

Abstract
Background
Metaphyseal dysplasia without hypotrichosis (MDWH) is a rare form of chondrodysplasia with no extraskeletal manifestations. MDWH is caused by RMRP mutations, but it is differentiated from the allelic condition cartilage-hair hypoplasia (CHH), which in addition to chondrodysplasia is characterised by thin hair, immunodeficiency and increased risk of malignancy. The long-term outcome of MDWH remains unknown.
Objective
We diagnosed severe agranulocytosis in a subject with RMRP mutations and normal hair. Based on this observation, we hypothesised that MDWH may, similar to CHH, associate with immune deficiency and malignancy.
Methods
We collected clinical and laboratory data for a cohort of 80 patients with RMRP mutations followed for over 30 years and analysed outcome data for those with features consistent with MDWH.
Results
In our cohort, we identified 10 patients with skeletal but no extraskeletal features during preschool age. Eight of these patients developed malignancy or clinically significant immunodeficiency during follow-up. Two of them died during chemotherapy for malignancy. At the time of the first extraskeletal manifestation, patients were school aged, 20, 43 and 50 years old. Laboratory signs of immunodeficiency (impaired lymphocyte proliferative responses) were demonstrated in four patients before the onset of symptoms. The patient outside this cohort, who had RMRP mutations, skeletal dysplasia, normal hair and severe agranulocytosis at 18 years of age, underwent haematopoietic stem cell transplantation.
Conclusions
MDWH can present with severe late-onset extraskeletal manifestations and thus should be reclassified and managed as CHH.
Keywords: agranulocytosis, cartilage-hair hypoplasia, immunodeficiency, malignancy, rmrp
Introduction
Mutations in RMRP (mitochondrial RNA-processing endoribonuclease) cause a spectrum of autosomal recessive skeletal dysplasias with variable severity. These are currently classified as cartilage-hair hypoplasia (CHH, MIM #250250), anauxetic dysplasia-1 (ANXD-1, MIM #607095) and metaphyseal dysplasia without hypotrichosis (MDWH, MIM #250460). CHH is the most common presentation and is characterised by metaphyseal chondrodysplasia, hair hypoplasia, variable immunodeficiency and anaemia.1 2 In addition, patients with CHH have a high risk of cancer often resulting in premature death.3 Clinical manifestations of CHH vary considerably, even among siblings.1
ANXD represents the most severe end of the RMRP skeletal disease spectrum. It is a rare clinical entity with extremely severe short stature—tallest reported patient having a height of 74 cm at 16 years—and can be caused by mutations in RMRP (ANXD-1) or POP1 (ANXD-2, MIM #602486).4–6 In contrast to the mainly metaphyseal involvement seen in CHH and MDWH, vertebral bodies, pelvis and femoral epiphyses are also affected in ANXD. Apart from mild mental retardation, no extraskeletal manifestations have been reported. The oldest reported individual with ANXD-1 was 16 years old and no data on long-term disease course are available.
MDWH has been described as an isolated skeletal dysplasia in which hair hypoplasia, immunodeficiency and other extraskeletal manifestations are absent.7 Only 11 patients with MDWH have been previously reported, aged 5–11.5 years, and their clinical course beyond childhood is unknown.7–10
RMRP mutations in CHH, ANXD and MDWH differ and possibly have different biological consequences, explaining distinct clinical presentation. Mutated RMRP in ANXD-1 results in normal processing of messenger RNA (mRNA), but impaired ribosomal RNA (rRNA) cleavage, while in CHH mRNA cleavage is also impacted.5 It has been proposed that rRNA and mRNA cleavage activity correlate with the degree of bone dysplasia and immunodeficiency, respectively.11
Our recent observation of severe bone marrow failure in an adult patient with MDWH-like presentation in childhood and the large variability in disease severity in our CHH cohort2 12 prompted us to evaluate long-term outcome in patients with RMRP-associated skeletal dysplasia who have normal hair and absent extraskeletal manifestations in early childhood.
