

SUMMARY
Although viral infections elicit robust interferon-γ (IFNγ) and long-lived antibody secreting cell (ASC) responses, the roles for IFNγ and IFNγ-induced transcription factors (TFs) in ASC development are unclear. We showed that B cell intrinsic expression of IFNγR and the IFNγ-induced TF T-bet were required for T-helper 1 cell induced differentiation of B cells into ASCs. IFNγR-signaling induced Blimp1 expression in B cells but also initiated an inflammatory gene program that, if not restrained, prevented ASC formation. T-bet did not affect Blimp1 upregulation in IFNγ-activated B cells but instead regulated chromatin accessibility within the Ifng and Ifngr2 loci and repressed the IFNγ-induced inflammatory gene program. Consistent with this, B cell intrinsic T-bet was required for formation of long-lived ASCs and secondary ASCs following viral, but not nematode, infection. Therefore, T-bet facilitates differentiation of IFNγ-activated inflammatory effector B cells into ASCs in the setting of IFNγ, but not IL-4, induced inflammatory responses.
Keywords: T-bet, B cell differentiation, IFNγ, antibody secreting cells, inflammation
INTRODUCTION
The interferon-γ (IFNγ) inducible T-box transcription factor (TF), T-bet, regulates the activation, proliferation, differentiation, lifespan and effector functions of T cells (Lazarevic et al., 2013) by modulating gene expression through its interactions with histone-modifying enzymes and other master regulator TFs, like Bcl6 and Blimp1 (Oestreich and Weinmann, 2012; Xin et al., 2016). T-bet promotes T helper-1 (Th1) development by activating effector cell gene programs (Zhu et al., 2012) and by repressing alternate cell fate (Lazarevic et al., 2013) and type I interferon (IFN)-induced inflammatory gene programs (Iwata et al., 2017). Although T-bet is required for IFNγ-driven switching to the IgG2c (B6) or IgG2a (BALB/c) isotype in mice (Peng et al., 2002), remarkably little is known about whether T-bet can also influence B cell fate decisions. However, recent studies showing that T-bet expressing B cells are expanded in aging, chronically infected and autoimmune mice and humans (Jenks et al., 2018; Karnell et al., 2017; Knox et al., 2017; Lau et al., 2017; Naradikian et al., 2016; Rubtsov et al., 2011; Rubtsova et al., 2017; Wang et al., 2018) suggest that T-bet may influence B cell transcriptional programming and cell fate decisions. In support of this idea, autoantibody (Ab) responses in some Systemic Lupus Erythematosus (SLE) prone mice are dependent on B cell intrinsic T-bet expression (Rubtsova et al., 2017) and expansion of an unusual population of T-bet expressing CD11c+CXCR5neg IgDnegCD27neg (DN2) human B cells correlates with disease severity in a subset of SLE patients (Jenks et al., 2018; Wang et al., 2018). Moreover, T-bet+ DN2 cells (Jenks et al., 2018; Wang et al., 2018) as well as T-bet+ CD27+CD21lo activated human memory B cells (Knox et al., 2017; Lau et al., 2017) exhibit phenotypic, molecular and functional similarities to Ab secreting cell (ASC) precursors.
Given the association between T-bet expression and pre-ASC formation, we tested whether T-bet was required for commitment to the ASC lineage. We showed that ASC development by B cells activated in vitro in the presence of IFNγ-producing T cells required B cell intrinsic expression of T-bet and the IFNγR. T-bet, despite facilitating IFNγ-dependent ASC development, was not required for IFNγ-induced upregulation of ASC programming TFs, like Blimp1, IRF4 and XBP1 (Nutt et al., 2015). Instead, T-bet repressed an IFNγ-induced inflammatory gene program that was incompatible with ASC differentiation. Moreover, we found that B cell intrinsic T-bet expression was required for long-lived ASC formation following primary infection with influenza virus and memory B cell differentiation into ASCs following influenza challenge infection. By contrast, T-bet expressing B cells were not required for ASC differentiation following a Th2-inducing nematode infection. Therefore, unlike the core TFs that are required for ASC commitment in all settings (Nutt et al., 2015), T-bet promotes ASC development by preventing B cells from assuming an alternate inflammatory effector cell fate in response to IFNγ, which is produced in response to some but not all pathogens and autoantigens.
RESULTS
The Blimp1-dependent ASC gene program is enhanced in Th1-activated B cells.
We reported that cultures of Th1 cell-stimulated B cells (Be1 cells) contained more secreted Ab than cultures of Th2 cell-stimulated B cells (Be2 cells) (Harris et al., 2005b). To test whether ASC development was enhanced in the Be1 cultures, we subdivided the day 4 Be1 cells into 4 discrete populations using the ASC markers CD138 and CD93 (Fig. 1A) and measured Ab production by the sort-purified cells. We found that the CD138+CD93+ subset produced the most Ab, as measured by Ab secretory rates, following re-culture of an equivalent number of cells for 5 hours (Fig. 1B), or by ELISPOT (Fig. 1C). Next, we quantitated CD138+CD93+ ASCs and Ab secretory rates in day 4 Be1 and Be2 cultures. CD138+CD93+ cells were more prevalent in day 4 Be1 cultures (Fig. 1D–E) and day 4 Be1 cells produced more Ab than day 4 Be2 cells (Fig. 1F), indicating enhanced ASC formation in Be1 cultures.
Figure 1. ASC development is preferentially initiated in Th1 cell-primed B cells.
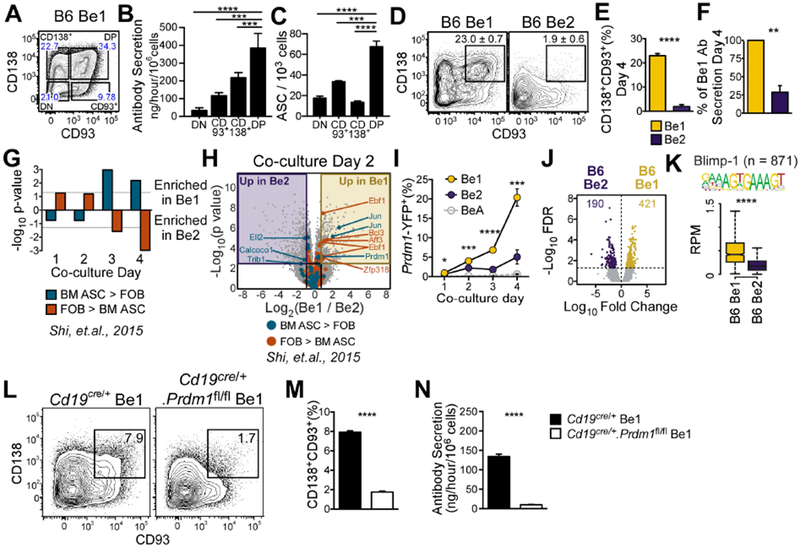
(A-C) Identification of ASCs in sort-purified Be1 cell subsets (A) divided using CD138 and CD93. Ab secretory rates (B) and ELISPOT (C) analyses of each subset.
(D-F) Identification (D) and enumeration (E) of CD138+CD93+ ASCs in Be1 and Be2 cultures. Ab secretory rates (F) of Be1 and Be2 cells shown as % of Be1 Ab secretion.
(G-H) Gene set expression analysis (GSEA) for differentially expressed TF genes in BM ASCs vs FOB cells (Shi et al., 2015) in days 1-4 Be1 and Be2 microarray (MA) data (G). Day 2 volcano plot (H) highlighting TF genes significantly (FDR<0.05, ≥1.75 FC) upregulated in BM ASCs or FOB (Shi et al., 2015).
(I-N) Analysis of Blimp1 in Be1 and Be2 cells.
(I) Enumeration of Blimp-1 reporter (YFP+) expressing Be1, Be2, and control BeA cells generated from Blimp-1 reporter mice by flow. Be1 vs Be2 p values shown.
(J) Volcano plot of day 2 B6 Be1 and Be2 cell ATAC-seq data showing 611 DAR (FDR<0.05).
(K) Chromatin accessibility within 100bp surrounding Blimp-1 binding motifs in Day 2 Be1 and Be2 cells by ATAC-seq. n= number of motif-containing DARs analyzed. p = 3.8×10−90.
(L-N) Identification (L) and quantification (M) of CD138+CD93+ ASCs in day 4 Be1 cultures containing control (Cd19cre/+) or Blimp-1 deficient (Cd19cre/+.Prdm1fl/fl) B cells. Day 4 Ab secretory rates (N).
Data representative of ≥2 independent experiments (A-E, I, L-N), representative pooled data from 4 independent experiments (F), 7 independent experimental samples/timepoint/group (MA) or 3 independent experimental samples/group (ATAC-seq). Data presented as mean + SD of ≥3 experimental replicates (B-C, E, I, M-N); mean ± SEM of 4 independent experiments (F); bar plot of nominal p values (G) or box and whisker plots (showing interquartile range and upper and lower limit) (K). p values determined using one-way ANOVA (B-C) or Student’s t test (E-F, I, K, M-N). See Supplemental STAR Methods for description of Be1 and Be2 cultures, DAR identification, and statistical analyses of GSEA and MA datasets. *p<0.05, **p≤0.01 ***p≤0.001, ****p≤0.0001, “ns” not significant. See also Figure S1 and Supplemental Tables 1–2.
To determine when ASC lineage commitment occurs Be1 cells, we used Gene Set Enrichment analysis (GSEA, (Subramanian et al., 2005)) and evaluated when TFs that are differentially expressed between bone marrow (BM) ASCs and follicular B cells (FOB) (Shi et al., 2015) were enriched in the Be1 transcriptome (Table S1, Fig. S1A). Since many ASC-specific TFs were significantly enriched in the Be1 transcriptome by day 3 (Fig. 1G, Fig. S1B), we examined day 2 cells and found that only two TFs, Prdm1 (Blimp1) and Jun, were upregulated at this timepoint in Be1 cells relative to Be2 cells (Fig. 1H, Fig S1C). Consistent with this, day 2 Be1 cells generated from Blimp1-reporter mice (Rutishauser et al., 2009) expressed detectable amounts of Blimp1 (Fig. 1I). Next, we performed ATAC-seq (Buenrostro et al., 2015) on day 2 Be1 and Be2 cells (Table S2). We identified 611 differentially accessible regions (DAR) (Fig. 1J) and observed significant enrichment (p=3.77 × 10−90) of accessible Blimp1-binding motifs in the day 2 Be1 cells compared to the Be2 cells (Fig. 1K). Finally, we observed few CD138+CD93+ ASCs (Fig. 1L–M) and significantly decreased Ab secretory rates in Be1 cultures containing Prdm1−/− Be1 cells (Fig. 1N). Thus, antigen and Th1 cell activated B cells rapidly upregulate Blimp1, undergo chromatin remodeling at Blimp1 binding sites and differentiate in a Blimp1-dependent fashion into CD138+CD93+ ASCs.
IFNγ controls early Blimp1 expression and ASC development in Be1 cells.
To identify the upstream TFs that might promote early Blimp1 expression in Be1 cells, we examined day 2 Be1 and Be2 ATAC-seq and transcriptome data sets using the PageRank (PR) algorithm (Yu et al., 2017) (Table S3, Fig. S2) and Ingenuity Pathway Analysis (IPA, (Kramer et al., 2014). We also identified the differentially expressed genes (DEG) and DAR in the day 2 datasets. Fourteen of the 357 PR-predicted TFs were also identified in at least 2 of the other 3 analyses and 2 of these TFs, T-bet (Tbx21) and Irf1, were identified in each analysis (Fig. 2A, Table S3). Expression of Tbx21 and Irf1 was induced in Be1 cells within one day (Fig. 2B–C) and by day 2 chromatin accessible regions containing binding motifs for T-bet (Fig. 2D) and IRF1, including the ISRE, EICE and AICE binding sites (Fig. 2E℃G), were significantly enriched in Be1 cells. Expression of Tbx21 and Irf1 was ablated in IFNγR1-deficient (Ifngr1−/−) Be1 cells (Fig. 2H), demonstrating that expression of these TFs was controlled by IFNγ. To test whether Be1 differentiation was dependent on IFNγ signals, we examined Prdm1 expression in Ifngr1−/− Be1 cells. Prdm1 amounts were significantly lower in day 2 Ifngr1−/− Be1 cells compared to day 2 B6 Be1 and Be2 cells (Fig. 2I). Moreover, expression of Prdm1 and other ASC promoting TFs, like Irf4, Pou2af1 and Xbp1, remained low even out to day 4 in Ifngr1−/− Be1 cells (Fig. 2J) and these B cells did not form CD138+CD93+ ASCs or secrete Ab (Fig. 2K–M). Thus, IFNγ controls rapid upregulation of Tbx21, Irf1 and Prdm1 in Be1 cells and is necessary for the development of Be1 ASCs.
Figure 2. IFNγR signals control Be1 differentiation into ASCs.
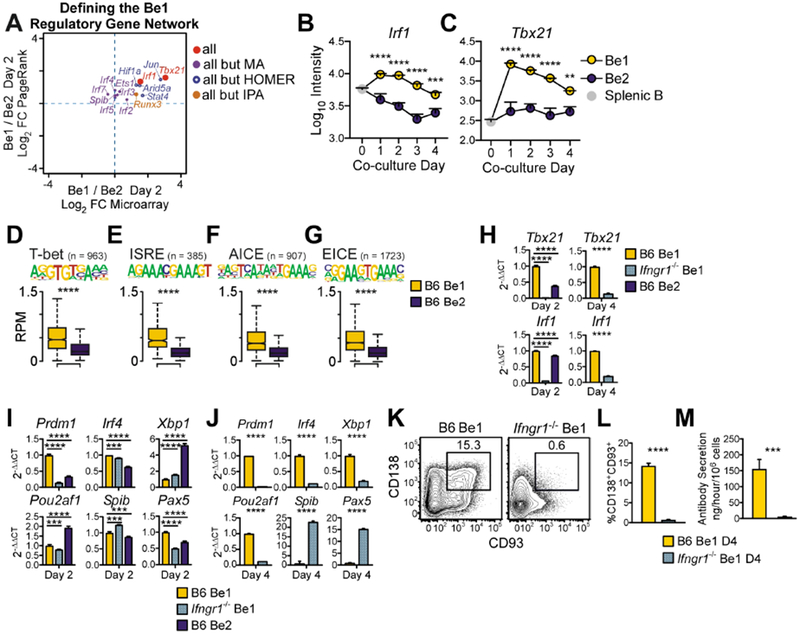
(A) TF regulators of the day 2 Be1 gene network as predicted by HOMER motif, Ingenuity Pathway (IPA) upstream regulator, PageRank (PR) and DEG analyses using day 2 Be1 and Be2 MA and ATAC-seq data. TFs predicted by all 4 analyses or 3 of 4 analyses are shown as MA Log2 FC vs PR Log2 FC.
(B-C) MA expression of Irf1 (B) and Tbx21 (C) by day 0 (splenic B) and day 1-4 Be1 and Be2 cells. Be1 vs Be2 p values shown.
(D-G) Chromatin accessibility within 100bp surrounding T-bet (D) and IRF (E-G) binding motifs in Day 2 Be1 and Be2 cells by ATAC-seq. n= number of motif-containing DAR analyzed. p values 1.6×10−68 (D), 3.1×10−49 (E), 5.9×10−74 (F), and 5×10−137 (G).
(H-J) qPCR analysis (see STAR Methods) of day 2 (H-I) or day 4 (H,J) B6 Be1, Ifngr1−/− Be1 or B6 Be2 cells.
(K-M) Identification (K) and quantification (L) of CD138+CD93+ ASCs in day 4 B6 and Ifngr1−/− Be1 cultures. Day 4 Ab secretory rates (M).
Data in H-M representative of 2 (H-I) or ≥3 (J-M) independent experiments. Shown as the mean + SD of ≥ 3 PCR (H-J) or experimental (L-M) replicates. MA data shown as mean ± SEM (B-C) of 7 experiments. ATAC-seq data shown as box and whisker plots (D-G) of 3 independent experimental samples/group. p values determined by Student’s t test (D-G, J, L-M), one-way ANOVA (H-I), or two-way ANOVA (B-C). **p≤0.01 ***p♤0.001, ****p≤0.0001, “ns” not significant. See also Figure S2 and Supplemental Table 3.
