 2 mL of anti-FLAG resin (Sigma) at 4 °C for 1.5 hours. Resin was washed with 3 column volumes of wash buffer (50 mM Tris pH 7.4, 400 mM KCl, 5 mM MgCl2, 0.5% Triton X-100) 3 times for 10 minutes with rotation followed by one wash with elution buffer without FLAG peptide (25 mM HEPES pH 7.4, 150 mM KCl, 2 mM MgCl2). Protein was eluted with 2 mL of elution buffer containing 0.2 mg/mL 3XFLAG peptide for ten minutes with rotation. Elution was repeated, and resin was drained with a final 2 mL of elution buffer. Protein was concentrated and exchanged into storage buffer (20 mM HEPES pH 7.4, 250 mM KCl, 1 mM MgCl2, 5% glycerol, 0.5 mM TCEP) before flash freezing.
A plasmid encoding p97-FLAG for bacterial expression was made through the addition of a FLAG tag to a plasmid encoding untagged p97 in a pET24b backbone. Protein was expressed in Rosetta(DE3)pLysS as previously described (Xue et al., 2016). Harvested cells were resuspended in lysis buffer (50 mM Tris pH 7.4, 150 mM KCl, 5 mM MgCl2, 5% glycerol) with protease inhibitors (Roche) and sonicated. Clarified lysate was incubated with 1 mL anti-FLAG resin (Sigma) and purified as described above. After elution, concentrated protein was purified by gel filtration (Superose 6) in storage buffer before flash freezing.
Substrate preparation
His6- UbG76V- UbG76V -mEos3.2 was constructed by replacing GFP in the previously published His6-UbG76V-UbG76V-GFP substrate (Blythe et al., 2017) with a PCR amplicon of mEos3.2 (Addgene plasmid #87030), using BamHI and NotI restriction sites. Protein was expressed in BL21(DE3) overnight at 18 °C, cells were lysed by sonication in 50 mM Tris pH 8.0, 250 mM NaCl, 20 mM imidazole, 2 mM betamercaptoethanol, and protease inhibitors (Roche), and clarified lysate was incubated with nickel-NTA resin for 2 hours at 4°C. After elution from the resin with 200 mM imidazole, protein was spin-concentrated and dialyzed overnight into 50 mM HEPES pH 7.0, 150 mM NaCl, 2 mM betamercaptoethanol. Sample was irradiated with 380 nm LED light (Thorlabs) for one hour at 4°C to induce the backbone cleavage of mEOS3.2. After filtration, protein was purified on a Superdex 200 column (20 mM HEPES pH 7.4, 100 mM KCl, 3 mM MgCl2), and fractions were pooled, concentrated and flash frozen. Ubiquitination and purification of the ubiquitinated substrate was carried out as previously described (Blythe et al., 2017), except that yeast ubiquitin was used. Final substrate showed ~40% photocleavage as measured by the absorbance at 507 and 572 nm (Zhang et al., 2012). Concentrations of substrate referred to in this paper correspond to the concentration of photocleaved (red) protein.
Unfoldase assays
Unfolding reactions contained final concentrations of 20 nM substrate, 400 nM p97 hexamer, and 800 nM UN, and were carried out in assay Buffer (20 mM HEPES pH 7.4, 150 mM KCl, 20 mM MgCl2, 1 mM TCEP, 1 mg/mL BSA) supplemented with an ATP regeneration system (5 mM ATP, 30 mM creatine phosphate, 50 μg/mL creatine phosphokinase). Proteins were preincubated in a 96-well plate (Costar 3694) for 15 minutes at 37 °C before the addition of the ATP regeneration system to initiate the reaction. Fluorescence was read on a PHERAstar Plus (BMG Labtech) or Synergy Neo2 (BioTek) plate reader using a 540 nm excitation filter and 590 nm emission filter. Data were normalized to the first reading and fit to a single exponential decay to determine the rate of substrate unfolding.