Methods
The index patient with MDWH was recruited to the study after diagnosis of agranulocytosis. The main study cohort involved 80 Finnish patients with genetically confirmed RMRP-related skeletal dysplasia, who were initially identified and underwent interview, clinical examination, radiographic assessment and blood sampling in 1985–1991. A follow-up visit was conducted in 2011–2015, when patients were contacted and invited for an interview, clinical assessment and blood tests. We also collected health information for the 80 patients from two Finnish National Medical Databases. The Care Register for Health Care has since 1969 recorded the activities of health centres, hospitals and other institutions providing inpatient or home nursing care. Outpatient primary healthcare data were also derived from the Register of Primary Healthcare Visits, which covers all health centres in Finland since 2011. Database information included health service providers, dates of the visits, diagnoses, as well as diagnostic and therapeutic procedures. Based on database information, we then collected and reviewed all patients’ detailed health records for inpatient and outpatient visits. Additionally, data on malignancies were obtained from the Finnish Cancer Register and mortality data from the Statistics Finland.
Birth lengths were compared with normal Finnish population13 and SD scores (SDS), adjusted for gestational age, were used for analysis.
Statistical analysis was performed with IBM SPSS Statistics software V.23. The Fisher’s exact test and regression analyses were applied as appropriate.
Results
Case report
We identified a patient, followed at our institution for skeletal dysplasia with normal hair and absent extraskeletal manifestations. She was born at term measuring 3110 g (−1.0 SDS) and 46 cm (−2.4 SDS). She developed progressive short stature (figure 1), for which growth hormone therapy at 6–7 years of age was ineffective. Skeletal dysplasia was suspected and confirmed at 7 years of age when radiographs revealed skeletal changes consistent with mainly epiphyseal dysplasia, particularly in the hands and hips (figure 2). Very mild metaphyseal changes were also noticed, and they became less apparent with age (figure 2). She underwent several surgeries for leg deformity (valgus in knees, varus in feet) and also developed significant scoliosis. She had beautiful long thick hair (figure 2). In addition to a few episodes of otitis media, her childhood history of infections was remarkable only for severe varicella, molluscum contagiosum and skin warts. She had mild lymphopenia, but no further immunological evaluations were regarded indicated. At 19 years, a skeletal dysplasia gene panel revealed compound heterozygous variants in the RMRP gene, n.155G>T and n.-5delins21 (NCBI reference sequence: NR_003051.3), confirming a RMRP-related skeletal dysplasia (figure 3).
Figure 1.
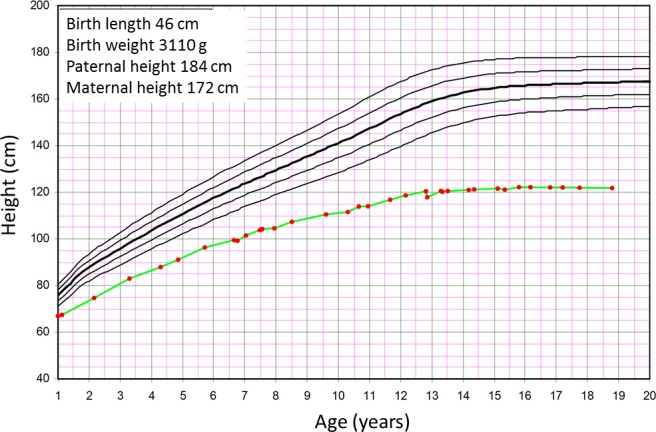
Growth in a patient with RMRP mutations, showing a typical pattern of growth failure progressing particularly during the first year of life and during puberty. The patient received growth hormone therapy from 6 to 7 years of age, with no effect on growth.
Figure 2.

Photographs and radiographs of the index patient. At 7 years, she had short stature with short limbs, valgus deformity of the knees and long, thick hair (A). Facial features, eye lashes and eye brows were normal (B). Radiographs indicated no metaphyseal irregularity at the wrist or hand (C) or the knee (D), although subtle widening of the metaphyseal area was observed in the distal femur and proximal tibia. Epiphyses in the hand (C) and knee (D) were small and flat. The patient developed significant scoliosis (E). Small and flat epiphyses were also seen at the hip (F) and by adulthood the femoral necks were short and the proximal femurs deformed (G).