T-bet control IFNγ-dependent ASC development but does not regulate early expression of Blimp1.
Since T-bet is known to regulate Blimp1 expression in T cells (Oestreich et al., 2012; Xin et al., 2016) and Blimp1 was required for Be1 ASC formation, we tested whether Be1 differentiation was T-bet dependent. We found that ASC formation and Ab secretion (Fig. 3A–C) was significantly impaired in Tbx21−/− Be1 cultures. Consistent with this, GSEA using RNA-seq data (Table S4) from day 4 B6 and Tbx21−/− Be1 cells revealed that the transcriptome of Tbx21−/− Be1 cells was not enriched for ASC-specific TFs (Shi et al., 2015) and was instead enriched in FOB TFs (Fig. 3D–E, Fig. S3A). To examine whether T-bet facilitates ASC development by promoting Blimp1 expression or activity, we analyzed quantitative PCR and ATAC-seq (Table S2) data from day 2 B6 and Tbx21−/− Be1 and Be2 effectors. We identified 561 DAR between day 2 B6 Be1 and Tbx21−/− Be1 cells but only 30 DAR between day 2 B6 Be2 and Tbx21−/− Be2 cells (Fig. 3F). In addition, we observed significantly increased chromatin accessibility in the 100 bp immediately surrounding T-bet consensus binding motifs in B6 Be1 cells relative to Tbx21−/− Be1 cells and B6 Be2 cells (Fig. 3G, Fig. S3B). However, chromatin accessibility near Blimp1-binding motifs was unchanged (Fig. 3G, Fig. S3B) and Prdm1 mRNA expression was equivalent (Fig. 3H) between the day 2 B6 and Tbx21−/− Be1 cells. Similarly, expression of other ASC-inducing TFs (Nutt et al., 2015) (Fig. 3H) and chromatin accessibility near binding sites for any of these TFs (Fig. 3G, Fig. S3B) was only modestly affected in the day 2 Tbx21−/− Be1 cells. Thus, T-bet appeared to promote IFNγ-dependent ASC formation via a distinct mechanism.
Figure 3. T-bet controls Be1 differentiation but does not regulate early ASC programming.
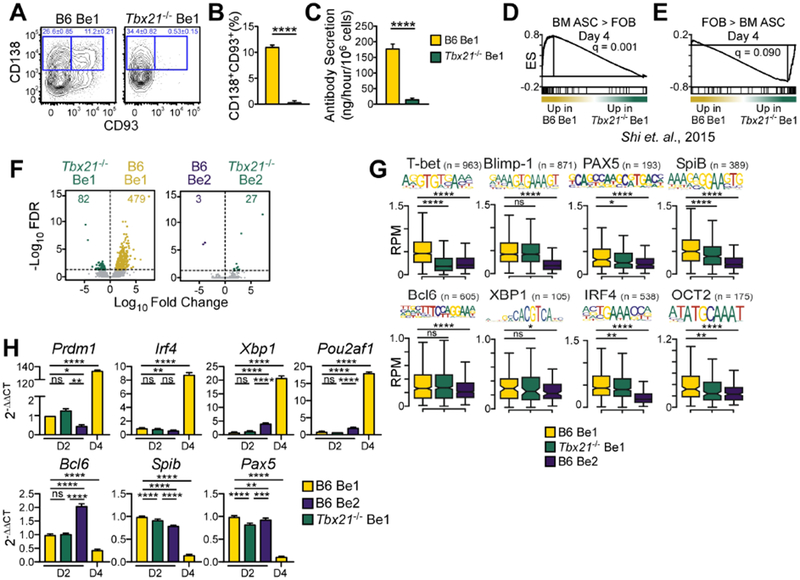
(A-C) Identification (A) and enumeration (B) of CD138+CD93+ ASCs in day 4 B6 and Tbx21−/− Be1 cultures. Day 4 Ab secretory rates (C).
(D-E) GSEA enrichment plots for differentially expressed TF genes in BM ASC (D) vs FOB (E) cells (Shi et al., 2015) in day 4 B6 and Tbx21−/− Be1 RNA-seq data. Enrichment score (ES).
(F) Day 2 ATAC-seq volcano plots showing DAR (FDR <0.05) between B6 Be1 and Tbx21−/− Be1 (561 DAR, left) and B6 Be2 and Tbx21−/− Be2 (30 DAR, right).
(G) Chromatin accessibility within 100bp surrounding the indicated TF binding motifs in 2 B6 Be1, B6 Be2 and Tbx21−/− Be1 cells by ATAC-seq. n= number of motif-containing DARs analyzed. p values provided in Fig. S3B.
(H) qPCR analysis (see STAR Methods) of days 2 or 4 Tbx21−/− Be1, B6 Be1, and B6 Be2 cells.
RNA-seq and ATAC-seq data from 3 independent experiments. Data representative of 2 (H) or ≥3 (A-C) independent experiments. Shown as the mean ± SD of ≥ 3 experimental replicates. Statistical significance determined by Student’s t test (B-C, G), one way-ANOVA (H) or FDR q analysis (D-F). *p<0.05, **p≤0.01, ***p≤0.001 or ****p≤0.0001 “ns” not significant. See also Figure S3 and Supplemental Tables 2 and 4.
Tbx21−/− Be1 cells maintain an activated inflammatory gene signature.
Although T-bet was not required to induce early Prdm1 expression in Be1 cells, we identified >2000 DEG between day 4 Tbx21−/− and B6 Be1 cells (Table S4). To understand which T-bet regulated genes were responsible for ASC development, we first examined the IPA-predicted upstream regulator TFs in B6 Be1 and Be2 cells and Tbx21−/− Be1 cells (Table S5). As expected, IFNγ-induced TFs, like STAT1, T-bet and IRF family members, were predicted to be activated (positive Z-score) by day 1 in Be1 cells compared to Be2 cells (Fig. 4A, Table S5). Moreover, by day 2, downstream inflammatory and anti-viral gene targets of STAT1, IRF3 and IRF7 were more highly expressed in Be1 cells compared to day 2 Be2 cells (Fig. 4B). However, expression of these genes declined by day 4 in Be1 cells and the predicted Z-scores for STAT1, IRF3 and IRF7 shifted from activated to inhibited (negative Z-score) (Fig. 4B). By contrast, the majority of the downstream gene targets of IRF3, IRF7 and STAT1 were more highly expressed in day 4 Tbx21−/− Be1 cells compared to day 4 Be1 cells (Fig. 4B) and IRF3, STAT1 and IRF7, which were the top IPA-identified upstream regulators of the day 4 Tbx21−/− Be1 transcriptome (Fig. 4C, Table S5), were predicted to be activated in these cells (Fig. 4C). The time-dependent downmodulation of the inflammatory gene signature in the B6 Be1 cells was not limited to downstream targets of IRF3, IRF7 and STAT1 as mRNA expression of multiple TFs from the IRF (Fig. 4D), STAT (Fig. 4E) and NF-κB (Fig. 4F) families also declined by day 4 in the B6 Be1 cells. Similarly, mRNA expression of receptors that activate NF-κB and IRF, including members of the TLR (Fig. 4G) and TNFR (Fig. 4H) families, decreased by day 4 in B6 Be1 cells. By contrast, expression of many STAT, IRF and NF-κB TFs (Fig. 4D–F) as well as receptors and ligands that activate NF-κB and IRF signaling, including TLRs (Fig. 4G) and TNF and TNFR family members (Fig. 4H) was increased in day 4 Tbx21−/− Be1 cells relative to B6 Be1 cells. The enhanced inflammatory gene signature observed in the Tbx21−/− Be1 cells was not due to the lack of ASCs in the Tbx21−/− Be1 cultures as we observed similar results comparing the transcriptomes of day 4 Tbx21−/− Be1 cells and the non-ASCs present in the day 4 Be1 cultures (Fig. S4A–G, Table S5). Thus, an inflammatory “effector-like” transcriptional signature, which is transiently observed in B6 Be1 cells, is maintained in Tbx21−/− Be1 cells.
Figure 4. T-bet represses inflammatory gene expression in Be1 cells.
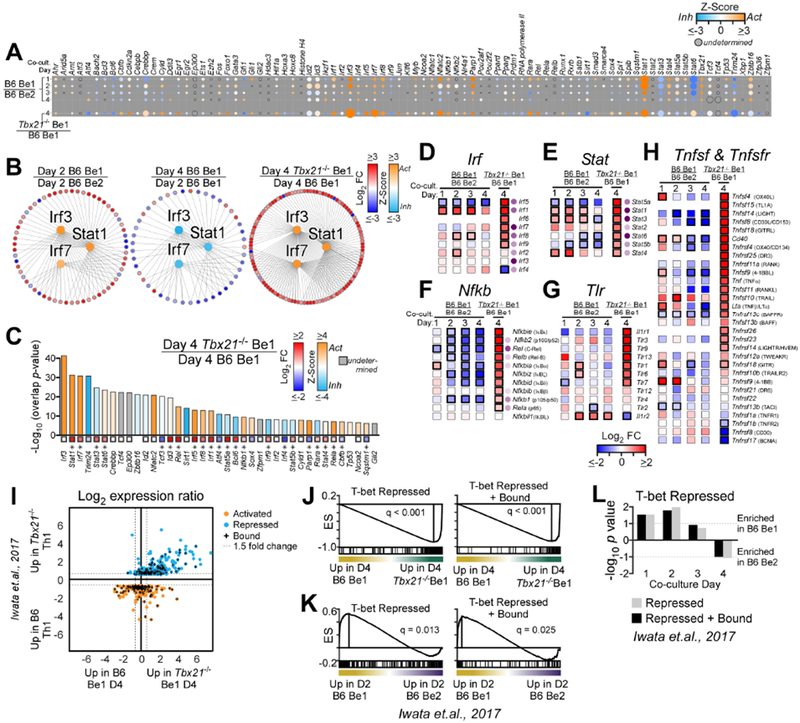
(A) Activation Z-score heatmap for IPA-predicted upstream regulator TFs in B6 Be1 over B6 Be2 (MA) or Tbx21−/− Be1 over B6 Be1 (RNA-seq). Circle size proportional to −log10 overlap p value.
(B) Predicted upstream regulators (center; Irf3, Irf7, Statl, colored by Z-score) and target genes (outer circle, colored by FC) in Day 2 B6 Be1 over Day 2 B6 Be2 (left), Day 4 B6 Be1 over Day 2 B6 Be1 (middle) and Day 4 Tbx21−/− Be1 over Day 4 B6 Be1 (right).
(C) IPA-predicted upstream regulators from day 4 Tbx21−/− Be1 over day 4 B6 Be1, showing activation Z-score (bars) and FC (squares with *=FDR <0.05).
(D-H) mRNA expression heatmaps (squares with black borders =FDR<0.05) of Irf (D), Stat (E), Nfkb (F), Tlr (G), Tnfsf and Tnfsfr (H) family members in B6 Be1 over B6 Be2 (MA) or Tbx21−/− Be1 over B6 Be1 (RNA-seq). Purple circles show −log10 overlap p values from IPA upstream regulator analysis described in (A) with darker shades increasing in significance. Protein names provided in parentheses.
(I) Intersection of T-bet regulated genes in day 3 Th1 cells (Iwata et al., 2017) with genes expressed by day 4 Tbx21−/− Be1 or B6 Be1 cells (RNA-seq data, n=497). Shown as FC expression in day 3 Tbx21−/− Th1 over B6 Th1 cells (Y axis) vs FC in day 4 Tbx21−/− Be1 over B6 Be1 cells (X axis). Activated (orange), repressed (blue) and direct targets of T-bet (“+”) in Th1 cells (Iwata et al., 2017) are indicated.
(J-L) GSEA enrichment plots for T-bet repressed or T-bet repressed+bound gene target in day 4 Th1 cells (Iwata et al., 2017) in day 4 B6 and Tbx21−/− Be1 cells by RNA-seq (J) or in B6 Be1 and B6 Be2 cells by MA. Enrichment score (ES). Bar plot (L) shows GSEA nominal p values.
Analysis includes 3 (RNA-seq) or 7 (MA) samples/group/timepoint. Statistical significance assessed using overlap p value (A-H) or GSEA FDR value analysis (J-L). Genes with an overlap p value <0.05 by IPA (see STAR methods for description) were predicted to be upstream regulators. See also Figure S4 and Supplemental Table 5.
T-bet is a transcriptional repressor in Be1 cells.
Although T-bet is well-characterized as a transcriptional activator of T cell effector development (Zhu et al., 2012) its role as a repressor is less appreciated. However, a recent report (Iwata et al., 2017), showing that commitment to the Th1 cell lineage requires T-bet directed repression of IFNα-driven inflammatory gene expression, suggested that T-bet might promote ASC development by repressing the inflammatory transcriptional program in Be1 cells. We therefore compared our RNA-seq data set with published B6 and Tbx21−/− Th1 cell RNA-seq and ChIP-seq data (Iwata et al., 2017) and found that many of the T-bet repressed target genes Th1 cells were also repressed in a T-bet dependent fashion in Be1 cells (Fig. 4I). Indeed, GSEA revealed that the day 4 Tbx21−/− Be1 transcriptome was significantly enriched relative to day 4 B6 Be1 transcriptome in targets that are normally repressed by T-bet in Th1 cells (Fig. 4J). Moreover, this T-bet dependent repression of inflammatory genes in Be1 cells was time dependent as expression of known targets of T-bet repression declined in Be1 cells between days 1-3 (Fig. 4K–L, Fig. S4H). Thus, deletion of T-bet in Be1 cells resulted in the sustained expression of inflammatory and anti-viral genes, including many already described (Iwata et al., 2017) targets of T-bet repression in T cells.
Sustained TLR and NF-κB signaling in Be1 cultures prevents ASC development.
Tbx21−/− Be1 cells sustained expression of many inflammatory genes and TFs, including NF-κB and NF-κB activators, that are normally suppressed during Be1 differentiation. Since NF-κB can regulate B cell development, activation and ASC formation (Gerondakis and Siebenlist, 2010; Klein and Heise, 2015), we assessed whether sustained NF-κB activation blocked differentiation of IFNγ-activated Be1 cells. We found that maintaining NF-κB activity in Be1 cultures, by adding the NF-κB activator betulinic acid (Kasperczyk et al., 2005) beginning on day 2, significantly suppressed the formation of ASCs and Ig-secreting cells (Fig. 5A–B, Fig. S5A) in the Be1 cultures. Next, we exposed Be1 cells, beginning on day 2, to NF-κB-activating TLR ligands. We found that ASC development declined significantly (Fig. 5C–D, Fig. S5B). This was not due to reduced cell recovery as the Be1 cells proliferated equally well in the TLR7 and TLR9 ligand exposed cultures (data not shown). Thus, sustained TLR and NF-κB signaling is sufficient to suppress Be1 ASC development, suggesting that ASC differentiation is likely to be impaired in B cells that cannot downmodulate the inflammatory gene network that activates NF-κB.
Figure 5. T-bet supports Be1 ASC formation by repressing the IFNγR-regulated inflammatory gene program.