In supplementary figures, experiments were carried out with a few modifications. For bacterially-made proteins, unfoldase assays used a modified assay buffer (50 mM Tris pH 7.4, 5 mM KCl, 20 mM MgCl2, 1 mM EDTA, 0.5 mM TCEP, 0.01% Triton X-100) (Chou et al., 2014) with 20 nM substrate, 250 nM p97 hexamer, 500 nM UN, and 2 mM ATP. For mammalian-made proteins, unfoldase assays used the modified assay buffer with 20 mM KCl and ATP regeneration system. For Figure S3A, experiments were carried out at 37 °C with 250 nM p97 and 500 nM UN. For Figure S3B, experiments were carried out at 25 °C with 2 mM of p97 and UN.
ATPase assays
Proteins (100–400 nM p97) were diluted in assay buffer and preincubated in a 96-well plate (Costar 3695) for 10 minutes at 37 °C be fore the addition of ATPase mix (5 mM ATP, 1 mM NADH, 7.5 mM phosphoenolpyruvate, 12 U/mL pyruvate kinase, and 18 U/mL lactate dehydrogenase (Sigma P0294)). For the measurement of working ATPase rates, final concentrations were 50 nM p97, 250 nM UN, and 250 nM substrate. Fifteen minutes after addition of ATP regeneration system, A340 was monitored for 10 minutes and the ATPase rate was determined by linear regression. A wild-type p97 control was included in every plate and used to normalize ATPase rates.
FRET-based UN-binding assays
Assays were carried out at room temperature in assay buffer supplemented with 2 mM ADP, 2 mM ATPΥS, or ATP regeneration system. Equilibrium binding assays were measured with 50 nM UTRITCN and varying concentrations of Cy5p97 by monitoring the TRITC donor fluorescence at 576 nm after excitation at 540 nm in a fluorimeter (Photon Technology International, Inc). Data were fit to a parabolic binding equation. Kinetic constants were measured using an AutoSF-120 stopped flow fluorimeter (KinTek) with an excitation wavelength of 540 nm and an emission filter of 576 ± 31 nm. For measurements of kon, the final concentrations were 50 nM UTRITCN and 50–400 nM Cy5p97. Data were fit to single exponential decay equation to determine kobs, and linear regression of kobs versus [Cy5p97] were used to calculate kon. For measurements of koff, 100 nM UTRITCN and 500 nM Cy5p97 were incubated before mixing with 5 μM UN (final dilution 2X). Data were fit to a single exponential association equation to extract koff. For the FRET-based competition binding experiments, final concentrations were 25 nM UTRITCN, 25 nM Cy5p97-A232E, and various amounts of unlabeled p97 protein. Fluorescence intensity was measured on a Synergy Neo2 plate reader (BioTek) using a 540 nm excitation filter and 590 nm emission filter. FRET efficiencies were calculated from a control UTRITCN sample, and percent binding inhibition was calculated relative to a UTRITCN-Cy5p97-A232E sample. Inhibition curves were fit to an IC50 equation, and Ki was calculated using the known KD of UN-binding to p97-A232E.
Protomer exchange assay
A mixture of 250 nM p97-FLAG and 250 nM of Cy5p97-His or 250 nM of Cy5p97-His alone was incubated for 10 minutes at 25 °C with 25 μL anti-FLAG resin (Sigma) in 100 μL exchange buffer (50 mM Tris pH 7.4, 150 mM NaCl, 10 mM MgCl2, 0.01% Triton X-100, 5 mM ATP, 1X ATP regeneration system). Resin was washed three times with 500 μL of exchange buffer without ATP, and resin was boiled in SDS-PAGE sample buffer to elute. Samples were run on a 7% SDS-PAGE gel and imaged on a ChemiDoc (BioRad).