Figure 3.

Sanger ECG of two different regions of the RMRP gene demonstrating (arrows) variants in the index patient and her parents.
Evaluation for fatigue at 18 years after an episode of rhinosinusitis and conjunctivitis uncovered pancytopenia (haemoglobin 93 g/L (normal range 117–155), platelets 109×109/L (normal range 150–360), white cell count 1.2×109/L (normal range 3.4–8.2) and no detectable neutrophils. While anaemia and thrombocytopenia were mild and did not require therapy, agranulocytosis persisted. The patient was lymphopenic (0.7×109/L (normal range 1.3–3.6), however, immunoglobulin levels appeared normal. Bone marrow examination revealed normal age-correlated cellularity (70%) but absent granulopoiesis and remarkable over-representation of lymphocytes (93% CD3+ cells, of which 60% were also CD8+). There was no clonal expansion of T cell receptor γ chain gene and the findings were not consistent with malignancy.
During the following year, agranulocytosis persisted despite treatment with filgrastim or prednisolone and the patient required multiple hospitalisations for bacterial and viral infections. She, therefore, received a haematopoietic stem cell transplant (HSCT) from a human leukocyte antigen (HLA) -identical unrelated donor after a reduced-toxicity conditioning with treosulfan, fludarabine and antithymocyte globulin. Neutrophils engrafted on day 20 post-HSCT, and bone marrow showed normal cellularity and total donor chimerism 2 months post-HSCT. She continued to experience viral and bacterial infections and was diagnosed with sinusoid obstruction syndrome 7 months post-transplant. Despite normal bone marrow cellularity and full chimerism, her peripheral blood cell counts remain below normal limits at 11 months after HSCT, which potentially stems from multifactorial aetiology.
Cohort of CHH patients with normal hair
In the cohort of 80 patients with confirmed biallelic RMRP mutations, we identified 10 subjects fulfilling the criteria for MDWH: in the preschool age, they presented with metaphyseal dysplasia, normal hair and absent extraskeletal manifestations (table 1). The remaining 70 patients had a clinical presentation typical for CHH. When clinical symptoms, radiographic features and laboratory results were compared, the 10 patients with MDWH did not differ significantly from the rest of the cohort, except for birth length. Their mean birth length was −2.03 SDS (95% CI −1.09 to −2.98 SDS), compared with the rest of the cohort with mean length of −3.06 SDS (95% CI −2.97 to −3.03 SDS) (p=0.031, B coefficient=0.647).
Table 1.
Clinical, laboratory and outcome characteristics of 10 patients fulfilling the criteria of metaphyseal dysplasia without hypotrichosis in early childhood
| P 1 | P 2 | P 3 | P 4 | P 5 | P 6 | P 7 | P 8 | P 9 | P 10 | |
| Mutation* | n.71A>G/ 71A>G |
n.71A>G/ 263G>T |
n.71A>G/ 71A>G |
n.71A>G/ 263G>T |
n.71A>G/ 71A>G |
n.71A>G/ 263G>T |
n.71A>G/ 71A>G |
n.71A>G/ 71A>G |
n.71A>G/ 71A>G |
n.71A>G/ 71A>G |
| Age at recruitment, years | 4.3 | 0.6 | 6.6 | 10.2 | 14.2 | 15.3 | 17.1 | 26.4 | 23.8 | 34.3 |
| Age at the latest follow-up, years | 20.9 | 28.9 | 34.8 | 37.6 | 44.9 | 46.3 | 48.1 | 50.8 | 54.6 | 65.4 |
| Duration of follow-up, years | 16.6 | 28.3 | 28.2 | 27.4 | 30.7 | 31.0 | 31.0 | 24.4 | 30.8 | 31.1 |
| Age at the first extraskeletal manifestation | 20.7 | N/A | N/A | 7–17 | 7–17 | 9 | Adulthood | 50.5 | 7–17 | 43.3 |