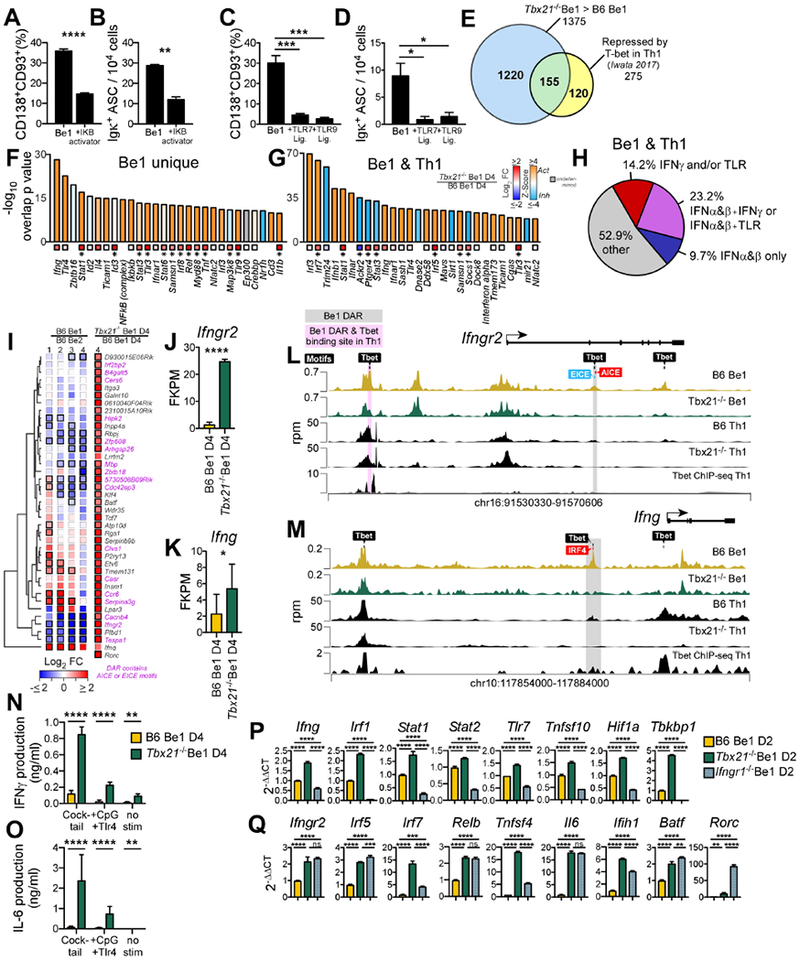
(A-D) NF-κB activator (Betulinic acid, (A-B)), R848 (C-D), CpG (C-D) or Vehicle (A-D) was added to day 2 B6 Be1 cultures. ASCs enumerated by flow (A, C) or ELISPOT (B, D) on day 4.
(E) Venn diagram showing T-bet repressed genes in Th1 cells ((Iwata et al., 2017), n=275) and in Be1 cells (n=1375). Indicated are genes unique to Be1 cells (1220, blue), unique to Th1 cells (120, yellow), or shared (155, green).
(F-G) IPA predicted regulators of T-bet repressed genes identified in (E) that are unique to Be1 cells (F) or shared between Be1 and Th1 cells (G), showing overlap p value (see STAR Methods) and activation Z-score (bars) and FC expression in day 4 Tbx21−/− Be1 over B6 Be1 (squares with *=FDR <0.05,).
(H) IPA analysis of the 155 T-bet repressed genes shared between Be1 and Th1 cells to identify genes that are induced (activated) by IFNα (or IFNβ) ± IFNγ ± TLR signals. Data shown as % of targets that are activated by individual or multiple upstream regulators.
(I) mRNA expression heatmap of T-bet repressed genes, based on FC>2, p< 0.05 in day 4 Tbx21−/− Be1 over B6 Be1 cells by RNA-seq, that also map to T-bet binding motif containing DARs (FDR <0.05 by ATAC-seq in in day 2 B6 and Tbx21−/− Be1 cells, n=40). FC B6 Be1 over B6 Be2 (MA) or Tbx21−/− Be1 over B6 Be1 (RNA-seq) is shown. Bold borders indicate FDR<0.05. Genes with DARs containing AICE or EICE binding motifs noted in pink. Genes (rows) clustered based on Euclidean distance of FC and complete linkage. Rorc was not detected in Be1 or Be2 samples.
(J-K) RNAseq expression (FPKM) for Ifngr2 (J) and Ifng (K) in day 4 Tbx21−/− Be1 and B6 Be1 cells.
(L-M) Chromatin accessibility (rpm, reads per million) by ATAC-seq, within Ifngr2 (L) and Ifng (M) loci in day 2 B6 Be1 (gold) and Tbx21−/− Be1 (green) cells and day 4 B6 and Tbx21−/− Th1 cells (black). Shaded boxes indicate DAR (FDR<0.05) in Be1 only (grey) or Be1 and Th1 cells (pink,(Zhu et al., 2012)). Binding motifs for T-bet, AICE, EICE, and IRF4 are indicated.
(N-O) IFNγ (N) and IL-6 (O) production by Day 4 Tbx21−/− Be1 and B6 Be1 cells before or after stimulation with anti-Ig F(ab’)2+anti-CD40+LPS+CpG (cocktail) or LPS+CpG.
(P-Q) qPCR analysis (described in STAR Methods) of Day 2 Tbx21−/−, Ifngr1−/− and B6 Be1 cells.
Data shown representative of 2 (B, N-O, P-Q) or 3 independent experiments (A, C-D) and reported as mean + SD of 3-4 experimental replicates (A, C), duplicate dilutions (B, D, N-O), or PCR triplicates (P-Q). Analysis includes 3 (RNA-seq) or 7 (MA) samples/group/timepoint. Statistical analysis performed using Student’s t test (A-B, J-K), one-way ANOVA (C-D, P-Q), or two-way ANOVA (N-O). Genes with an overlap p value <0.05 by IPA (see STAR methods for description) were predicted to be upstream regulators. *p<0.05, **p≤0.01, ***p≤0.001 or ****p≤0.0001 or “ns” not significant. See also Figure S5 and Supplemental Tables 6–7.
T-bet regulates IFNγR signaling in Be1 cells by repressing Ifng and Ifngr2 expression.
T-bet facilitates Th1 cell commitment by repressing Ifna expression and preventing initiation of an IFNα-driven autocrine inflammatory loop (Iwata et al., 2017). Since Ifna and the Ifnarwere not overexpressed in Tbx21−/− Be1 cells (data not shown), we hypothesized that T-bet might facilitate ASC commitment in IFNγ-activated B cells by inhibiting expression of inflammatory genes that are normally induced by IRF and NF-κB TFs following activation with IFNγ or intracellular TLR ligands. We therefore performed IPA upstream regulator analysis on the T-bet repressed genes that were either unique to Be1 cells or shared between Be1 and Th1 cells (Iwata et al., 2017) (Fig. 5E, Tables S6–S7). In agreement with our hypothesis, IFNγ and TLRs were predicted by IPA to be the top upstream regulators of the 1220 T-bet repressed genes that were unique to Be1 cells (Fig. 5F, Table S7). Furthermore, many of the genes that were repressed by T-bet in both Th1 and Be1 cells (Fig. 5G, Table S7) were also predicted by IPA to be downstream targets of IFNγ and/or TLR signaling (Fig. 5H, Table S7). Thus, both unique targets of T-bet repression in B cells and shared targets of T-bet repression in Be1 and Th1 cells can be induced by IFNγR and/or TLR signaling.
Next, we determined the overlap between T-bet repressed genes in day 4 Be1 cells (Table S6) with genes containing T-bet dependent DAR in day 2 Be1 cells (Table S2). We identified 40 genes that were repressed in a T-bet-dependent fashion in Be1 cells and mapped to T-bet dependent DAR that also contained one or more consensus T-bet binding motifs (Fig. 5I). The DAR from 17 of these genes contained AICE binding motifs for AP1-IRF complexes or EICE binding sites for Ets-IRF complexes (Fig. 5I), suggesting that many of the T-bet repressed genes in Be1 cells may be co-regulated by IFNγ-induced IRF TFs. Two of the genes identified in this analysis included Ifngr2 and Ifng (Fig. 5I–K). Both loci contained DAR with T-bet binding motifs (Fig. 5L–M) that also co-localized with binding motifs for IRF family members. To address whether these T-bet regulated chromatin accessible regions in the Ifngr2 (Fig. 5L) or the Ifng (Fig. 5M) loci were conserved between Be1 and Th1 cells, we compared the day 2 Be1 cell ATAC-seq data with day 4 Th1 cell ATAC-seq data and published Th1 cell T-bet ChIP-seq data (Zhu et al., 2012). We found DAR that were shared between Be1 and Th1 cells and DAR that were specific to either Be1 or Th1 cells. The Be1 unique DAR in the Ifngr2 locus contained T-bet, EICE and AICE binding motifs while the Be1 unique DAR in the Ifng locus contained IRF4 and T-bet binding motifs (Fig. 5M). Chromatin accessibility in the B cell unique Ifng and Ifngr2 DAR increased in a T-bet dependent fashion (Fig. 5L–M), despite the fact that the genes were downregulated in a T-bet dependent fashion in Be1 cells (Fig. 5J–K), These results suggest that T-bet orchestrates chromatin opening in loci undergoing repression and may repress gene expression either through direct binding or by indirectly facilitating recruitment of other repressors to these loci.
T-bet tunes expression of IFNγR regulated inflammatory genes.
Since T-bet dampened inflammatory gene expression in Be1 cells, we predicted that Tbx21−/− Be1 cells would make enhanced inflammatory responses following activation with ligands of TLR or TNFR family receptors. Consistent with this, significantly more IFNγ (Fig. 5N) and IFNγ-induced cytokines, like IL-6 (Fig. 5O), were produced by day 4 Tbx21−/− Be1 cells stimulated with TLR7 + TLR9 ligands or TLR ligands plus anti-CD40 and anti-Ig. Given this result, we examined whether T-bet prevented establishment of an IFNγR-induced inflammatory feedback loop. We found that inflammatory genes like Ifng, Irf1, Stat1, Tlr7, Tnfsf10 and Hif1a (Fig. 5P) were expressed at significantly higher amounts in day 2 Tbx21−/− Be1 cells and at significantly reduced amounts in day 2 Ifngr1−/− Be1 cells when compared to control day 2 B6 Be1 cells. Therefore, expression of these IFNγ-inducible genes appeared to be restrained by T-bet. However, other genes, like Ifngr2, Irf5, Relb, Il6, Batf and Rorc were more highly expressed in both Ifngr1−/− and Tbx21−/− Be1 cells relative to B6 Be1 cells (Fig. 5Q), suggesting that IFNγR signaling could also repress, in a T-bet dependent fashion, expression of inflammatory genes and alternate fate-specifying TFs in Be1 cells. Collectively, these data show that T-bet does not directly initiate IFNγ-dependent ASC programming. Instead, T-bet blocks inappropriate activation of the IFNγ-induced inflammatory gene program and prevents establishment of alternate effector cell fates in IFNγ-activated B cells.
T-bet+ B cells regulate primary ASC responses to viral infection.
To test whether B cell intrinsic expression of T-bet was required for ASC development in vivo, we first infected ZsGreen (ZsG) T-bet reporter mice (Zhu et al., 2012) with influenza A/Puerto Rico/8/1934 H1N1 (PR8) virus or the parasite Heligmosomoides polygyrus (Hp) and characterized the T-bet expressing B cells. Consistent with our in vitro Be1 and Be2 data (Fig. S6A), ZsG (T-bet) was expressed by LN FOB, ASC and germinal center B cells (GCB) from the flu but not Hp-infected mice (Fig. 6A–B). ZsG was also expressed by flu nucleoprotein (NP) (Allie et al., 2019) specific GCB, ASCs and memory B cells (Fig. 6C–H). Although ZsG could be detected in early NP-specific plasmablasts (Fig. 6C), by day 60 post-infection only a fraction of splenic ASCs (Fig. 6D, Fig. S6B) and few of the bone marrow (BM) long-lived ASCs (LL-ASCs) continued to express ZsG (Fig. 6E, Fig. S6C). By contrast, most flu NP+ memory LN B cells continued to express ZsG (Fig. 6F–G, Fig. S6D) as well as intracellular T-bet protein (Fig. 6H).
Figure 6. T-bet expressing B cells regulate humoral immunity to influenza.
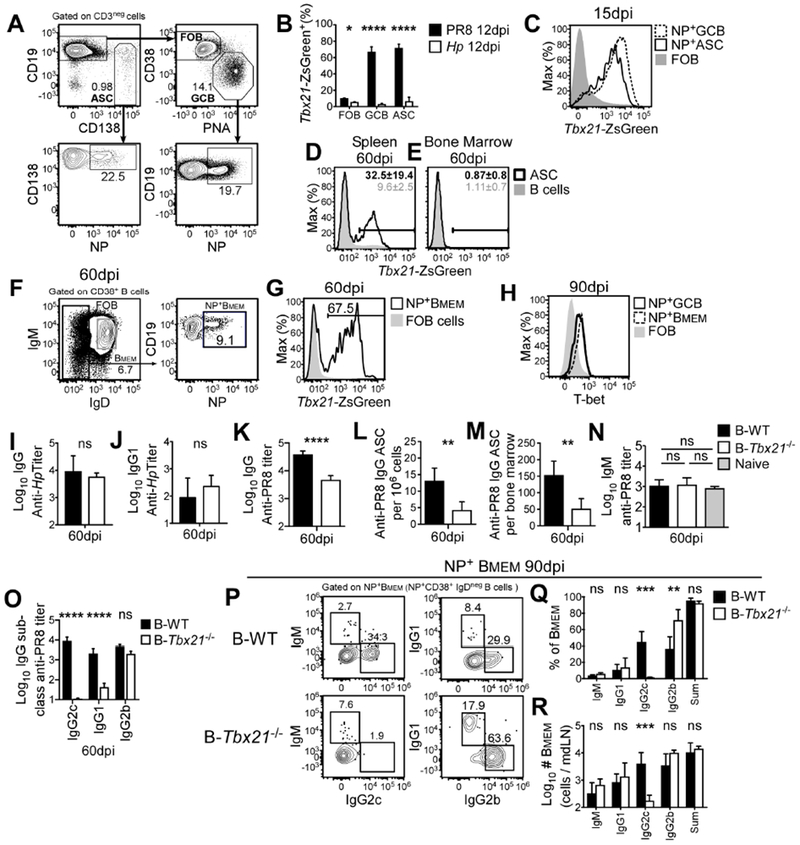
Tbx21 reporter (A-G), B6 (H) and BM chimeric mice, generated with 80% μMT BM + 20% B6 BM (B-WT) or 80% μMT BM + 20% Tbx21−/−BM (B-Tbx21−/−) (I-R), were infected with PR8 influenza virus (A-H, K-R) or Hp (B, I-J).
(A-H) Gating strategy (A) for ASC, FOB, and GCB cells (top) and for flu nucleoprotein (NP) specific ASC and GCB (bottom) in mdLN 15 days post-PR8 infection. Frequencies (B) of T-bet reporter (ZsG+) ASC, GCB and FOB B cells from draining LN (gated as in A) day 12 post-PR8 or Hp infection. T-bet reporter expression (C) by mdLN NP+ ASC and NP+ GCB (gated as in A) day 15 day post-PR8 infection. T-bet reporter expression day 60 post-PR8 infection in splenic (D) and BM (E) ASCs and mdLN NP-specific switched memory B cells (F-G). Intracellular staining day 90 post-PR8 infection for T-bet (H) in mdLN NP+ GCB, NP+ switched memory B cells and FOB cells.
(I-O). Titers of Hp-specific IgG (I) Hp-specific IgG1 (J) and PR8-specific IgG ((K), all subclasses) in B-WT and B-Tbx21−/− mice 60 days post-Hp (I-J) or PR8 (K) infection. Frequency (L) and number (M) of PR8-specific BM IgG+ ASCs (ELISPOT) and titers of NP+ IgM (N), IgG2c, IgG1, and IgG2b (O) in B-WT and B-Tbx21−/− mice 60 days after infection.
(P-R) Gating strategy (P) to identify Ig isotype of NP+ memory B cells, gated on NP+ CD38+IgDneg B cells. Frequency (Q) and number (R) of NP+ mdLN memory B cells from B-WT and B-Tbx21−/− chimeras day 90 post-infection.
Data representative of 2 (C-G, I-J, P-R) or ≥ 3 (A-B, H, L-N) independent experiments with 3-5 (A-J, L-M, P-R), or 7-10 (N) mice/group and reported as mean ± SD. Data (K, O) pooled from 5 independent experiments and shown as or mean ± SEM of 26-29 mice/group. p values determined using Student’s t test, *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001 or “ns” not significant. See also Figure S6.
To address whether T-bet expression by B cells was required for ASC development in vivo, we generated BM chimeras that selectively lacked T-bet in all B cells (B-Tbx21−/− mice) or were T-bet sufficient in all lineages (B-WT mice) (Fig. S6E) and measured day 60 Hp- and flu-specific responses. Although Hp-specific IgG and IgG1 (Fig. 6I–J) responses were similar in both groups, flu-specific IgG Ab (Fig. 6K) and BM ASC (Fig. 6L–M) responses were significantly decreased in the B-Tbx21−/− chimeras. Thus, T-bet expressing B cells facilitate LL-ASC and Ab responses to an IFNγ-inducing viral infection but not to an IL-4-dominated nematode infection.