Cryo-EM data analysis
The p97-A232E·UN complex was formed by mixing 5 μM p97-A232E and 7.5 μM UN in assay buffer with 10 mM ATP and 0.05% NP-40, and incubating for 5 mins at 37 °C, before applying it to glow-discharged C-F lat holey carbon grids (CF-2/1–3C-T, EMS). The samples for p97 and p97-A232E in the presence of ATP or ADP were prepared by mixing 1 mM p97 or p97-A232E with 5 mM ATP or ADP, incubating on ice for 5 minutes, and applying the protein to glow-discharged C-Flat holey carbon grids (CF-2/1–3C-T, EMS). Samples were plunge-frozen using a vitrobot (FEI Company) and imaged on either a Titan Krios TEM operated at 300 keV (FEI Company) for the p97-A232EÚN complex or a Talos Arctica TEM operated at 200 keV (FEI Company) for the nucleotide-bound p97 variants. Images were collected as described in Table S1. Dose-fractionated imaging was performed by automated collection methods using SerialEM (Mastronarde, 2005). Whole-frame drift correction was performed via Motioncor2 (Zheng et al., 2017), removing the first 2 frames of the data, with dose weighting applied. Micrographs were CTF corrected using CTFFIND4 (Rohou and Grigorieff, 2015) in Relion (Scheres, 2012).
For p97-A232E·UN data processing, 381,811 single particles were automatically selected from 5898 micrographs using Gautomatch (K. Zhang, MRC LMB (www.mrc-lmb.cam.ac.uk/kzhang)). Initial 2D classification was performed to assess data quality and remove contamination, accounting for 68% of the particles. All 3D classification and refinement steps were performed with Relion (Scheres, 2012). For the p97-A232E·UN data, the classification and refinement scheme and resulting data are depicted (Figure S4). A 3D map of p97-ATP was low pass filtered to 60 Å and used as an initial model for the 3D classification of the remaining 122,626 particles, sorted into 16 classes. The particles from the best three classes were combined, for a final total of 50,035 particles. Both CtfRefinement and Bayesian polishing were applied to this particle set, and the resulting refinement was sharpened using postprocessing (Figure S4)(Scheres, 2012). The final structure was refined to an estimated 4.26 Å (Figure S4).
For the nucleotide-bound p97 datasets, single particles were automatically selected using Relion Laplacian autopicker (Scheres, 2012). 2D classification was performed in Relion and contamination was removed. Back projections were generated for the 3D models of p97-ATPyS (EMD:3297) and p97-ADP (EMD:3299) (Banerjee et al., 2016), using e2project3d.py from EMAN2 (Tang et al., 2007). 2D averages were matched to back projections of both ATPyS- and ADP-bound p97 using SPIDER (Shaikh et al., 2008) to determine the correct NTD assignment. Averages lacking strong NTD density were classified as ‘mobile.’
Quantification and statistical analysis
Statistical details of experiments can be found in figure legends. Data were fit as described in Prism (GraphPad).
Data and code availability
The Cryo-EM map for the p97-A232E mutant bound to cofactors UFD1L and NPLOC4 has been deposited to EMDB with accession code EMD-20730.
Supplementary Material
Highlights.
p97 mutants exhibit accelerated NPLOC4·UFD1L (UN)-dependent substrate unfolding
Mutations increase UN affinity but do not affect ATPase activity during unfolding
p97 mutant N-terminal domain conformations are decoupled from nucleotide state
Data support a gain-of-function model for p97·UN in multisystem proteinopathy
Acknowledgements
We thank Rebecca Voorhees for advice on mammalian protein purification; Xiawei Huang for help with cloning; and the Deshaies and Martin labs for helpful discussion. A.M. is an HHMI investigator, R.J.D. was an HHMI investigator, and this work was funded by HHMI, the National Institutes of Health (R01-GM094497 to A.M.), and a gift from Amgen to Caltech. S.N.G. is a Howard Hughes Medical Institute Fellow of the Damon Runyon Cancer Research Foundation, DRG-2342–18.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
E.E.B., S.N.G. and A.M. declare no competing interests. R.J.D. is currently Senior Vice President of Global Research at Amgen.