| First extraskeletal manifestation | Hodgkin disease | Pollen allergy | Pollen allergy | Recurrent Sin | Recurrent Sin requiring sinus puncture | Recurrent Sin requiring sinus surgery at 8 years | Recurrent Sin requiring sinus punctures at 37 years | Neuroendocrine carcinoma | Recurrent Sin with polyposis requiring sinus punctures | Recurrent Sin, requiring sinus surgery at 61 years |
| Other extraskeletal manifestations in adulthood | None | None | None | Recurrent otitis media since 31 years, skin warts, pollen allergy | Pollen allergy, bronchiectasis since 43 years | Pollen allergy | Skin warts | Pollen allergy | Maxillary polyposis requiring polypectomy at 14 years | Recurrent pneumonia since 55 years and bronchiectasis since 60 years |
| Severity of growth failure† | Severe | Mild | Mild | Moderate | Mild | Mild | Mild | Mild | Moderate | Mild |
| Adult height group, cm | 120–130 | 130–140 | 130–140 | 130–140 | 130–140 | 140–150 | 130–140 | 130–140 | 120–130 | 120–130 |
| Lymphocyte subtypes‡ | N/A | Low CD19+at 0.6 and 26 years | Normal | Normal | Normal | Low CD3+ and CD4+ at 44 years | Normal | N/A | Low CD3+, CD4+ and CD8+ at 36 and 52 years | Normal |
| Lymphocyte proliferation§ | N/A | Abnormal at 0.6 years | Normal | Abnormal at 10 years | N/A | Abnormal at 2 years | Abnormal at 7 years | Abnormal at 12 years | Abnormal at 10 years | N/A |
| IgA, M and G levels¶ | N/A | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal IgA and IgG, low IgM at 60 years |
| SAD** | N/A | N/A | Yes, at 32 years | N/A | N/A | None at 44 years | None at 45 years | N/A | Yes, at 52 years | None at 62 years |
| Outcome | Died at 20 years during cancer treatment | Alive | Alive | Alive | Alive | Alive | Alive | Died at 50 years during cancer treatment | Alive | Alive |
*Reference sequence: NR_003051.3.
†Growth failure classified based on CHH-specific growth charts as described in.20
‡Lymphocyte subtypes included CD3+, CD4+, CD8+, CD19+ and CD16/56+ absolute cell numbers measured by flow cytometry as described in.2
§Lymphocyte proliferation measured to PHA, ConA and PWM as described in.20
¶Measured as described in.2
**Specific antibody deficiency was defined as inadequate antibody response to Pneumovax: a fourfold rise in antibody titres and post-immunisation antibody levels ≥0.35 µg/mL to <70% of serotypes, measured as described previously in.2
CHH, cartilage-hair hypoplasia; NA, not available; PHA, phytohaemagglutinin; SAD, specific antibody deficiency.
Among the 10 patients with MDWH, we identified eight patients who despite skeletal-only presentation during childhood, later developed extraskeletal manifestations (table 1). Their clinical course was unremarkable except for short-stature until the first extraskeletal symptoms at 7–17, 20, 43 and even 50 years of age. Two patients developed malignancy (Hodgkin lymphoma and neuroendocrine carcinoma), and both deceased during chemotherapy. Another six patients developed signs of immunodeficiency including recurrent episodes of otitis media, rhinosinusitis and pneumonia, as well as bronchiectasis.
Two patients with MDWH are alive and well at 28 and 34 years, with no extraskeletal manifestations apart from pollen allergy. One of them has been diagnosed with asymptomatic specific antibody deficiency, another has low levels of peripheral B cells (table 1).
Systematic laboratory assessments of immunological parameters prior to the onset of extraskeletal manifestations have not been performed outside research visits, reflecting mild initial clinical course of the patients. Abnormal lymphocyte proliferative responses were detected in four patients before the development of clinical immunodeficiency and malignancy.