Although flu-specific Ab and ASC responses were significantly decreased in B-Tbx21−/− mice, humoral immunity was not completely ablated in these mice. Since B cell intrinsic T-bet is required for isotype-switch to IgG2a (Peng et al., 2002), we assessed the isotype distribution of the NP-specific Ab and memory B cell responses to flu to determine whether the loss of IgG2c B cells was sufficient to account for the decline in flu-specific Abs in B-Tbx21−/− mice. As expected (Barnett et al., 2016; Baumgarth et al., 1999; Peng et al., 2002; Wang et al., 2012), the flu-specific IgM Ab response was short-lived in both groups of mice (Fig. 6N). Consistent with a requirement for T-bet in switching to IgG2c, NP-specific IgG2c Abs (Fig. 6O) and day 60 IgG2c+ NP+ memory B cells (Fig. S6F, Fig. 6P–R) were missing from B-Tbx21−/− mice. By contrast, both the NP-specific IgG2b Ab (Fig. 6O) and the NP+ IgG2b+ memory B cell (Fig. 6P–R) responses were intact in the B-Tbx21−/− mice. However, despite normal frequencies and numbers of IgG1+ flu NP+ memory B cells in B-Tbx21−/− mice (Fig. 6P–R), the NP-specific IgG1 Ab response was significantly decreased in B-Tbx21−/− mice (Fig. 6O). These data therefore indicate that T-bet not only regulates switching to IgG2c following flu infection but also directs the formation of IgG1 long-lived flu-specific Ab responses.
ASC recall responses to flu require T-bet+ memory B cells.
Although the total number of flu NP+ memory B cells was not altered in B-Tbx2T−/− mice (Fig. 6R), T-bet was expressed by many memory B cells (Fig. 6G–H). To address whether T-bet+ memory B cells contributed to secondary ASC formation, we infected Tbx21fl/fl.hCD20-TAM-cre mice (Fig. 7A, Fig. S7A) and control B6 mice with PR8 virus. On day 90 post-infection, we exposed the Tbx21fl/fl.hCD20-TAM-cre mice and B6 controls to tamoxifen (TAM) to selectively and inducibly (Khalil et al., 2012) delete Tbx21 from B cells in the flu memory Tbx21fl/fl.hCD20-TAM-cre mice (Fig. 7A, Fig. S7A). Eight days after the last TAM treatment, we examined expression of CXCR3 (Fig. 7B), a known T-bet target gene (Zhu et al., 2012), in B and T lineage cells. TAM treatment of memory Tbx21fl/fl.hCD20-TAM-cre mice did not affect CXCR3 expression by T cells (Fig. 7C) but did cause significant reductions in the frequencies of CXCR3+ B cells and CXCR3+NP+ memory B cells (Fig. 7D–E, Fig. S7B). However, inducible deletion of T-bet in the CD20+ compartment did not affect the number of total LN cells, B cells or T cells (data not shown) or the number of LN NP+ memory B cells (Fig. 7F, Fig. S7B). Therefore, continued expression of T-bet by memory B cells is not required for short-term maintenance of the flu NP+ memory B cell pool.
Figure 7. ASC development after flu challenge infection requires T-bet+ memory B cells.
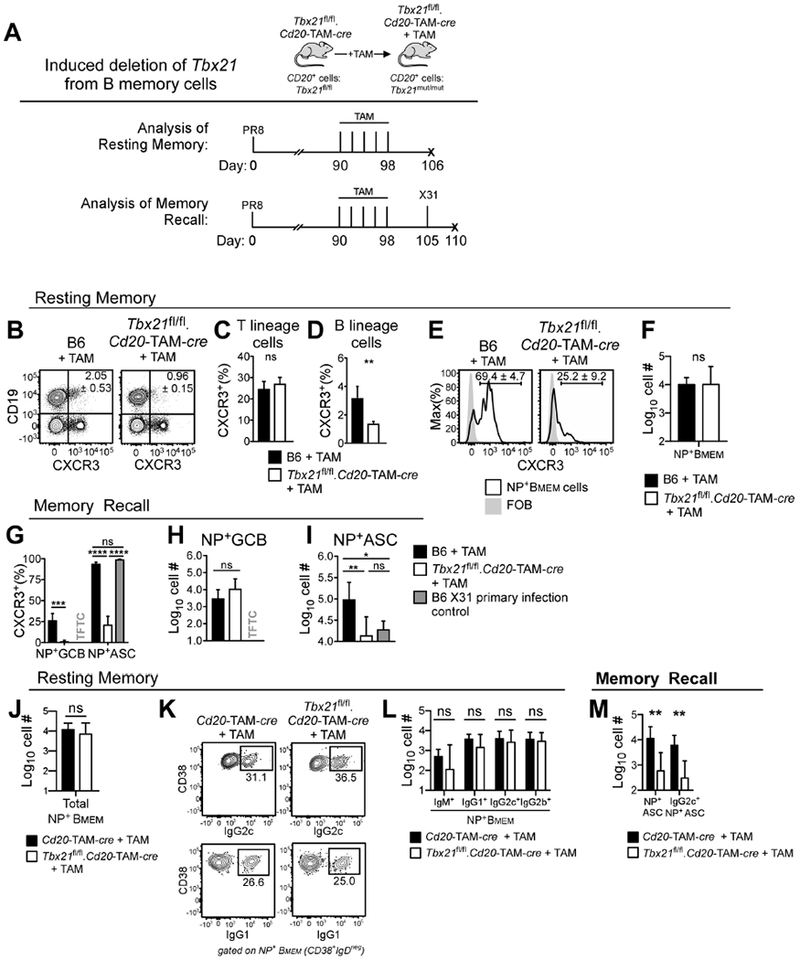
Experimental design (A) showing tamoxifen (TAM) treatment of day 90 PR8 flu memory Tbx21fl/fl.hCD20-TAM-cre (B-M), hCD20-TAM-cre (J-M) and B6 (B-I) mice in order to inducibly delete T-bet from B cells in Tbx21fl/fl.hCD20-TAM-cre mice. Mice analyzed (resting memory, B-F, J-L) 8 days following last TAM treatment or challenged with X31 influenza and analyzed 5 days later (memory recall, G-I, M).
(B-E) Enumeration of CXCR3+ mdLN cells following TAM treatment, showing flow plots (B, E) and frequency of CXCR3+ T cells (C), B cells (D) and NP+CD38+IgDneg memory B cells (Bmem).
(F) Enumeration of NP-specific Bmem cells following TAM-treatment.
(G-I) Enumeration of mdLN CXCR3+ NP+ GCB and ASCs (G) and NP+ GCB (H) and ASCs (I) in day 5 primary X31 infected B6 mice and X31-challenged, TAM-treated B6 and Tbx21fl/fl.hCD20-TAM-cre flu memory mice.
(J-L) Enumeration of mdLN NP+ memory (NP+CD38+IgDneg) B cells (J) 8 days post-TAM treatment of Tbx21fl/fl.hCD20-TAM-cre and hCD20-TAM-cre mice. Identification (K) and enumeration (L) of IgM, IgG 1, IgG2c and IgG2b expressing NP+ Bmem cells.
(M) Enumeration of total and IgG2c+ NP+ mdLN ASCs in TAM-treated memory Tbx21fl/fl .hCD20-TAM-cre and hCD20-TAM-cre mice 5 days after X31 challenge.
Representative data from one of 2 (K-M) or 3 (B-J) independent experiments shown as the mean + SD of 3-6 mice/group. p values determined using one-way ANOVA (G (for NP+ ASCs), I) or Student’s t test (all others). *p<0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001 or “ns” not significant. “TFTC” too few to count (<10 cells /sample). See also Figure S7.
Next, we infected naïve B6 mice (“X31 primary” mice) and the TAM-treated B6 and Tbx21fl/fl.hCD20-TAM-cre PR8 flu memory mice with X31 (H3N2) influenza (Fig. 7A, Fig. S7A). Since the H1-specific Abs generated during the primary PR8 infection do not neutralize the H3 X31 virus, we were able to productively infect the PR8 memory mice and follow the response to NP, which is conserved between both viruses (Kees and Krammer, 1984). Similar to our prior experiment, T-bet was efficiently and specifically deleted in B cells from the TAM-treated Tbx21fl/fl.hCD20-TAM-cre mice as measured by decreased CXCR3 expression by total B cells (Fig. S7C–D) and NP+ GCB and ASCs (Fig. 7G, Fig. S7F–G) on day 5 post-X31 challenge. However, CXCR3 continued to be expressed by T lineage cells (Fig. S7E). NP+ GCB cells, while not yet detectable in the X31 primary infected mice (Fig. 7H), were present in equal numbers in LNs from X31-challenged TAM-treated B6 and Tbx21fl/fl.hCD20-TAM-cre mice (Fig. 7H, Fig. S7F). However, the number of NP+ ASCs in the challenged memory Tbx21fl/fl.hCD20-TAM-cre mice was decreased 10-fold compared to the X31-challenged PR8 memory B6 animals (Fig. 7I, Fig. S7G). Thus, T-bet expression by memory B cells regulates the differentiation of reactivated flu-specific memory B cells.
Secondary flu-specific IgG2c ASC responses require T-bet+ memory B cells.
Since our data showed that T-bet was not required for the maintenance of the memory B cell pool (Fig. 7F), we used the Tbx21−/−.hCD20-TAM-cre mice to address whether T-bet regulates the differentiation of flu NP+ memory B cells into IgG2c-producing ASCs. Consistent with our earlier results, TAM treatment of day 90 PR8 memory hCD20-TAM-cre controls and Tbx21fl/fl.hCD20-TAM-cre mice had no impact on the number of flu NP+ memory B cells (Fig. 7J, Fig. S7H). This was true whether we looked at the NP+ IgM-expressing IgDnegCD38+ memory B cells or the IgG1 and IgG2c NP+ memory B cells (Fig. 7K–L, Fig. S7H). Next, we challenged the mice with heterologous X31 virus and measured the ASC response five days post-challenge. Again, we observed a significant reduction in the number of NP+ ASCs in the Tbx21fl/fl.hCD20-TAM-cre mice compared to control animals (Fig. 7M, Fig. S7I). Moreover, IgG2c NP+ ASCs, which were easily detected in the challenged control group, were significantly decreased in the TAM-treated Tbx21fl/fl hCD20-TAM-cre mice (Fig. 7M, Fig. S7I). Therefore, T-bet controls the differentiation of flu-specific memory B cells into IgG2c+ ASCs. Taken together, these data support the conclusion that T-bet expression by B cells does facilitate the development of ASC and Ab responses following primary and secondary flu infections. However, T-bet, unlike Blimp1 or IRF4 (Nutt et al., 2015), is not a master regulator of ASC development since some types of ASC responses remain intact in the absence of T-bet-expressing B cells. The importance of the T-bet controlled ASC developmental pathways in health and disease is discussed.
Discussion
In IFNγ-activated T cells, T-bet regulates cell fate decisions by activating lineage-specific programs and repressing alternate fates and inflammatory feedback loops (Iwata et al., 2017; Lazarevic et al., 2013). Although T-bet is expressed by human DN2 and memory pre-ASCs (Jenks et al., 2018; Lau et al., 2017; Wang et al., 2018) and T-bet expressing B cells are required for autoAb responses in some autoimmune mice (Peng et al., 2002; Rubtsova et al., 2017) the role for T-bet in ASC lineage commitment had not been well-studied. We previously reported that cognate encounters between antigen-presenting B cells and IFNγ-producing Th1 cells results in rapid upregulation of T-bet (Harris et al., 2005a) and robust Ab production (Harris et al., 2005b). This result was initially quite puzzling as at that time Th2 cytokines were thought to promote B cell differentiation (Randolph et al., 1999) and IFNγ was thought to induce B cell apoptosis (Bernabei et al., 2001). However, more recent publications revealing that IFNγR, STAT1 and T-bet expressing B cells are required for autoAb responses in some mouse models of autoimmunity (Domeier et al., 2016; Jackson et al., 2016; Rubtsova et al., 2017; Thibault et al., 2008), suggested that IFNγ signaling might also be important for ASC development. Our data showing that IFNγ-producing T cells not only induced IFNγ- and T-bet-dependent B cell differentiation but were even more effective in promoting ASC development that IL-4 producing T cells demonstrated that potent B cell fate cues can be provided by an inflammatory cytokine that is often associated with viral infection and autoimmunity.
Although we fully expected to find that T-bet induced B cell differentiation by promoting TFs that initiate ASC commitment, we realized that T-bet did not regulate the early expression or activity of any of the well-described ASC-associated TFs (Nutt et al., 2015) including Blimp1, which can be modulated in a T-bet dependent fashion in T cells (Oestreich et al., 2012; Xin et al., 2016). Likewise, T-bet did not function to repress TFs like Pax5 that maintain B cell identity and prevent ASC differentiation (Nutt et al., 2015). Instead, we found that IFNγ, rather than T-bet, was responsible for early induction of Prdm1 and that T-bet functioned to repress anti-viral and inflammatory genes that are known downstream targets of type I and type II IFN signaling (Pollard et al., 2013). These results were similar to data (Iwata et al., 2017) showing that T-bet prevents Ifna expression and represses initiation of an autocrine Type I IFN inflammatory circuit in developing Th1 cells. Although expression of Ifna, Ifnb and Ifnar was not impacted in Tbx21−/− Be1 cells, we observed that expression of Ifng and Ifngr2 was higher in Tbx21−/− Be1 cells relative to B6 Be1 cells and that Tbx21−/− Be1 cells produced more IFNγ following TLR-stimulation. These data suggested that T-bet might repress an IFNγ-induced autocrine or paracrine inflammatory circuit in differentiating B cells. In fact, IPA revealed that many of the >1000 T-bet repressed genes in Be1 cells were predicted to be activation targets of IFNγR and or TLR signaling. Since engagement of IFNγR, TLR and TNF family receptors can activate IRF and NF-κB TFs (Hiscott, 2007; Rickert et al., 2011; Schroder et al., 2004), we postulate that a key function of T-bet in B cells is to restrain IRF and NF-κB directed transcriptional programs and prevent the establishment of feedforward inflammatory circuits. Furthermore, we predict that once NF-κB and IRF inflammatory loops are initiated in Tbx21−/− Be1 cells, additional IFNγR signaling may not be required to sustain the inflammatory phenotype, as several TNF family ligand and receptor pairs capable of driving NF-κB inflammatory loops (Rickert et al., 2011) were upregulated in Tbx21−/− Be1 cells.
Our data showed that T-bet prevented sustained activation of TFs from the STAT and IRF and NF-κB families. While expression of many Nfkb family members declined rapidly in Be1 cells compared to Be2 cells, Tbx21−/− Be1 cells maintained high expression of Nfkb family members relative to B6 Be1 cells. Enforced NF-κB activation in the B6 Be1 cells, either through addition of a NF-κB activator or exogenous TLR ligands to the Be1 cultures, significantly suppressed ASC formation. Thus, sustained NF-kB activation was sufficient to prevent IFNγ-induced B cell differentiation. This was somewhat unexpected since many of the cues that drive B cell activation and proliferation promote NF-κB activity (Hoffmann and Baltimore, 2006). Moreover, deletion or mutation of some NF-κB family members, specifically within mature B cell compartment, is reported to impair ASC commitment in some settings (Grossmann et al., 2000; Heise et al., 2014; Kaisho et al., 2001). However, a recent publication shows that c-REL (Rel), which is overexpressed and predicted by IPA to be in an activated state in Tbx21−/− Be1 cells, blocks ASC differentiation in response to TLR ligands (Roy et al., 2019). Furthermore, we found that two IPA-predicted activation targets of c-REL, Bach2 and CD40, were down-regulated in a T-bet dependent fashion in differentiating Be1 cells. Given that downmodulation of both Bach2 and CD40 is required for ASC development (Igarashi et al., 2014; Lee et al., 2005; Randall et al., 1998), it is tempting to speculate that one way T-bet facilitates ASC formation is by preventing sustained NF-κB and c-REL signaling that promotes continued expression of pro-proliferation and anti-differentiation genes like Cd40 and Bach2.