References
- Al-Obeidi E, Al-Tahan S, Surampalli A, Goyal N, Wang AK, Hermann A, Omizo M, Smith C, Mozaffar T, and Kimonis V (2018). Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin. Genet 93, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, et al. (2015). Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 28, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhzaouy K, Strucksberg K-H, Tung SM, Tangavelou K, Stumpf M, Faix J, Schröder R, Clemen CS, and Eichinger L (2012). Heteromeric p97/p97R155C Complexes Induce Dominant Negative Changes in Wild-Type and Autophagy 9-Deficient Dictyostelium strains. PLoS ONE 7, e46879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. (2016). 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe EE, Olson KC, Chau V, and Deshaies RJ (2017). Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci 114, E4380–E4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar NO, and Rapoport TA (2017). Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 169, 722–735.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar NO, Kim KH, Ji Z, Wales TE, Svetlov V, Nudler E, Engen JR, Walz T, and Rapoport TA (2018). Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1–Npl4. Nat. Struct. Mol. Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, and Meyer H (2018). VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 69, 182–194. [DOI] [PubMed] [Google Scholar]
- Bulfer SL, Chou T-F, and Arkin MR (2016). p97 Disease Mutations Modulate Nucleotide-Induced Conformation to Alter Protein–Protein Interactions. ACS Chem. Biol 11, 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia WS, Chia DX, Rao F, Bar Nun S, and Geifman Shochat S (2012). ATP Binding to p97/VCP D1 Domain Regulates Selective Recruitment of Adaptors to Its Proximal N-Domain. PLoS ONE 7, e50490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching JK, Elizabeth SV, Ju J-S, Lusk C, Pittman SK, and Weihl CC (2013). mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum. Mol. Genet 22, 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T-F, Bulfer SL, Weihl CC, Li K, Lis LG, Walters MA, Schoenen FJ, Lin HJ, Deshaies RJ, and Arkin MR (2014). Specific Inhibition of p97/VCP ATPase and Kinetic Analysis Demonstrate Interaction between D1 and D2 ATPase Domains. J. Mol. Biol 426, 2886–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney I, Han H, Stewart MG, Carson RH, Hansen DT, Iwasa JH, Price JC, Hill CP, and Shen PS (2019). Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science eaax0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer SK, Neumann M, Lu H, Wright AC, and Taylor JP (2010). Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet 19, 1741–1755. [DOI] [PubMed] [Google Scholar]
- Dai R-M, Chen E, Longo DL, Gorbea CM, and Li C-CH (1998). Involvement of Valosin-containing Protein, an ATPase Co-purified with IkBa and 26 S Proteasome, in Ubiquitin-Proteasome-mediated Degradation of IkBa. J. Biol. Chem 273, 3562–3573. [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, and Brunger AT (2003). Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat. Struct. Mol. Biol 10, 856–863. [DOI] [PubMed] [Google Scholar]
- Dong KC, Helgason E, Yu C, Phu L, Arnott DP, Bosanac I, Compaan DM, Huang OW, Fedorova AV, Kirkpatrick DS, et al. (2011). Preparation of Distinct Ubiquitin Chain Reagents of High Purity and Yield. Structure 19, 1053–1063. [DOI] [PubMed] [Google Scholar]
- Ewens CA, Kloppsteck P, Förster A, Zhang X, and Freemont PS (2010). Structural and functional implications of phosphorylation and acetylation in the regulation of the AAA+ protein p97. Biochem. Cell Biol 88, 41–48. [DOI] [PubMed] [Google Scholar]
- Fernández-Sáiz V, and Buchberger A (2010). Imbalances in p97 co-factor interactions in human proteinopathy. EMBO Rep 11, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillen CM, and Forbush B (1999). Functional interaction of the K-Cl cotransporter (KCC1) with the Na-K-Cl cotransporter in HEK-293 cells. Am. J. Physiol.-Cell Physiol 276, C328–C336. [DOI] [PubMed] [Google Scholar]