Discussion
We tested our hypothesis that MDWH is not a separate disease entity by exploring the long-term outcome data in a large cohort on patients with RMRP mutations and skeletal dysplasia and demonstrated that subjects with MDWH can indeed develop late-onset immunodeficiency and malignancy, characteristic for CHH. Our index patient with biallelic RMRP mutations, skeletal dysplasia, normal hair and largely absent extraskeletal manifestations in childhood, who developed severe bone marrow failure by early adulthood confirms the variable course and the importance of long-term surveillance.
We describe 10 Finnish patients from our large (n=80) CHH cohort who first presented with skeletal dysplasia only, thus fulfilling the criteria for MDWH. However, eight of them developed malignancy or clinical immunodeficiency later during follow-up. Thus, we suggest that MDWH represents cases of CHH with a risk for late-onset extraskeletal symptoms.
The 10 patients with MDWH presentation differed from the rest of the CHH cohort only by a less profound growth failure at birth. Although it would be tempting to associate milder growth failure with less severe extraskeletal manifestations, our present and previous studies contradict this by describing severe and even fatal complications of immunodeficiency in some patients with very mild growth failure.12
Genotype-phenotype correlations in cases with RMRP mutations remain poorly understood.11 12 Possible explanations include polymorphisms in RMRP gene and non-allelic modifiers.11 14 Almost all patients in our cohort were homozygous or heterozygous for the founder n.71A>G mutation. However, the patient with agranulocytosis was compound heterozygous for two very rare RMRP mutations n.155G>T and n.-5delins21. The n.155G>T has previously been identified in compound heterozygous state with the founder mutation n.71G>A in a Finnish CHH patient.15 In addition, another variant in the same position, n.155G>C has been found in compound heterozygous state with a 15 bp duplication at position −24 in the promoter region in a patient with CHH, Omenn syndrome and CD8 lymphopenia.16 Regarding the other mutation, almost the same insertion, differing from the identified variant only by two nucleotides, is a 20 bp duplication, n.-6_−25dup20 (n.5_−24dup20 using NR_003051.3 transcript), described by Hermanns et al.17 The RMRP variant n.-6_−25dup20 was found in two Belgian CHH patients (siblings, identical mutations) in compound heterozygous state with both, n.91G>A and n.101C>T (n.92G>A and n.102C>T according to NR_003051.3) with unexplained functional implication. Both patients had clinical diagnosis of CHH. The n.-6_−25dup20 duplication leads to reduced (but not absent) activity of the RMRP promoter compared with the wild type promoter, suggesting that RMRP transcription is decreased but not abolished.18
The spectrum of diseases caused by RMRP mutations incorporates skeletal dysplasia of various severity and diverse extraskeletal manifestations. Clinical classification into MDWH, CHH and ANXD is based on the degree of skeletal dysplasia, the presence of hair hypoplasia and other extraskeletal manifestations. However, no long-term follow-up data have until now been published for patients with RMRP mutations. Therefore, it has been unclear, whether MDWH or ANXD patients develop clinically significant immunodeficiency or malignancies later in life. Our index patient had skeletal characteristics overlapping with ANXD (significant hip and spine involvement) and the findings were not typical for CHH or MDWH. In contrast, the patients with MDWH presented with radiographic findings typical for CHH.19
Patients with CHH present with extremely variable clinical features, and even short-stature can be absent in childhood.11 In the largest clinical series of 108 individuals with CHH, hair was normal in 7% and no susceptibility to infections could be shown in 44% of the patients.1 Thus, some of the patients could be designated to have MDWH. On the other hand, severe growth failure up to −11 SD that is in the range of ANXD has also been reported in subjects with CHH,1 raising the question of whether MDWH, CHH and ANXD are actually all the same disease of variable severity.