In addition to repressing expression of Nfkb family members, T-bet also downmodulated genes, like Irf1 and Statl, that are induced in an Ifngr1-dependent fashion in Be1 cells. These data suggested that IFNγR signals induce an inflammatory interferon stimulated gene (ISG) transcriptional program and simultaneously engage a T-bet mediated negative feedback loop to tune the magnitude and duration of ISG expression. However, genes such as Baft, Rorc, Ifih1 and Il6 were highly expressed in both Tbx21−/− and Ifngr−/− Be1 cells, suggesting IFNγ and T-bet cooperate to repress some genes. Since we identified overlap between T-bet repressed genes and DAR with EICE or AICE binding motifs, one possible mechanism is through recruitment of repressive IRF complexes by T-bet. Given that IRF TFs were identified by PageRank, IPA and HOMER as putative upstream regulators of the Be1 transcriptome network, we speculate that T-bet, perhaps through its capacity to recruit chromatin modifying enzymes to DNA (Miller and Weinmann, 2010), may increase chromatin accessibility and allow for binding of IFNγ-induced IRF-containing repressive TF complexes. Regardless, the data support a model in which IFNγ and T-bet cooperate to prevent expression of alternate cell fate-specifying transcriptional programs in the activated Be1 cells – similar to the role that T-bet plays in cementing commitment to the Th1 cell lineage (Oestreich and Weinmann, 2012).
Our in vivo experiments using B-Tbx21−/− chimeras demonstrated that B cell intrinsic T-bet was required for the development of a primary IgG1 long-lived Ab response and the secondary IgG2c ASC response to influenza virus but was not required for the development or maintenance of flu-specific memory B cells. This result initially appeared inconsistent with a previous study reporting that T-bet was required for maintenance of IgG2c memory B cells (Wang et al., 2012). However, this study, which actually evaluated the memory B cell recall response following in vivo reactivation with BCR ligands, showed reduced formation of secondary ASCs following reactivation of memory IgG2c cells. Thus, this result is very consistent with our data showing that inducible deletion of T-bet in memory cells greatly impairs the IgG2c ASC recall response to flu. However, our experiments, which also examined memory cell maintenance under steady state following T-bet deletion, demonstrated that T-bet was not required for memory cell maintenance, at least over a 10-day period. This result is similar to CD8 T cells where T-bet is not required for memory formation (Joshi et al., 2007) but does regulate memory cell differentiation to secondary effectors (Joshi et al., 2011).
While our in vivo data clearly showed a role for B cell intrinsic T-bet in regulating some primary and secondary ASC responses to influenza, our data also indicated that B cell intrinsic expression of T-bet is dispensable for the IgG1 Ab response to Hp and the IgG2b Ab response to influenza. These data indicated that, unlike Blimp1 or IRF4, T-bet is not a universal ASC lineage commitment factor, instead T-bet regulates IFNγ-induced ASC differentiation. We believe that these results are very consistent with our in vitro data that suggested that T-bet primarily functions to prevent IFNγ-activated B cells from being “locked into” an effector inflammatory cell fate that is not compatible with commitment to the ASC lineage. If this is correct, then T-bet should be completely dispensable when B cells are activated in an environment with few IFNγ-producing cells or have received previous programming signals that render the B cells non-responsive to IFNγ signals. We think this is likely to be the case for the B cells responding to Hp infection as the response to this pathogen is dominated by IL-4 producing Th2 and Tfh2 cells (Leon et al., 2012). While we cannot yet explain why flu-specific IgG2b+ ASC formation is intact in the B-Tbx21−/− mice, TGFβ is the cytokine most often associated with IgG2b class-switch recombination (Deenick et al., 2005; McIntyre et al., 1993; Sellars et al., 2009) and it is known, at least in T cells, that TGFβ potently suppresses IFNγ signaling (Lin et al., 2005). Thus, it is possible that IgG2b+ B cells are unable to respond to IFNγ signaling and differentiate in an IFNγ and T-bet independent manner. In summary, in vivo activated B cells must integrate a complex array of microenvironmental cues during the processes of class switch recombination, proliferation and differentiation. We propose that T-bet, while not a universal regulator of B cell differentiation, acts in a cytokine-dependent manner in the settings of virus infection and autoimmunity to finely tune the IFN-induced inflammatory gene network and allow B cells to transition from an activated inflammatory “effector” cell to a terminally differentiated ASC.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frances Lund (flund@uab.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and generation of bone marrow chimeras.
All experimental animals were bred and maintained in the UAB animal facilities. All procedures involving animals were approved by the UAB Institutional Animal Care and Use Committee and were conducted in accordance with the principles outlined by the National Research Council. Mouse strains used in these experiments include the following: CD45.1+OT-II (intercrossed C57BL/6-Tg(TcraTcrb)425Cbn/J and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1+ B6 mice), B6.129S7-Ifngr1tm1Agt/J (Ifngr1−/−), B6.129P2(C)-Cd19tm1(Cre)Cgn/J (Cd19Cre/+), Cd19Cre/+.Prdm1fl/fl (intercrossed B6.129-Prdm1tm1Clme/J and Cd19Cre/+ mice), hCD20-TAM-cre, Tbx21fl/fl.hCD20-TAM-cre (intercrossed hCD20-TAM-cre and B6.129-Tbx21tm2Srnr/J mice), B6.Blimp1-YFP reporters and B6.T-bet-ZsGreen reporters. Blimp-1 (Rutishauser et al., 2009) and T-bet (Zhu et al., 2012) reporter mice were obtained from Dr. Meffre (Yale University) and Dr. Zhu (NIH), respectively. hCD20-TAM-cre mice (Khalil et al., 2012) were obtained from Mark Shlomchik (University of Pittsburgh) and all other strains were obtained from Jackson Laboratory. Bone marrow (BM) chimeric mice were generated by irradiating B cell deficient μMT (B6.129S2-Ighmtm1Cgn/J) recipient animals with 950 Rads from a high-energy X-ray source, delivered in a split dose 4 hrs apart, and then reconstituting the recipients with 107 BM cells by retro-orbital injection. BM cell mixtures were as follows: 80% μMT BM + 20% Tbx21−/− BM (B-Tbx21−/− chimeras) or with 80% μMT BM + 20% B6 (C57BL/6J) BM (B-WT). BM chimeras were used in experiments 8-12 weeks post-reconstitution. Both male and female mice were used in this study. Within each experiment, animals were matched for age, 8-12 weeks at time zero, and sex. No differences were observed between cohorts of male versus female mice.
Infections and tamoxifen exposure.
BM chimeric mice were infected (i.n.) with a sublethal dose (1.5 × 104 VFU) of the H1N1 influenza virus, A/PR/8/34 (PR8) or by gavage with 200 H. polygyrus (Hp) L3 larvae. To terminate Hp infection, 10 mg pyrantel pamoate (Pin-X, Quartz) was administered by gavage 28 days post infection. In some experiments, mice were given a primary infection with PR8, allowed to recover for ≥90 days, injected i.p. 5 times over 8 days with tamoxifen (Sigma, 200 μl of 10 mg/ml drug dissolved in 10% ethanol and 90% corn oil) and then analyzed or challenged i.n. (1.25×106 VFU) with the heterosubtypic H3N2 influenza virus, A/Aichi/68 (X31) before analysis.
T and B cell effector generation and stimulation.
Th1 and Th2 cells were generated in vitro as previously described (Harris et al., 2005a). Briefly, splenic CD4+ CD45.1+ OT-II TCR Tg T cells were purified by positive selection (Miltenyi Biotec) and cultured in complete medium in the presence of platebound anti-CD3 (2 μg/ml) and anti-CD28 (5 μg/ml) and either IL-12 (2 ng/ml) and anti-IL-4 (11B11, 20 μg/ml) (Th1 cell conditions) or IL-4 (50 units/ml) and anti-IFNγ (XMG1.2, 10 μg/ml) (Th2 cell promoting conditions). T cells were transferred into new plates after 48-72 hrs and cultured for an additional 48 hours in media supplemented with IL-2 (20 units/ml). Polarized Th1 and Th2 cells were treated with mitomycin C, washed and then co-cultured at a 1:1 ratio with positively selected (Miltenyi Biotec) CD19+ splenic B cells in complete B cell media supplemented with OT-II peptide (5 μM), anti-IgM F(ab’)2 (10 μg/ml), and IL-2 (20 Unit/ml). “Be1” refers to B cells from cultures containing Th1 cells, “Be2” refers to B cells from cultures containing Th2 cells, and “BeA” refers to cells cultured under the same conditions but without T cells. In some experiments IKKα & IKBα activator, betulinic acid at 10 μg/ml; TLR7 agonist, R848 at 12.5μg/ml, (InvivoGen); TLR9 agonist, CpG ODN1826 at 12.5 μg/ml, (InvivoGen); or vehicle, DMSO (Sigma) were added to cultures at day 2, prior to analysis on day 4. For analysis of the cytokines made by effector B cell subsets, B cells were collected, purified by positive selection using B220 microbeads and MACS, assessed for purity by FACS and then restimulated at 1×106 cells/ml with TLR ligands (CpG + LPS) or with a restimulation cocktail (LPS + CpG + anti-IgM + anti-CD40 (10 μg/ml) for 24h. Supernatants were collected and tested for IFNγ and IL-6 using Luminex array beads.
METHOD DETAILS
ELISPOT and Ab secretory rate assay.
Day 4 B effector cells were harvested, washed and recultured in fresh media for 5-6 hr at 1 × 106 live cells/ml. Secreted Ab was quantified using an anti-Kappa ELISA (Southern Biotechnology) and a Kappa standard (Sigma). Secretory rates were reported as ng Kappa chain secreted/hour/106 cells. For ELISPOT, Day 4 B effector cells were harvested, washed and in some experiments sort-purified before being recultured in duplicate in fresh media for 5 hrs on multiscreen cellulose filter ELISPOT plates (Millipore) coated with goat anti-mouse kappa light chain (Southern Biotech). Bound Ab was detected with AP-conjugated goat anti-mouse Ig(H+L) Ab (Southern Biotech) and the AP substrate 5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt and nitro-blue tetrazolium chloride (BCIP/NBT, Moss Substrates). ELISPOTS were counted using a dissecting microscope and imaged using S6 Ultra-V Analyzer (Cellular Technology Limited).
Hp and flu Ab titers.
Immune serum samples were serially diluted in ELISA plates coated with purified PR8 virus proteins (Lee et al., 2005) or Hp extract (Harris et al., 2000). Bound Ab was detected using HRP-conjugated goat anti-mouse heavy chain IgG, IgG 1, IgG2c or IgG2b-specific Abs (Southern Biotechnology) and ABTS substrate followed by oxalic acid stop. Absorbance values at 405nm (OD) were read and endpoint titers were determined using the average OD from naive samples as baseline.
Ex vivo ELISPOT.
BM cells were isolated from flu-infected mice (2 tibia + 2 femur/mouse), run over lymphocyte separation media gradient (Corning, 1.077-1.080g/ml) to remove dead cells, serially diluted in duplicate in complete media and incubated for 5 hr at 37°C on multiscreen cellulose filter ELISPOT plates (Millipore) coated with purified PR8 virus protein. Bound Ab was detected with AP-conjugated goat anti-mouse heavy chain-specific pan-IgG Ab (Jackson ImmunoResearch) and the AP substrate BCIP/NBT (Moss Substrates). ELISPOTS were counted using a dissecting microscope and imaged using S6 Ultra-V Analyzer (Cellular Technology Limited).
Cell isolation, flow cytometry analysis and cell sorting.
Spleen and BM single cell suspensions were prepared by gently disrupting tissue on fine wire mesh then red blood cells were lysed. LN single cell suspensions prepared by gently disrupting tissue between glass slides. Cell suspensions were filtered through 70μm nylon mesh then incubated in FcR blocking mAb 2.4G2 (10 μg/ml). Cells were stained with fluorochrome-conjugated Abs, PNA or recombinant flu nucleoprotein (NP) B cell tetramers, prepared as previously described (Allie et al., 2019), and 7-aminoactinomycin D (7AAD, Sigma). For IgG isotype staining, immunoglobulin antibodies were stained in a separate step prior to other cell surface markers and all incubations were performed in staining media supplemented with 5% goat serum (Invitrogen) and 5% rat serum (Invitrogen). For IgG isotype staining cells were stained with the LIVE/DEAD fixable stain (ThermoFisher) before fixation with 10% neutral buffered formalin (Sigma). For intracellular T-bet staining, stained cells were incubated with LIVE/DEAD fixable stain, then fixed and permeabilized with the TF staining buffer set (eBioscience) before intracellular staining. Stained cells were analyzed using a FACSCanto II (BD Bioscience) and Attune NxT (Invitrogen, ThermoFisher) or were sort-purified with a FACSAria (BD Biosciences) located in the UAB Comprehensive Flow Cytometry Core. Antibodies used in this study include: anti-mouse CD19 (6D5), B220 (RA3-6B2), CD93 (AA4.1), CD138 (281-2), CD38 (90), CXCR3 (CXCR3-173), IgM (II-41), IgD (AMS 9.1 and 11-26c.2a), IgG1 (RMG1-1), IgG2b (RMG2b-1 and polyclonal goat anti-mouse), IgG2c (polyclonal goat antimouse), CD45.1 (A20), CD45.2 (104), CD8 (53-6.7), CD3 (17A2), CD4 (GK1.5 and RM4-5) and T-bet (4B10). Monoclonal Abs were obtained from BioLegend, E-Bioscience and Southern Biotech.
Quantitative RT-PCR.
TRIzol (ThermoFisher) or RNeasy (Qiagen) was used to isolate total RNA from sort-purified B effector cells (CD19+CD4negCD45.2+CD45.1neg). RNA quantity and quality were assessed using the Nanodrop 6000 and the Agilent 2100 Bioanalyzer. cDNA was generated from total RNA using SuperScript II double stranded cDNA synthesis kit (Invitrogen) with random hexamers according to manufacturer’s protocols. Real-time PCR was performed using TaqMan Gene Expression Master Mix, with the following parameters on a Roche LightCycler 480: 50°C for 2 min, 95 °C for 10 min, followed by 45 two-step cycles at 95°C for 15 sec and at 60° C for 1 min. Applied Biosystems pre-designed TaqMan Gene Expression Assays were used for real-time PCR (Batf Mm00479410_m1, Bcl6 Mm00477633_m1, Cxcl10 Mm00445235_m1, Ets1 Mm01175819_m1, Gapdh Mm99999915_g1, Hif1a Mm00468869_m1, Ifih1 Mm00459183_m1, Ifng Mm01168134_m1, Ifngr2 Mm01210592_m1, Il6 Mm00446190_m1, Irf4 Mm00516431_m1, Irf5 Mm00496447_m1, Irf7 Mm00516788_m1, Jun Mm00495062_s1, Pax5 Mm00435501_m1, Pou2af1 Mm004488326_m1, Prdm1 Mm00476128_m1, Relb Mm00485664_m1, Rorc Mm01261022_m1, Runx3 Mm00490666_m1, Spib Mm03048233_m1 Stat2 Mm00490880_m1, Stat4 Mm00448881_m1, Tbkbp1 Mm00446590_m1, Tbx21 Mm00450960_m1, Tlr7 Mm00446590_m1, Tnfsf4 Mm00437214_m1, Tnfsf10 Mm01283606_m1, Xbp1 Mm00457359_m1). B effector gene expression analyses included three experimental replicates/group. At least three independent experiments were performed for each analysis. For quantification of gene expression, each sample was normalized to expression of an endogenous control gene, Gapdh for Be1. The fold change in expression of each gene compared to a control sample, set at 1.0 was calculated as 2− ddCT. Samples with CT values above 32 were considered negative.
Affymetrix array sample preparation.
Total RNA was purified (TRIzol) from day 1-4 Be1 and Be2 cells (n= 7 independent experimental samples/timepoint/group) and converted to biotin-labeled cRNA using the Affymetrix one-cycle cDNA synthesis and IVT kit. Labeled cRNA was fragmented to an average size of 35 to 200 bases by incubation at 94°C for 35 min. Hybridization (16 hr), washing, and staining of the Affymetrix GeneChip® Mouse Genome U430 Plus 2.0 Array was conducted according to manufacturer specifications.