- Halawani D, LeBlanc AC, Rouiller I, Michnick SW, Servant MJ, and Latterich M (2009). Hereditary Inclusion Body Myopathy-Linked p97/VCP Mutations in the NH2 Domain and the D1 Ring Modulate p97/VCP ATPase Activity and D2 Ring Conformation. Mol. Cell. Biol 29, 4484–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann P, and Schindelin H (2011). The Structural and Functional Basis of the p97/Valosin-containing Protein (VCP)-interacting Motif (VIM): mutually exclusive binding of cofactors to the N-terminal domain of p97. J. Biol. Chem 286, 38679–38690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann P, and Schindelin H (2016). Characterization of an Additional Binding Surface on the p97 N-Terminal Domain Involved in Bipartite Cofactor Interactions. Structure 24, 140–147. [DOI] [PubMed] [Google Scholar]
- Hänzelmann P, and Schindelin H (2017). The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97. Front. Mol. Biosci 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann P, Buchberger A, and Schindelin H (2011). Hierarchical Binding of Cofactors to the AAA ATPase p97. Structure 19, 833–843. [DOI] [PubMed] [Google Scholar]
- Huang R, Ripstein ZA, Rubinstein JL, and Kay LE (2019). Cooperative subunit dynamics modulate p97 function. Proc. Natl. Acad. Sci 116, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson RL, Pye VE, Simpson P, Meyer HH, Zhang X, Freemont PS, and Matthews S (2007). Detailed Structural Insights into the p97-Npl4-Ufd1 Interface. J. Biol. Chem 282, 21361–21369. [DOI] [PubMed] [Google Scholar]
- Ju JS, and Weihl CC (2010). Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: a disorder of autophagy. Hum. Mol. Genet 19, R38–R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J-S, Fuentealba RA, Miller SE, Jackson E, Piwnica-Worms D, Baloh RH, and Weihl CC (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol 187, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Fukushi J, Hori S, Matsuda N, Okatsu K, Kakiyama Y, Kawawaki J, Kakizuka A, and Tanaka K (2013). Different dynamic movements of wild-type and pathogenic VCPs and their cofactors to damaged mitochondria in a Parkin-mediated mitochondrial quality control system. Genes Cells 18, 1131–1143. [DOI] [PubMed] [Google Scholar]
- Kirchner P, Bug M, and Meyer H (2013). Ubiquitination of the N-terminal Region of Caveolin-1 Regulates Endosomal Sorting by the VCP/p97 AAA-ATPase. J. Biol. Chem 288, 7363–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le LTM, Kang W, Kim J-Y, Le OTT, Lee SY, and Yang JK (2016). Structural Details of Ufd1 Binding to p97 and Their Functional Implications in ER-Associated Degradation. PLOS ONE 11, e0163394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J-M, Wu H, Zhang W, Blackburn MR, and Jin J (2014). The p97-UFD1L-NPL4 Protein Complex Mediates Cytokine-Induced IkappaBalpha Proteolysis. Mol. Cell. Biol 34, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno A, Noguchi M, Fukushi J, Motohashi Y, and Kakizuka A (2010). Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Genes Cells 911–922. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, and Warren G (2000). A complex of mammalian Ufd1 and Npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J 19, 2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian A, Donkervoort S, Dec E, Badadani M, Katheria V, Rana P, Nguyen C, Mukherjee J, Caiozzo V, Martin B, et al. (2011). The Multiple Faces of Valosin-Containing Protein-Associated Diseases: Inclusion Body Myopathy with Paget’s Disease of Bone, Frontotemporal Dementia, and Amyotrophic Lateral Sclerosis. J. Mol. Neurosci 45, 522–531. [DOI] [PubMed] [Google Scholar]
- Niwa H, Ewens CA, Tsang C, Yeung HO, Zhang X, and Freemont PS (2012). The Role of the N-Domain in the ATPase Activity of the Mammalian AAA ATPase p97/VCP. J. Biol. Chem 287, 8561–8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski MM, Williams C, Dong KC, and Martin A (2019). The Cdc48 unfoldase prepares well-folded protein substrates for degradation by the 26S proteasome. Commun. Biol 2, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, et al. (2017). VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36, 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MV, Williams DR, Cocklin S, and Loll PJ (2017). Interaction between the AAA+ ATPase p97 and its cofactor ataxin3 in health and disease: Nucleotide-induced conformational changes regulate cofactor binding. J. Biol. Chem 292, 18392–18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma JM, Liu X, Reichermeier KM, Moradian A, Sweredoski MJ, Hess S, and Deshaies RJ (2017). Composition and Regulation of the Cellular Repertoire of SCF Ubiquitin Ligases. Cell 171, 1326–1339.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, Tang W, Winton MJ, Neumann M, Trojanowski JQ, et al. (2010). TDP-43 Mediates Degeneration in a Novel Drosophila Model of Disease Caused by Mutations in VCP/p97. J. Neurosci 30, 7729–7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz D, Vuk M, Kirchner P, Bug M, Schütz S, Hayer A, Bremer S, Lusk C, Baloh RH, Lee H, et al. (2011). Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat. Cell Biol 13, 1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saracino D, Clot F, Camuzat A, Anquetil V, Hannequin D, Guyant-Maréchal L, Didic M, Guillot-Noël L, Rinaldi D, Latouche M, et al. (2018). Novel VCP mutations expand the mutational spectrum of frontotemporal dementia. Neurobiol. Aging [DOI] [PubMed] [Google Scholar]