Labelling mildly affected patients with RMRP mutations as having MDWH can negatively affect the management and follow-up of such patients, blunting the clinicians away from potential extraskeletal complications. Our study demonstrates the development of clinically significant immunodeficiency and malignancies in previously asymptomatic short-statured patients. Therefore, careful and regular clinical assessment should be organised for individuals with RMRP mutations even in the absence of extraskeletal manifestations on presentation.
We demonstrate serious late-onset extraskeletal manifestations in patients with MDWH and suggest that these cases should be reclassified and managed as CHH.
Footnotes
Contributors: SV and OM planned the research. SV collected and analysed the data and drafted the manuscript. AC performed the genetic analysis. SV, AC, MT, UW-K and OM edited and finalised the manuscript and approved the final version.
Funding: The study was funded by the Sigrid Jusélius Foundation (OM), the Academy of Finland (OM), the Folkhälsan Research Foundation (OM), the Novo Nordisk Foundation (OM), the Helsinki University Hospital Research Funds (MT and OM), the Swedish Childhood Cancer Foundation (OM), the Foundation for Pediatric Research (MT and OM) and the Doctoral School in Health Sciences at the University of Helsinki (SV).
Competing interests: UW-K declares consultancy to Pfizer, Amgen, Novartis and Sanofi. OM declares consultancy to Kyowa Kirin, Alexion and Sandoz.
Patient consent for publication: Obtained.
Ethics approval: This study is part of our research programme on skeletal dysplasias in Finland, approved by the Institutional Research Ethics Committee at Helsinki University Hospital, Finland.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: All data relevant to the study are included in the article or uploaded as online supplementary information.
References
- 1. Mäkitie O, Kaitila I. Cartilage-hair hypoplasia--clinical manifestations in 108 Finnish patients. Eur J Pediatr 1993;152:211–7. 10.1007/BF01956147 [DOI] [PubMed] [Google Scholar]
- 2. Kostjukovits S, Klemetti P, Valta H, Martelius T, Notarangelo LD, Seppänen M, Taskinen M, Mäkitie O. Analysis of clinical and immunologic phenotype in a large cohort of children and adults with cartilage-hair hypoplasia. J Allergy Clin Immunol 2017;140:612–4. 10.1016/j.jaci.2017.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taskinen M, Ranki A, Pukkala E, Jeskanen L, Kaitila I, Mäkitie O. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet A 2008;146A:2370–5. 10.1002/ajmg.a.32478 [DOI] [PubMed] [Google Scholar]
- 4. Horn D, Rupprecht E, Kunze J, Spranger J. Anauxetic dysplasia, a spondylometaepiphyseal dysplasia with extreme dwarfism. J Med Genet 2001;38:262–5. 10.1136/jmg.38.4.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thiel CT, Horn D, Zabel B, Ekici AB, Salinas K, Gebhart E, Rüschendorf F, Sticht H, Spranger J, Müller D, Zweier C, Schmitt ME, Reis A, Rauch A. Severely incapacitating mutations in patients with extreme short stature identify RNA-processing endoribonuclease RMRP as an essential cell growth regulator. Am J Hum Genet 2005;77:795–806. 10.1086/497708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glazov EA, Zankl A, Donskoi M, Kenna TJ, Thomas GP, Clark GR, Duncan EL, Brown MA. Whole-Exome re-sequencing in a family quartet identifies Pop1 mutations as the cause of a novel skeletal dysplasia. PLoS Genet 2011;7:e1002027 10.1371/journal.pgen.1002027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verloes A, Pierard GE, Le Merrer M, Maroteaux P. Recessive metaphyseal dysplasia without hypotrichosis. A syndrome clinically distinct from McKusick cartilage-hair hypoplasia. J Med Genet 1990;27:693–6. 10.1136/jmg.27.11.693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castriota-Scanderbeg A, Dallapiccola B, Mingarelli R, Kozlowski K. Distinctive metaphyseal chondrodysplasia simulating cartilage hair hypoplasia. Am J Med Genet 2001;99:289–93. 10.1002/ajmg.1212 [DOI] [PubMed] [Google Scholar]