RNA-seq sample preparation
500 ng of total RNA (TRIzol) from three biological replicates of day B6 and Tbx21−/− Be1 cells, and one replicate of CXCR3+CCR6+ B6 Be1 and CXCR3+CCR6neg B6 Be1 cells, was used as input for the Illumina TruSeq RNA-seq library kit. Following quality assessment on a bioanalyzer, RNA-seq libraries were pooled and sequenced on one lane of a HiSeq2500 using 50 bp paired-end chemistry as previously described (Barwick et al., 2016).
ATAC-seq sample preparation.
ATAC-seq was performed on Be1 and Be2 cells as previously described (Buenrostro et al., 2015) and Th1 and Th2 cells as previously described (Chisolm et al., 2017). Nuclei from 50,000 cells were extracted and incubated with Tn5 transposase (Illumina) for 30 minutes at 37°C. DNA samples were purified with the MinElut Kit (Qiagen). Library amplification was performed using Nextara primers with Next High-Fidelity 2× PCR Master Mix (New England BioLabs), followed by purification with the PCR Purification Kit (Qiagen). The libraries were sequenced using a 1×50bp paired end run at the UAB Heflin Genomics Center.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of all experiments including tests used, n, and number of experimental repeats are provided in figure legends. FlowJo (Tree Star) used for flow cytometric analyses. Prism graphpad used for statistical analyses and graphing except where indicated. Details of transcriptomics library generation and statistical analysis are provided below.
Affymetrix array analysis.
The signal value of each probe set (gene) was calculated using the Microarray Suite 5 (MAS5) algorithm and normalized using global scaling which set the average signal intensity of an array to 750. MAS5 values were log2-transformed and a p value for each probe set was calculated using an unpaired t-test. Positive FDR and q values were computed using Matlab (The Mathworks Inc., Natick MA) based on the method of Storey (Storey, 2002). FC data reported as log2 expression ratio of indicated groups.
RNA-seq analysis.
Sequencing reads were quality checked using the FASTX-Toolkit and mapped to the mm9 genome using TopHat2 (Kim et al., 2013) with the default settings and the mm9 UCSC Known Gene table as a reference transcriptome. HOMER software (Heinz et al., 2010) was used to summarize reads in transcripts using the analyzeRepeats.pl script with the following options ‘-strand both –count exons –condenseGenes –noadj’. Genes that contained 2 or more reads in at least 3 samples were deemed expressed (13,924 of 24,016) and used as input for edgeR (Robinson et al., 2010) to identify differentially expressed genes using the HOMER script ‘getDiffExpression.pl –repeats’. Following edgeR analysis, p-values for genes with 2-fold change or greater were false-discovery rate (FDR) corrected using the Benjamini-Hochberg method. Genes with a FDR of <0.05 and fold change ≥ 2 were considered significant between B6 and Tbx21−/− Be1 cells. Expression data was normalized to reads per kilobase per million mapped reads (FPKM) using the analyzeRepeats.pl script with the following options “-strand both – count exons – condenseGenes – rpkm’. Data visualization was performed using custom scripts and the R/Bioconductor package, which are available upon request. FC data reported as log2 expression ratio of indicated groups.
GSEA.
Gene set enrichment analysis (GSEA) was performed using the GSEA program (http://software.broadinstitute.org/gsea/index.jsp). For analysis of Be1 and Be2 Affymetrix microarray data, non-log transformed expression data from Be1 (n=7) and Be2 (n=7) samples were submitted to GSEA. Analysis metrics (Subramanian et al., 2005) are summarized briefly as follows: Probe sets were collapsed to genes, which were then ranked by the signal-to-noise metric based on the Be1 vs. Be2 phenotype comparison. Nominal p-values were calculated empirically using 1000 random phenotype label permutations to produce a null distribution of enrichment score. When comparing against a database of large numbers of gene sets, enrichment scores were normalized to account for differences in set sizes, and false discovery rates were computed to control for multiple comparisons. For GSEA analysis of RNA-seq data, all detected genes were ordered by their score (−log10 of the p-value from edgeR multiplied by the sign of the fold change) from most upregulated in Tbx21−/− Be1 cells to most downregulated in Tbx21−/− Be1 cells and used as input for the GSEA Preranked analysis, where nominal p-values are based on gene set permutations.
Bioinformatic identification of predicted upstream regulatory TFs.
To define the TFs that participate in the Be1 transcriptional network we integrated four datasets including HOMER motif analysis, Ingenuity Pathway Analysis (IPA) upstream regulator analysis (Kramer et al., 2014), PageRank analysis (Yu et al., 2017), and differential expression of each factor. Putative Be1 network TFs were selected based on being predicted as a Be1 TF in all four or three of the four different analyses. For the HOMER motif analysis, TF motifs specifically enriched in either Be1 or Be2 cells were first defined. Next, where specific motifs matched multiple factors, the motif was assigned to a TF family (i.e., IRF3 = IRF/ISRE or cJun = AP-1). TFs overlapping the Be1 list were annotated. For PageRank analysis the log2 fold change (logFC) of the Be1 versus the Be2 PageRank statistic was computed. TFs with positive logFC values were assigned to the Be1 network and negative logFC values the Be2 network. Significant expression changes at day 2 by MA analysis was determined based on FDR < 0.05 and absolute value log2 ratio of > 1. For IPA upstream regulator analysis TFs were grouped into Be1 or Be2 and annotated accordingly. Activation Z-scores were used to characterize regulators as activated or inhibited based on the observed pattern of up-/down-regulation of the target molecules compared with expected directions of changes documented in Ingenuity’s curated database. The Z-score captures the degree to which the directions of changes of the individual DEGs in the regulator’s target set is consistent with a its activated or inhibited state based on the IPA curated, expected influences of the regulator on each target. Statistical analysis of the IPA upstream regulator analysis was measured using overlap p-value as previously described (Kramer et al., 2014). Briefly, the overlap p-value (bar height after −log10 transform) characterizes the enrichment of a regulator’s target set within a DEG set based on Fisher’s exact test, which can suggest involvement of a regulator even if that regulator is not a DEG. Genes with an overlap p value <0.05 by IPA were predicted to be upstream regulators. Unless otherwise indicated, all IPA upstream regulator analyses were performed using only direct interactions.
ATAC-seq analysis.
Data processing was performed as previously described (Scharer et al., 2016). Specifically, raw sequencing data was mapped to the mm9 version of the mouse genome using Bowtie (Langmead et al., 2009). with the default settings. Duplicate reads were marked using the Picard Tools MarkDuplicates function (http://broadinstitute.github.io/picard/) and eliminated from downstream analyses. Enriched accessible peaks were identified using the HOMER findPeaks.pl script with setting “-style dnase”. Genomic annotations were computed for ATAC-seq peaks using the HOMER annotatePeaks.pl script and gene expression data annotated using the Entrez ID as a reference. Significantly differential accessible loci (DAR) were identified by the following steps. First, a composite list of all peaks occurring in any sample were obtained using the HOMER mergePeaks.pl script resulting in 62,925 unique regions. Next, the unnormalized read counts for all peaks were annotated for each sample from the bam file using the Genomic Ranges (Lawrence et al., 2013) R/Bioconductor package. This matrix was used as input for edgeR (Robinson et al., 2010) and a pairwise differential analysis was performed between all groups. p-values were FDR corrected for multiple testing using the Benjamini-Hochberg method. Peaks with an FDR < 0.05 were called significant. De novo motif enrichment was determined for peaks that mapped to DAR for the indicated comparison using the HOMER findMotifsGenome.pl script with the default settings. Locations of individual specific motifs (Blimp-1, T-bet, ISRE, AICE, EICE, PAX5, SpiB, Bcl6, XBP1, IRF4, OCT2) were identified in peaks using the findMotifs.pl script with the “-m <factor> –mbed” and ATAC-seq reads in the 100 bp surrounding each motif were annotated for each sample. All other analyses and data display was performed using R/Bioconductor with custom scripts that are available upon request.
DATA AND SOFTWARE AVAILABILITY
The Microarray data have been deposited in the Gene Expression Omnibus (GEO) database under ID code GSE84948. The RNA-seq data have been deposited in the GEO database under ID code GSE83697. The ATAC-seq data have been deposited in the GEO database under ID code GSE118984. Software used in transcriptomics analysis is detailed in METHODS DETAILS and KEY RESOURCES TABLE. Custom scripts for RNA-seq and ATAC-seq data display in R/Bioconductor are available by request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD3 | BD, Biolegend | Clone 17A2 |
| anti-mouse CD4 | Biolegend | Clone GK1.5 |
| anti-mouse CD4 | BD | Clone RM4-5 |
| anti-mouse CD8 | BD, Biolegend | Clone 53-6.7 |
| anti-mouse CD16/32 (Fc Block) | BD | Clone 2.4G2 |
| anti-mouse CD19 | Biolegend | Clone 6D5 |
| anti-mouse CD28 | BD | Clone 37.51 |
| anti-mouse CD38 | eBioscience | Clone 90 |
| anti-mouse B220 | BD, eBioscience | Clone RA3-6B2 |
| anti-mouse CD45.1 | BD, eBioscience | Clone A20 |
| anti-mouse CD45.2 | BD, eBioscience | Clone 104 |
| anti-mouse CD93 | Biolegend, eBioscience | Clone AA4.1 |
| anti-mouse CD138 | BD, Biolegend | Clone 281-2 |
| anti-mouse CXCR3 | eBioscience | Clone CXCR3-173 |
| anti-mouse IL-4 | BioXCell | Clone 11B11 |
| anti-mouse IFNγ | BioXCell | Clone XMG1.2 |
| anti-mouse IgM F(ab’)2 | Southern Biotech | Polyclonal, goat |
| anti-mouse IgM | eBioscience | Clone II-41 |
| anti-mouse IgD | BD | Clone AMS 9.1 |
| anti-mouse IgD | Biolegend | Clone 11-26c.2a |
| anti-mouse heavy chain-specific IgG1 | Biolegend | Clone RMG1-1 |
| anti-mouse heavy chain-specific IgG2b | Biolegend | Clone RMG2b-1 |
| anti-mouse heavy chain-specific IgG2b | Southern Biotech | Polyclonal, goat |
| anti-mouse heavy chain-specific IgG2c | Southern Biotech | Polyclonal, goat |
| anti-mouse kappa light chain | Southern Biotech | Polyclonal, goat |
| anti-mouse Ig, heavy and light chain | Southern Biotech | Polyclonal, goat |
| anti-mouse T-bet | eBioscience | Clone 4B10 |
| anti-mouse heavy chain-specific pan-IgG | Jackson ImmunoResearch | Cat#315-056-046 |
| Bacterial and Virus Strains | ||
| H1N1 influenza virus, A/PR/8/34 (PR8) | (Allie et al., 2019) | |
| H3N2 influenza virus, A/Aichi/68 (X31) | (Allie et al., 2019) | |
| Heligmosomoides polygyrus (Hp) | (Leon et al., 2012) | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pin-X, pyrantel pamoate | Quartz Pharmaceuticals | NDC 13893-691-49 |
| Tamoxifen | Sigma | Cat#T5648 |
| Mitomycin C | Sigma | Cat#M4287 |
| OVA(323-339) peptide (OT-II) | New England Peptide | Cat# BP10-910 |
| Betulinic acid | Abcam | Cat#AB120654 |
| R848 | InvivoGen | Cat#tlrl-r848 |
| CpG ODN1826 | InvivoGen | Cat#tlrl-1826 |
| recombinant IL-2 | PreproTech | Cat#212-12 |
| recombinant IL-4 | PreproTech | Cat#214-14 |
| recombinant IL-12 | Genetics Institute | |
| PNA (lectin) Pacific Blue conjugate | Sigma | L0881 |
| recombinant flu nucleoprotein tetramer | (Allie et al., 2019) | |
| purified PR8 virus proteins | (Lee et al., 2005) | |
| Heligmosomoides polygyrus (Hp) extract | (Harris et al., 2000) | |
| 7-aminoactinomycin D (7AAD) | Sigma | Cat#A9400-1MG |
| LIVE/DEAD Fixable Dead Cell Stains | ThermoFisher | Cat#L34960 |
| Neutral Buffered Formalin Solution, 10% | Sigma | Cat#HT501128 |
| Alkaline phosphatase substrate BCIP/NBT | Moss Substrates | Cat#NBIM-1000 |
| Lymphocyte separation media | Corning | Cat#B003L38 |
| TRIzol Reagent | ThermoFisher | Cat#15596026 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | Cat#4369016 |
| TaqMan Gene Expression Assay Batf | Applied Biosystems | Mm00479410_m1 |
| TaqMan Gene Expression Assay Bcl6 | Applied Biosystems | Mm00477633_m1 |
| TaqMan Gene Expression Assay Cxcl10 | Applied Biosystems | Mm00445235_m1 |
| TaqMan Gene Expression Assay Ets1 | Applied Biosystems | Mm01175819_m1 |
| TaqMan Gene Expression Assay Gapdh | Applied Biosystems | Mm99999915_g1 |
| TaqMan Gene Expression Assay Hif1a | Applied Biosystems | Mm00468869_m1 |
| TaqMan Gene Expression Assay Ifih1 | Applied Biosystems | Mm00459183_m1 |
| TaqMan Gene Expression Assay Ifng | Applied Biosystems | Mm01168134_m1 |
| TaqMan Gene Expression Assay Ifngr2 | Applied Biosystems | Mm01210592_m1 |
| TaqMan Gene Expression Assay Il6 | Applied Biosystems | Mm00446190_m1 |
| TaqMan Gene Expression Assay Irf4 | Applied Biosystems | Mm00516431_m1 |
| TaqMan Gene Expression Assay Irf5 | Applied Biosystems | Mm00496447_m1 |
| TaqMan Gene Expression Assay Irf7 | Applied Biosystems | Mm00516788_m1 |
| TaqMan Gene Expression Assay Jun | Applied Biosystems | Mm00495062_s1 |
| TaqMan Gene Expression Assay Pax5 | Applied Biosystems | Mm00435501_m1 |
| TaqMan Gene Expression Assay Pou2af1 | Applied Biosystems | Mm004488326_m1 |
| TaqMan Gene Expression Assay Prdm1 | Applied Biosystems | Mm00476128_m1 |
| TaqMan Gene Expression Assay Relb | Applied Biosystems | Mm00485664_m1 |
| TaqMan Gene Expression Assay Rorc | Applied Biosystems | Mm01261022_m1 |
| TaqMan Gene Expression Assay Runx3 | Applied Biosystems | Mm00490666_m1 |
| TaqMan Gene Expression Assay Spib | Applied Biosystems | Mm03048233_m1 |
| TaqMan Gene Expression Assay Stat2 | Applied Biosystems | Mm00490880_m1 |
| TaqMan Gene Expression Assay Stat4 | Applied Biosystems | Mm00448881_m1 |
| TaqMan Gene Expression Assay Tbkbp1 | Applied Biosystems | Mm00446590_m1 |
| TaqMan Gene Expression Assay Tbx21 | Applied Biosystems | Mm00450960_m1 |
| TaqMan Gene Expression Assay Tlr7 | Applied Biosystems | Mm00446590_m1 |
| TaqMan Gene Expression Assay Tnfsf4 | Applied Biosystems | Mm00437214_m1 |
| TaqMan Gene Expression Assay Tnfsf10 | Applied Biosystems | Mm01283606_m1 |
| TaqMan Gene Expression Assay Xbp1 | Applied Biosystems | Mm00457359_m1 |
| Critical Commercial Assays | ||
| eBioscience Foxp3 / Transcription Factor Staining Buffer Set | ThermoFisher | Cat#00-5523-00 |
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| Invitrogen SuperScript II Reverse Transcriptase | ThermoFisher | Cat#18064 |
| Next High-Fidelity Taq 2× Master Mix | New England BioLabs | Cat#M0270L |
| Nextera DNA Library Preparation Kit | Illumina | Cat#FC-121-1030 |
| TruSeq RNA Library Prep Kit | Illumina | Cat#RS-122-2001 |
| PCR Purification Kit | Qiagen | Cat#28104 |
| MinElut Kit | Qiagen | Cat#28004 |
| Affymetrix GeneChip Mouse Genome U430 Plus 2.0 Array | ThermoFisher | Cat#900497 |
| Affymetrix one-cycle cDNA synthesis kit | ThermoFisher | Cat#10752030 |
| Affymetrix IVT Labeling Kit | ThermoFisher | Cat#900449 |
| Deposited Data | ||
| Microarray data | this paper | GEO: GSE84948 |
| RNA-seq data | this paper | GEO: GSE83697 |
| ATAC-seq data | this paper | GEO: GSE118984 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6-Tg(TcraTcrb)425Cbn/J (OT-II) | Jackson Laboratory | |
| B6.SJL-Ptprca Pepcb/BoyJ (CD45.1+) | Jackson Laboratory | |
| B6.129S7-Ifngr1tm1Agt/J (Ifngr1−/−) | Jackson Laboratory | |
| B6.129P2(C)-Cd19tm1(Cre)Cgn/J (Cd19Cre/+) | Jackson Laboratory | |
| B6.129-Tbx2tm2Srnr/J (Tbx21Cl/fl) | Jackson Laboratory | |
| B6.129-Prdm1tm1Clme/J (Prdm1fl/fl) | Jackson Laboratory | |
| B6.129S2-Ighmtm1Cgn/J (μMT) | Jackson Laboratory | |
| hCD20-TAM-cre | Mark Shlomchik (Khalil et al., 2012) | |
| B6.Blimp1-YFP | Eric Meffre (Rutishauser et al., 2009) | |
| B6.T-bet-ZsGreen | Jinfang Zhu (Zhu et al., 2012) | |
| Software and Algorithms | ||
| Bowtie | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| FASTX-Toolkit | http://hannonlab.cshl.edu/fastx_toolkit/ | |
| FlowJo | Tree Star | https://www.flowjo.com/ |
| R/Bioconductor & edgeR | (Robinson et al., 2010) | https://www.bioconductor.org/ |
| Genomic Ranges R/Bioconductor package | (Lawrence et al., 2013) | |
| GraphPad Prism | GraphPad Software | |
| GSEA program | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/index.jsp |
| HOMER, analyzeRepeats.pl, annotatePeaks.pl, findPeaks.pl, findMotifs.pl, findMotifsGenome.pl, getDiffExpression.pl –repeats, and mergePeaks.pl scripts | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| Ingenuity Pathway Analysis (IPA) | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ | |
| IPA upstream regulator analysis | (Kramer et al., 2014) | |
| MATLAB | The Mathworks Inc. | https://www.mathworks.com/products/matlab.html |
| Microarray Suite 5 (MAS5) algorithm | Affymetrix | |
| PageRank analysis | (Yu et al., 2017) | |
| Picard Tools MarkDuplicates function | http://broadinstitute.github.io/picard/ | |
| TopHat2 | (Kim et al., 2013) | https://ccb.jhu.edu/software/tophat/index.shtml |
ADDITIONAL RESOURCES
Not applicable
Supplementary Material
Supplemental Table S1. Microarray (MA) dataset from sort-purified naïve B cells (Day 0) and days 1-4 paired mouse Be1 and Be2 cells. Related to Figures 1, 2, 4 and 5 and Supplemental Figures S1 and S4. (n = 7 independent experiments with 7 samples/timepoint/group). See Star Methods for description of culture conditions and MA protocol.