- Scheres SHW (2012). RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz AK, and Kay LE (2016). A Dynamic molecular basis for malfunction in disease mutants of p97/VCP. ELife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz AK, Rennella E, and Kay LE (2017). Exploiting conformational plasticity in the AAA+ protein VCP/p97 to modify function. Proc. Natl. Acad. Sci 114, E6822–E6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TR, Gao H, Baxter WT, Asturias FJ, Boisset N, Leith A, and Frank J (2008). SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc 3, 1941–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WK, and Xia D (2013). Altered Intersubunit Communication Is the Molecular Basis for Functional Defects of Pathogenic p97 Mutants. J. Biol. Chem 288, 36624–36635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WK, and Xia D (2016). Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front. Mol. Biosci 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Tang WK, Li D, Li C, Esser L, Dai R, Guo L, and Xia D (2010). A novel ATP-dependent conformation in p97 N-D1 fragment revealed by crystal structures of disease-related mutants. EMBO J 29, 2217–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WK, Zhang T, Ye Y, and Xia D (2017). Structural basis for nucleotide-modulated p97 association with the ER membrane. Cell Discov 3, 17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile CS, Witte MD, Blom AEM, Kundrat L, Ploegh HL, and Guimaraes CP (2013). Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc 8, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Ji Z, Wales TE, Bodnar NO, Ficarro SB, Marto JA, Engen JR, and Rapoport TA (2019). Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science eaax1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, and Kimonis VE (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet 36, 377–381. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Dalal S, Pestronk A, and Hanson PI (2006). Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet 15, 189–199. [DOI] [PubMed] [Google Scholar]
- Weith M, Seiler J, van den Boom J, Kracht M, Hülsmann J, Primorac I, del Pino Garcia J, Kaschani F, Kaiser M, Musacchio A, et al. (2018). Ubiquitin-Independent Disassembly by a p97 AAA-ATPase Complex Drives PP1 Holoenzyme Formation. Mol. Cell 72, 766–777.e6. [DOI] [PubMed] [Google Scholar]
- Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Rocker C, Salih A, Spindler K-D, and Nienhaus GU (2004). EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci 101, 15905–15910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Blythe EE, Freiberger EC, Mamrosh JL, Hebert AS, Reitsma JM, Hess S, Coon JJ, and Deshaies RJ (2016). Valosin-containing protein (VCP)–Adaptor Interactions are Exceptionally Dynamic and Subject to Differential Modulation by a VCP Inhibitor. Mol. Cell. Proteomics 15, 2970–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y (2003). Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol 162, 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, et al. (2012). Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat. Methods 9, 727–729. [DOI] [PubMed] [Google Scholar]
- Zhang T, Mishra P, Hay BA, Chan D, and Guo M (2017). Valosin-containing protein (VCP/p97) inhibitors relieve Mitofusin-dependent mitochondrial defects due to VCP disease mutants. ELife 6, e17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gui L, Zhang X, Bulfer SL, Sanghez V, Wong DE, Lee Y, Lehmann L, Lee JS, Shih P-Y, et al. (2015). Altered cofactor regulation with disease-associated p97/VCP mutations. Proc. Natl. Acad. Sci 112, E1705–E1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Cryo-EM map for the p97-A232E mutant bound to cofactors UFD1L and NPLOC4 has been deposited to EMDB with accession code EMD-20730.