- 9. Bonafé L, Schmitt K, Eich G, Giedion A, Superti-Furga A. RMRP gene sequence analysis confirms a cartilage-hair hypoplasia variant with only skeletal manifestations and reveals a high density of single-nucleotide polymorphisms. Clin Genet 2002;61:146–51. 10.1034/j.1399-0004.2002.610210.x [DOI] [PubMed] [Google Scholar]
- 10. Ridanpää M, Ward LM, Rockas S, Särkioja M, Mäkelä H, Susic M, Glorieux FH, Cole WG, Mäkitie O. Genetic changes in the RNA components of RNase MRP and RNase P in Schmid metaphyseal chondrodysplasia. J Med Genet 2003;40:741–6. 10.1136/jmg.40.10.741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thiel CT, Mortier G, Kaitila I, Reis A, Rauch A. Type and level of RMRP functional impairment predicts phenotype in the cartilage hair hypoplasia-anauxetic dysplasia spectrum. Am J Hum Genet 2007;81:519–29. 10.1086/521034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klemetti P, Valta H, Kostjukovits S, Taskinen M, Toiviainen-Salo S, Mäkitie O. Cartilage-Hair hypoplasia with normal height in childhood-4 patients with a unique genotype. Clin Genet 2017;92:204–7. 10.1111/cge.12969 10.1111/cge.12969 [DOI] [PubMed] [Google Scholar]
- 13. Pihkala J, Hakala T, Voutilainen P, Raivio K. Uudet suomalaiset sikion kasvukayrat [In Finnish]. Duodecim 1989;105:1540–6. [PubMed] [Google Scholar]
- 14. Notarangelo LD, Roifman CM, Giliani S. Cartilage-Hair hypoplasia: molecular basis and heterogeneity of the immunological phenotype. Curr Opin Allergy Clin Immunol 2008;8:534–9. 10.1097/ACI.0b013e328310fe7d [DOI] [PubMed] [Google Scholar]
- 15. Ridanpää M, Sistonen P, Rockas S, Rimoin DL, Mäkitie O, Kaitila I. Worldwide mutation spectrum in cartilage-hair hypoplasia: ancient founder origin of the major70A-->G mutation of the untranslated RMRP. Eur J Hum Genet 2002;10:439–47. 10.1038/sj.ejhg.5200824 [DOI] [PubMed] [Google Scholar]
- 16. Kavadas FD, Giliani S, Gu Y, Mazzolari E, Bates A, Pegoiani E, Roifman CM, Notarangelo LD. Variability of clinical and laboratory features among patients with ribonuclease mitochondrial RNA processing endoribonuclease gene mutations. J Allergy Clin Immunol 2008;122:1178–84. 10.1016/j.jaci.2008.07.036 [DOI] [PubMed] [Google Scholar]
- 17. Hermanns P, Tran A, Munivez E, Carter S, Zabel B, Lee B, Leroy JG. RMRP mutations in cartilage-hair hypoplasia. Am J Med Genet A 2006;140:2121–30. 10.1002/ajmg.a.31331 [DOI] [PubMed] [Google Scholar]
- 18. Hermanns P, Bertuch AA, Bertin TK, Dawson B, Schmitt ME, Shaw C, Zabel B, Lee B. Consequences of mutations in the non-coding RMRP RNA in cartilage-hair hypoplasia. Hum Mol Genet 2005;14:3723–40. 10.1093/hmg/ddi403 [DOI] [PubMed] [Google Scholar]
- 19. Mäkitie O, Marttinen E, Kaitila I. Skeletal growth in cartilage-hair hypoplasia. A radiological study of 82 patients. Pediatr Radiol 1992;22:434–9. [DOI] [PubMed] [Google Scholar]
- 20. Mäkitie O, Kaitila I, Savilahti E. Susceptibility to infections and in vitro immune functions in cartilage-hair hypoplasia. Eur J Pediatr 1998;157:816–20. 10.1007/s004310050943 [DOI] [PubMed] [Google Scholar]