Supplemental Table S2. ATAC-seq dataset from sort-purified day 2 paired B6 and Tbx21−/− Be1 and Be2 cells. Related to Figures 1, 2, 3 and 5 and Supplemental Figure S3. (n=3 independent experiments with 3 samples/group). See Star Methods for description of culture methods and ATAC-seq protocol.
Supplemental Table S3. Bioinformatic Identification of putative regulators of the day 2 Be1 and Be2 transcriptional network. Related to Figure 2 and Supplemental Figure S2. Day 2 Be1 and Be2 MA data (See Suppl. Table S1) and day 2 ATAC-seq data (See Suppl. Table S2) were analyzed using the PageRank (PR) algorithm to identify transcription factors (TF, n=357) predicted to regulate the day 2 Be1 and Be2 transcriptional networks. These TFs were queried using HOMER motif analysis, Ingenuity Pathway Analysis (IPA) upstream regulator analysis (Kramer et al., 2014), and differential expression of each gene. Putative Be1 network TFs were selected based on being predicted as a Be1 TF in all four or three of the four different analyses. See Star Methods for details.
Supplemental Table S4. RNA-seq dataset from sort-purified day 4 B6 and Tbx21−/− Be1 cells and day 4 B6 Be1 subsets. Related to Figures 3–5 and Supplemental Figures S3 and S4. (n=3 independent experiments with 3 B6 and Tbx21−/− Be1 samples/group and n= 1 experiment with 4 Be1 subsets based on expression of CXCR3 and CCR6). See Star Methods for description of culture conditions and RNA-seq protocol.
Supplemental Table S5. IPA predicted upstream regulators of transcriptome data sets. Related to Figure 4 and Supplemental Figure S4. Transcriptome data sets derived from days 1-4 paired mouse Be1 and Be2 cells (MA data see Suppl. Table S1) and day 4 B6 and Tbx21−/− Be1 cells (RNA-seq data see Suppl. Table S3). IPA predicted TF upstream regulators and downstream targets are shown. See Star Methods for details.
Supplemental Table S6. Compilation of genes that are inhibited by T-bet in day 4 Be1 cells and day 3 Th1 cells. Related to Figure 5. Genes inhibited by T-bet in day 4 Be1 cells defined as up in day 4 Tbx21−/− Be1 over day 4 B6 Be1 cells (see RNA-seq data in Suppl. Table S4). Genes inhibited by T-bet in day 3 Th1 cells defined by Iwata (Iwata et al., 2017) as T-bet repressed or T-bet repressed and bound.
Supplemental Table S7. IPA predicted upstream regulators of genes identified as T-bet inhibitory targets that are unique to Be1 cells (Be1 only) or shared between Be1 and Th1 cells (Be1 AND Th1). Related to Figure 5. IPA was used to identify predicted upstream regulators of T-bet repressed target genes (See Suppl. Table S6 for T-bet repressed targets). See Star Methods for details.
ACKNOWLEDGEMENTS
We thank Thomas Scott Simpler, Uma Mudunuru, Rebecca Burnham and Holly Bachus for technical support and Drs. Eric Meffre (Yale University), Shane Crotty (LIAI), Mark Shlomchik (Univ. Pittsburgh) for providing mouse strains that were used in the experiments. Boehringer Ingelheim Pharmaceutical Inc. (to FEL, SRB, and HJ) provided support for Affymetrix transcriptome analysis. Funding for all other experiments was provided by the US National Institutes of Health (NIH): P01 AI078907, R01 AI110508, (to FEL), P01 AI125180 (to FEL and ASW), U19 AI109962 (to FEL and TDR), R01 HL069409 and R01 AI097357 (to TDR), AI061061 (to ASW) and AI23733-01 (to JMB and CDS). SLS received grant support from the UAB Medical Scientist Training Program NIGMS T32GM008361. NIH P30 AR048311 and P30 AI027767 provided support for the UAB consolidated flow cytometry core and G20RR022807-01 provided support for the UAB Animal Resources Program X-irradiator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, and Randall TD (2019). The establishment of resident memory B cells in the lung requires local antigen encounter. Nature immunology 20, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, Gunn B, Chen D, Paley MA, Alter G, et al. (2016). Cutting Edge: B Cell-Intrinsic T-bet Expression Is Required To Control Chronic Viral Infection. Journal of immunology 197, 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwick BG, Scharer CD, Bally AP, and Boss JM (2016). Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nature immunology 17, 1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarth N, Herman OC, Jager GC, Brown L, Herzenberg LA, and Herzenberg LA (1999). Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proceedings of the National Academy of Sciences of the United States of America 96, 2250–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernabei P, Coccia EM, Rigamonti L, Bosticardo M, Forni G, Pestka S, Krause CD, Battistini A, and Novelli F (2001). Interferon-gamma receptor 2 expression as the deciding factor in human T, B, and myeloid cell proliferation or death. J Leukoc Biol 70, 950–960. [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, Leon B, Crossman DK, Murre C, Myers RM, and Weinmann AS (2017). CCCTC-Binding Factor Translates Interleukin 2- and alpha-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity 47, 251–267 e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deenick EK, Hasbold J, and Hodgkin PD (2005). Decision criteria for resolving isotype switching conflicts by B cells. European journal of immunology 35, 2949–2955. [DOI] [PubMed] [Google Scholar]
- Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, Cooper TK, Kitamura D, and Rahman ZS (2016). IFN-gamma receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. The Journal of experimental medicine 213, 715–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerondakis S, and Siebenlist U (2010). Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol 2, a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann M, O’Reilly LA, Gugasyan R, Strasser A, Adams JM, and Gerondakis S (2000). The anti-apoptotic activities of Rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J 19, 6351–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DP, Goodrich S, Gerth AJ, Peng SL, and Lund FE (2005a). Regulation of IFN-gamma production by B effector 1 cells: essential roles for T-bet and the IFN-gamma receptor. Journal of immunology 174, 6781–6790. [DOI] [PubMed] [Google Scholar]
- Harris DP, Goodrich S, Mohrs K, Mohrs M, and Lund FE (2005b). Cutting edge: the development of IL-4-producing B cells (B effector 2 cells) is controlled by IL-4, IL-4 receptor alpha, and Th2 cells. Journal of immunology 175, 7103–7107. [DOI] [PubMed] [Google Scholar]
- Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM, Johnson LL, Swain SL, and Lund FE (2000). Reciprocal regulation of polarized cytokine production by effector B and T cells. Nature immunology 1,475–482. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, and Klein U (2014). Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. The Journal of experimental medicine 211, 2103–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J (2007). Convergence of the NF-kappaB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev 18, 483–490. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, and Baltimore D (2006). Circuitry of nuclear factor kappaB signaling. Immunological reviews 210, 171–186. [DOI] [PubMed] [Google Scholar]
- Igarashi K, Ochiai K, Itoh-Nakadai A, and Muto A (2014). Orchestration of plasma cell differentiation by Bach2 and its gene regulatory network. Immunological reviews 261, 116–125. [DOI] [PubMed] [Google Scholar]
- Iwata S, Mikami Y, Sun HW, Brooks SR, Jankovic D, Hirahara K, Onodera A, Shih HY, Kawabe T, Jiang K, et al. (2017). The Transcription Factor T-bet Limits Amplification of Type I IFN Transcriptome and Circuitry in T Helper 1 Cells. Immunity 46, 983–991 e984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, Hou B, Buckner JH, and Rawlings DJ (2016). B cell IFN-gamma receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. The Journal of experimental medicine 213, 733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, Tomar D, Simon Z, Burgovsky R, Blalock EL, et al. (2018). A new B cell effector pathway with defective regulatioin of TLR7 signaling in human SLE. Immunity in press. [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Dominguez CX, Chen JH, Hand TW, and Kaech SM (2011). Increased numbers of preexisting memory CD8 T cells and decreased T-bet expression can restrain terminal differentiation of secondary effector and memory CD8 T cells. Journal of immunology 187, 4068–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisho T, Takeda K, Tsujimura T, Kawai T, Nomura F, Terada N, and Akira S (2001). IkappaB kinase alpha is essential for mature B cell development and function. The Journal of experimental medicine 193, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnell JL, Kumar V, Wang J, Wang S, Voynova E, and Ettinger R (2017). Role of CD11c(+) T-bet(+) B cells in human health and disease. Cellular immunology 321, 40–45. [DOI] [PubMed] [Google Scholar]
- Kasperczyk H, La Ferla-Bruhl K, Westhoff MA, Behrend L, Zwacka RM, Debatin KM, and Fulda S (2005). Betulinic acid as new activator of NF-kappaB: molecular mechanisms and implications for cancer therapy. Oncogene 24, 6945–6956. [DOI] [PubMed] [Google Scholar]
- Kees U, and Krammer PH (1984). Most influenza A virus-specific memory cytotoxic T lymphocytes react with antigenic epitopes associated with internal virus determinants. The Journal of experimental medicine 159, 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Cambier JC, and Shlomchik MJ (2012). B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science 336, 1178–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, and Heise N (2015). Unexpected functions of nuclear factor-kappaB during germinal center B-cell development: implications for lymphomagenesis. Curr Opin Hematol 22, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox JJ, Buggert M, Kardava L, Seaton KE, Eller MA, Canaday DH, Robb ML, Ostrowski MA, Deeks SG, Slifka MK, et al. (2017). T-bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau D, Lan L, Andrews SF, Henry C, Thatcher Rojas K, Neu KE, Huang M, Huang Y-P, DeKosky B, Palm A-KE, et al. (2017). Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Science Immunol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V, Glimcher LH, and Lord GM (2013). T-bet: a bridge between innate and adaptive immunity. Nature reviews. Immunology 13, 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BO, Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Makris M, Sprague F, Lund FE, and Randall TD (2005). CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. Journal of immunology 175, 5827–5838. [DOI] [PubMed] [Google Scholar]
- Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, and Lund FE (2012). Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nature immunology 13, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JT, Martin SL, Xia L, and Gorham JD (2005). TGF-beta 1 uses distinct mechanisms to inhibit IFN-gamma expression in CD4+ T cells at priming and at recall: differential involvement of Stat4 and T-bet. Journal of immunology 174, 5950–5958. [DOI] [PubMed] [Google Scholar]
- McIntyre TM, Klinman DR, Rothman P, Lugo M, Dasch JR, Mond JJ, and Snapper CM (1993). Transforming growth factor beta 1 selectivity stimulates immunoglobulin G2b secretion by lipopolysaccharide-activated murine B cells. The Journal of experimental medicine 177, 1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, and Weinmann AS (2010). Molecular mechanisms by which T-bet regulates T-helper cell commitment. Immunological reviews 238, 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naradikian MS, Hao Y, and Cancro MP (2016). Age-associated B cells: key mediators of both protective and autoreactive humoral responses. Immunological reviews 269, 118–129. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Hodgkin PD, Tarlinton DM, and Corcoran LM (2015). The generation of antibody-secreting plasma cells. Nature reviews. Immunology 15, 160–171. [DOI] [PubMed] [Google Scholar]
- Oestreich KJ, Mohn SE, and Weinmann AS (2012). Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nature immunology 13, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestreich KJ, and Weinmann AS (2012). T-bet employs diverse regulatory mechanisms to repress transcription. Trends in immunology 33, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SL, Szabo SJ, and Glimcher LH (2002). T-bet regulates IgG class switching and pathogenic autoantibody production. Proceedings of the National Academy of Sciences of the United States of America 99, 5545–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KM, Cauvi DM, Toomey CB, Morris KV, and Kono DH (2013). Interferon-gamma and systemic autoimmunity. Discov Med 16, 123–131. [PMC free article] [PubMed] [Google Scholar]
- Randall TD, Heath AW, Santos-Argumedo L, Howard MC, Weissman IL, and Lund FE (1998). Arrest of B lymphocyte terminal differentiation by CD40 signaling: mechanism for lack of antibody-secreting cells in germinal centers. Immunity 8, 733–742. [DOI] [PubMed] [Google Scholar]
- Randolph DA, Huang G, Carruthers CJ, Bromley LE, and Chaplin DD (1999). The role of CCR7 in TH1 and TH2 cell localization and delivery of B cell help in vivo. Science 286, 2159–2162. [DOI] [PubMed] [Google Scholar]
- Rickert RC, Jellusova J, and Miletic AV (2011). Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunological reviews 244, 115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy K, Mitchell S, Liu Y, Ohta S, Lin Y. s., Metzig MO, Nutt SL, and Hoffmann A (2019). A Regulatory Circuit Controlling the Dynamics of NFkB cRel Transitions B Cells from Proliferation to Plasma Cell Differentiation. Immunity 50, 616–628. e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, and Marrack P (2011). Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood 118, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, and Marrack P (2017). B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. The Journal of clinical investigation in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, and Kaech SM (2009). Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31, 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer CD, Blalock EL, Barwick BG, Haines RR, Wei C, Sanz I, and Boss JM (2016). ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naive SLE B cells. Sci Rep 6, 27030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, and Hume DA (2004). Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75, 163–189. [DOI] [PubMed] [Google Scholar]
- Sellars M, Reina-San-Martin B, Kastner P, and Chan S (2009). Ikaros controls isotype selection during immunoglobulin class switch recombination. The Journal of experimental medicine 206, 1073–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, Smyth GK, Hodgkin PD, Nutt SL, and Corcoran LM (2015). Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nature immunology 16, 663–673. [DOI] [PubMed] [Google Scholar]
- Storey JD (2002). A direct approach to false discovery rate. J. R. Stat. Soc. Ser. B 64, 479–498. [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault DL, Chu AD, Graham KL, Balboni I, Lee LY, Kohlmoos C, Landrigan A, Higgins JP, Tibshirani R, and Utz PJ (2008). IRF9 and STAT1 are required for IgG autoantibody production and B cell expression of TLR7 in mice. The Journal of clinical investigation 118, 1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang NS, McHeyzer-Williams LJ, Okitsu SL, Burris TP, Reiner SL, and McHeyzer-Williams MG (2012). Divergent transcriptional programming of class-specific B cell memory by T-bet and RORalpha. Nature immunology 13, 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, Rahman S, Zerrouki K, Hanna R, Morehouse C, et al. (2018). IL-21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T-bet(+) B cells in SLE. Nat Commun 9, 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin A, Masson F, Liao Y, Preston S, Guan T, Gloury R, Olshansky M, Lin JX, Li P, Speed TP, et al. (2016). A molecular threshold for effector CD8(+) T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nature immunology 17, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP, Pereira RM, Crotty S, Chang JT, Pipkin ME, et al. (2017). Epigenetic landscapes reveal transcription factors that regulate CD8(+) T cell differentiation. Nature immunology 18, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, et al. (2012). The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37, 660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1. Microarray (MA) dataset from sort-purified naïve B cells (Day 0) and days 1-4 paired mouse Be1 and Be2 cells. Related to Figures 1, 2, 4 and 5 and Supplemental Figures S1 and S4. (n = 7 independent experiments with 7 samples/timepoint/group). See Star Methods for description of culture conditions and MA protocol.
Supplemental Table S2. ATAC-seq dataset from sort-purified day 2 paired B6 and Tbx21−/− Be1 and Be2 cells. Related to Figures 1, 2, 3 and 5 and Supplemental Figure S3. (n=3 independent experiments with 3 samples/group). See Star Methods for description of culture methods and ATAC-seq protocol.
Supplemental Table S3. Bioinformatic Identification of putative regulators of the day 2 Be1 and Be2 transcriptional network. Related to Figure 2 and Supplemental Figure S2. Day 2 Be1 and Be2 MA data (See Suppl. Table S1) and day 2 ATAC-seq data (See Suppl. Table S2) were analyzed using the PageRank (PR) algorithm to identify transcription factors (TF, n=357) predicted to regulate the day 2 Be1 and Be2 transcriptional networks. These TFs were queried using HOMER motif analysis, Ingenuity Pathway Analysis (IPA) upstream regulator analysis (Kramer et al., 2014), and differential expression of each gene. Putative Be1 network TFs were selected based on being predicted as a Be1 TF in all four or three of the four different analyses. See Star Methods for details.
Supplemental Table S4. RNA-seq dataset from sort-purified day 4 B6 and Tbx21−/− Be1 cells and day 4 B6 Be1 subsets. Related to Figures 3–5 and Supplemental Figures S3 and S4. (n=3 independent experiments with 3 B6 and Tbx21−/− Be1 samples/group and n= 1 experiment with 4 Be1 subsets based on expression of CXCR3 and CCR6). See Star Methods for description of culture conditions and RNA-seq protocol.
Supplemental Table S5. IPA predicted upstream regulators of transcriptome data sets. Related to Figure 4 and Supplemental Figure S4. Transcriptome data sets derived from days 1-4 paired mouse Be1 and Be2 cells (MA data see Suppl. Table S1) and day 4 B6 and Tbx21−/− Be1 cells (RNA-seq data see Suppl. Table S3). IPA predicted TF upstream regulators and downstream targets are shown. See Star Methods for details.
Supplemental Table S6. Compilation of genes that are inhibited by T-bet in day 4 Be1 cells and day 3 Th1 cells. Related to Figure 5. Genes inhibited by T-bet in day 4 Be1 cells defined as up in day 4 Tbx21−/− Be1 over day 4 B6 Be1 cells (see RNA-seq data in Suppl. Table S4). Genes inhibited by T-bet in day 3 Th1 cells defined by Iwata (Iwata et al., 2017) as T-bet repressed or T-bet repressed and bound.
Supplemental Table S7. IPA predicted upstream regulators of genes identified as T-bet inhibitory targets that are unique to Be1 cells (Be1 only) or shared between Be1 and Th1 cells (Be1 AND Th1). Related to Figure 5. IPA was used to identify predicted upstream regulators of T-bet repressed target genes (See Suppl. Table S6 for T-bet repressed targets). See Star Methods for details.
Data Availability Statement
The Microarray data have been deposited in the Gene Expression Omnibus (GEO) database under ID code GSE84948. The RNA-seq data have been deposited in the GEO database under ID code GSE83697. The ATAC-seq data have been deposited in the GEO database under ID code GSE118984. Software used in transcriptomics analysis is detailed in METHODS DETAILS and KEY RESOURCES TABLE. Custom scripts for RNA-seq and ATAC-seq data display in R/Bioconductor are available by request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD3 | BD, Biolegend | Clone 17A2 |
| anti-mouse CD4 | Biolegend | Clone GK1.5 |
| anti-mouse CD4 | BD | Clone RM4-5 |
| anti-mouse CD8 | BD, Biolegend | Clone 53-6.7 |
| anti-mouse CD16/32 (Fc Block) | BD | Clone 2.4G2 |
| anti-mouse CD19 | Biolegend | Clone 6D5 |
| anti-mouse CD28 | BD | Clone 37.51 |
| anti-mouse CD38 | eBioscience | Clone 90 |
| anti-mouse B220 | BD, eBioscience | Clone RA3-6B2 |
| anti-mouse CD45.1 | BD, eBioscience | Clone A20 |
| anti-mouse CD45.2 | BD, eBioscience | Clone 104 |
| anti-mouse CD93 | Biolegend, eBioscience | Clone AA4.1 |
| anti-mouse CD138 | BD, Biolegend | Clone 281-2 |
| anti-mouse CXCR3 | eBioscience | Clone CXCR3-173 |
| anti-mouse IL-4 | BioXCell | Clone 11B11 |
| anti-mouse IFNγ | BioXCell | Clone XMG1.2 |
| anti-mouse IgM F(ab’)2 | Southern Biotech | Polyclonal, goat |
| anti-mouse IgM | eBioscience | Clone II-41 |
| anti-mouse IgD | BD | Clone AMS 9.1 |
| anti-mouse IgD | Biolegend | Clone 11-26c.2a |
| anti-mouse heavy chain-specific IgG1 | Biolegend | Clone RMG1-1 |
| anti-mouse heavy chain-specific IgG2b | Biolegend | Clone RMG2b-1 |
| anti-mouse heavy chain-specific IgG2b | Southern Biotech | Polyclonal, goat |
| anti-mouse heavy chain-specific IgG2c | Southern Biotech | Polyclonal, goat |
| anti-mouse kappa light chain | Southern Biotech | Polyclonal, goat |
| anti-mouse Ig, heavy and light chain | Southern Biotech | Polyclonal, goat |
| anti-mouse T-bet | eBioscience | Clone 4B10 |
| anti-mouse heavy chain-specific pan-IgG | Jackson ImmunoResearch | Cat#315-056-046 |
| Bacterial and Virus Strains | ||
| H1N1 influenza virus, A/PR/8/34 (PR8) | (Allie et al., 2019) | |
| H3N2 influenza virus, A/Aichi/68 (X31) | (Allie et al., 2019) | |
| Heligmosomoides polygyrus (Hp) | (Leon et al., 2012) | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pin-X, pyrantel pamoate | Quartz Pharmaceuticals | NDC 13893-691-49 |
| Tamoxifen | Sigma | Cat#T5648 |
| Mitomycin C | Sigma | Cat#M4287 |
| OVA(323-339) peptide (OT-II) | New England Peptide | Cat# BP10-910 |
| Betulinic acid | Abcam | Cat#AB120654 |
| R848 | InvivoGen | Cat#tlrl-r848 |
| CpG ODN1826 | InvivoGen | Cat#tlrl-1826 |
| recombinant IL-2 | PreproTech | Cat#212-12 |
| recombinant IL-4 | PreproTech | Cat#214-14 |
| recombinant IL-12 | Genetics Institute | |
| PNA (lectin) Pacific Blue conjugate | Sigma | L0881 |
| recombinant flu nucleoprotein tetramer | (Allie et al., 2019) | |
| purified PR8 virus proteins | (Lee et al., 2005) | |
| Heligmosomoides polygyrus (Hp) extract | (Harris et al., 2000) | |
| 7-aminoactinomycin D (7AAD) | Sigma | Cat#A9400-1MG |
| LIVE/DEAD Fixable Dead Cell Stains | ThermoFisher | Cat#L34960 |
| Neutral Buffered Formalin Solution, 10% | Sigma | Cat#HT501128 |
| Alkaline phosphatase substrate BCIP/NBT | Moss Substrates | Cat#NBIM-1000 |
| Lymphocyte separation media | Corning | Cat#B003L38 |
| TRIzol Reagent | ThermoFisher | Cat#15596026 |
| TaqMan Gene Expression Master Mix | Applied Biosystems | Cat#4369016 |
| TaqMan Gene Expression Assay Batf | Applied Biosystems | Mm00479410_m1 |
| TaqMan Gene Expression Assay Bcl6 | Applied Biosystems | Mm00477633_m1 |
| TaqMan Gene Expression Assay Cxcl10 | Applied Biosystems | Mm00445235_m1 |
| TaqMan Gene Expression Assay Ets1 | Applied Biosystems | Mm01175819_m1 |
| TaqMan Gene Expression Assay Gapdh | Applied Biosystems | Mm99999915_g1 |
| TaqMan Gene Expression Assay Hif1a | Applied Biosystems | Mm00468869_m1 |
| TaqMan Gene Expression Assay Ifih1 | Applied Biosystems | Mm00459183_m1 |
| TaqMan Gene Expression Assay Ifng | Applied Biosystems | Mm01168134_m1 |
| TaqMan Gene Expression Assay Ifngr2 | Applied Biosystems | Mm01210592_m1 |
| TaqMan Gene Expression Assay Il6 | Applied Biosystems | Mm00446190_m1 |
| TaqMan Gene Expression Assay Irf4 | Applied Biosystems | Mm00516431_m1 |
| TaqMan Gene Expression Assay Irf5 | Applied Biosystems | Mm00496447_m1 |
| TaqMan Gene Expression Assay Irf7 | Applied Biosystems | Mm00516788_m1 |
| TaqMan Gene Expression Assay Jun | Applied Biosystems | Mm00495062_s1 |
| TaqMan Gene Expression Assay Pax5 | Applied Biosystems | Mm00435501_m1 |
| TaqMan Gene Expression Assay Pou2af1 | Applied Biosystems | Mm004488326_m1 |
| TaqMan Gene Expression Assay Prdm1 | Applied Biosystems | Mm00476128_m1 |
| TaqMan Gene Expression Assay Relb | Applied Biosystems | Mm00485664_m1 |
| TaqMan Gene Expression Assay Rorc | Applied Biosystems | Mm01261022_m1 |
| TaqMan Gene Expression Assay Runx3 | Applied Biosystems | Mm00490666_m1 |
| TaqMan Gene Expression Assay Spib | Applied Biosystems | Mm03048233_m1 |
| TaqMan Gene Expression Assay Stat2 | Applied Biosystems | Mm00490880_m1 |
| TaqMan Gene Expression Assay Stat4 | Applied Biosystems | Mm00448881_m1 |
| TaqMan Gene Expression Assay Tbkbp1 | Applied Biosystems | Mm00446590_m1 |
| TaqMan Gene Expression Assay Tbx21 | Applied Biosystems | Mm00450960_m1 |
| TaqMan Gene Expression Assay Tlr7 | Applied Biosystems | Mm00446590_m1 |
| TaqMan Gene Expression Assay Tnfsf4 | Applied Biosystems | Mm00437214_m1 |
| TaqMan Gene Expression Assay Tnfsf10 | Applied Biosystems | Mm01283606_m1 |
| TaqMan Gene Expression Assay Xbp1 | Applied Biosystems | Mm00457359_m1 |
| Critical Commercial Assays | ||
| eBioscience Foxp3 / Transcription Factor Staining Buffer Set | ThermoFisher | Cat#00-5523-00 |
| RNeasy Micro Kit | Qiagen | Cat#74004 |
| Invitrogen SuperScript II Reverse Transcriptase | ThermoFisher | Cat#18064 |
| Next High-Fidelity Taq 2× Master Mix | New England BioLabs | Cat#M0270L |
| Nextera DNA Library Preparation Kit | Illumina | Cat#FC-121-1030 |
| TruSeq RNA Library Prep Kit | Illumina | Cat#RS-122-2001 |
| PCR Purification Kit | Qiagen | Cat#28104 |
| MinElut Kit | Qiagen | Cat#28004 |
| Affymetrix GeneChip Mouse Genome U430 Plus 2.0 Array | ThermoFisher | Cat#900497 |
| Affymetrix one-cycle cDNA synthesis kit | ThermoFisher | Cat#10752030 |
| Affymetrix IVT Labeling Kit | ThermoFisher | Cat#900449 |
| Deposited Data | ||
| Microarray data | this paper | GEO: GSE84948 |
| RNA-seq data | this paper | GEO: GSE83697 |
| ATAC-seq data | this paper | GEO: GSE118984 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6-Tg(TcraTcrb)425Cbn/J (OT-II) | Jackson Laboratory | |
| B6.SJL-Ptprca Pepcb/BoyJ (CD45.1+) | Jackson Laboratory | |
| B6.129S7-Ifngr1tm1Agt/J (Ifngr1−/−) | Jackson Laboratory | |
| B6.129P2(C)-Cd19tm1(Cre)Cgn/J (Cd19Cre/+) | Jackson Laboratory | |
| B6.129-Tbx2tm2Srnr/J (Tbx21Cl/fl) | Jackson Laboratory | |
| B6.129-Prdm1tm1Clme/J (Prdm1fl/fl) | Jackson Laboratory | |
| B6.129S2-Ighmtm1Cgn/J (μMT) | Jackson Laboratory | |
| hCD20-TAM-cre | Mark Shlomchik (Khalil et al., 2012) | |
| B6.Blimp1-YFP | Eric Meffre (Rutishauser et al., 2009) | |
| B6.T-bet-ZsGreen | Jinfang Zhu (Zhu et al., 2012) | |
| Software and Algorithms | ||
| Bowtie | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| FASTX-Toolkit | http://hannonlab.cshl.edu/fastx_toolkit/ | |
| FlowJo | Tree Star | https://www.flowjo.com/ |
| R/Bioconductor & edgeR | (Robinson et al., 2010) | https://www.bioconductor.org/ |
| Genomic Ranges R/Bioconductor package | (Lawrence et al., 2013) | |
| GraphPad Prism | GraphPad Software | |
| GSEA program | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/index.jsp |
| HOMER, analyzeRepeats.pl, annotatePeaks.pl, findPeaks.pl, findMotifs.pl, findMotifsGenome.pl, getDiffExpression.pl –repeats, and mergePeaks.pl scripts | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| Ingenuity Pathway Analysis (IPA) | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ | |
| IPA upstream regulator analysis | (Kramer et al., 2014) | |
| MATLAB | The Mathworks Inc. | https://www.mathworks.com/products/matlab.html |
| Microarray Suite 5 (MAS5) algorithm | Affymetrix | |
| PageRank analysis | (Yu et al., 2017) | |
| Picard Tools MarkDuplicates function | http://broadinstitute.github.io/picard/ | |
| TopHat2 | (Kim et al., 2013) | https://ccb.jhu.edu/software/tophat/index.shtml |