

Abstract
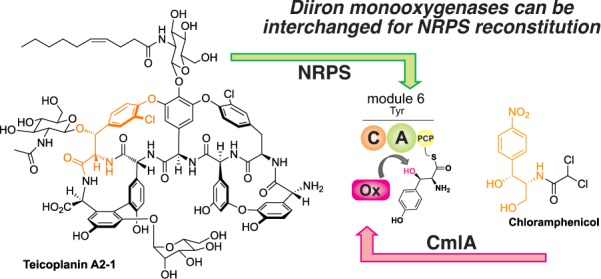
β-Hydroxylation plays an important role in the nonribosomal peptide biosynthesis of many important natural products, including bleomycin, chloramphenicol, and the glycopeptide antibiotics (GPAs). Various oxidative enzymes have been implicated in such a process, with the mechanism of incorporation varying from installation of hydroxyl groups in amino acid precursors prior to adenylation to direct amino acid oxidation during peptide assembly. In this work, we demonstrate the in vitro utility and scope of the unusual nonheme diiron monooxygenase CmlA from chloramphenicol biosynthesis for the β-hydroxylation of a diverse range of carrier protein bound substrates by adapting this enzyme as a non-native trans-acting enzyme within NRPS-mediated GPA biosynthesis. The results from our study show that CmlA has a broad substrate specificity for modified phenylalanine/tyrosine residues as substrates and can be used in a practical strategy to functionally cross complement compatible NRPS biosynthesis pathways in vitro.
Nonribosomal peptide synthetases (NRPSs) are mega-enzyme complexes that are responsible for the biosynthesis of many important peptide natural products with medical relevance.1 While most peptides formed through nonribosomal peptide synthesis are relatively short (<20 AA), they can be composed of a very wide array of monomers and are often extensively modified, which leads to this class of molecules having a broad range of potent biological activity (Figure 1).2 Peptide biosynthesis by NRPS assembly lines is most commonly based on the incorporation of single monomers into the growing peptide chain through the actions of repetitive catalytic units, which are known as modules. Each NRPS module is comprised of individual catalytic domains, each of which has a specific function in peptide biosynthesis. Adenylation domains (A) are required for substrate selection, activation, and covalent attachment of the activated residue onto the free thiol group of the peptidyl carrier protein (PCP)-bound phosphopantetheine cofactor. Condensation domains (C) are then responsible for peptide bond formation between two activated, PCP-bound substrates, with the specificity of peptide synthesis dictated by the combination of A- and C-domain specificity for residue structure and stereochemistry. Beyond the A, C, and PCP domains that together constitute a minimal NRPS extension module, these can be further enriched by additional domains such as epimerization (E) domains, which are responsible for altering the stereochemistry of peptide residues and thioesterase (TE) domains that cleave the mature peptide from the NRPS.1 In the case of glycopeptide antibiotic (GPA) biosynthesis, the final NRPS modules contain an additional, specialized oxidase recruitment (X) domain, which is required for the extensive cross-linking of aromatic peptide side chains during peptide maturation.3,4
Figure 1.

Examples of peptides biosynthesized by nonribosomal peptide synthesis assembly lines and incorporate amino acid residues that are β-hydroxylated by diiron enzymes during peptide assembly.
While the GPA-cyclization cascade serves as an impressive example of the modification of peptidyl–PCP intermediates during NRPS biosynthesis, the most common modification of NRPS bound intermediates in trans occurs toward aminoacyl PCP-bound substrates.5,6 These tailoring reactions, which include oxidation, hydroxylation, and halogenation, serve to introduce significant diversity into the products of NRPS assembly lines and offer tantalizing potential for biosynthetic redesign of complex NRPS biosynthetic pathways.7 Within GPA biosynthesis, the modification of PCP-bound tyrosine residues by both halogenases and hydroxylases is important for modulating the potency of these antibiotics,8 and as such, understanding the mechanism by which such trans-interacting enzymes operate is of significant interest as a prequel to biosynthetic redesign (Figure 2).9 β-Hydroxylation of amino acids found in GPA peptides can be introduced by one of two different mechanisms—either by a trans-interacting nonheme diiron monooxygenase that directly modifies tyrosine residues bound to PCP domains on the main NRPS (e.g., teicoplanin type GPAs, Tyr6 residue)5 or incorporation of β-hydroxytyrosine (Bht) residues through direct amino acid activation by an A-domain (e.g., vancomycin type, Tyr2 and Tyr6 residues).10−12 When Bht is formed offline from the main NRPS, a specific three enzyme system comprising a minimal A-PCP NRPS, cytochrome P450 β-hydroxylase, and specific thioesterase or other offloading enzyme, is found encoded in such clusters to generate the Bht precursor (e.g., nikkomycin and novobiocin biosynthesis).13−15 This shows the diversity of both selection mechanisms and enzyme types that can play a role in hydroxylation of PCP-bound amino acids.
Figure 2.

Alternate routes for the incorporation of β-hydroxy amino acids in NRPS-mediated peptide biosynthesis. (A) Hydroxylation of the Tyr6 residue by a diiron monooxygenase in type IV GPA biosynthesis occurs during peptide assembly with the residue covalently attached to module 6 of the NRPS. (B,C) Modification of amino acids by diiron monooxygenases during peptide assembly in bleomycin (His8) and lysobactin (Phe3, Leu4, and Asn10) biosynthesis. (D) Multiple oxidation of different amino acids in the biosynthesis of skyllamycin (Phe5, OMe-Tyr7, Leu11) by a cytochrome P450 monooxygenase. (E) Provision of Bht precursors for the NRPS assembly lines that form type I–III GPAs utilizes a P450-mediated hydroxylation of Tyr on a separate, dedicated NRPS module. (F) Activity of the diiron monooxygenase CmlA toward PCP-bound 4-aminophenylalanine in chloramphenicol biosynthesis.
While significant work has been invested in understanding the formation of Bht in GPA biosynthesis and related systems via the second (offline) pathway, few results have been reported for the nonheme diiron monooxygenase mediated route outside of a single gene disruption experiment in a GPA producer strain.16 In this work, we leveraged this amino acid modification route for teicoplanin biosynthesis using an in vitro NRPS reconstitution system recently developed in our laboratory.17 Using this system, we have demonstrated that the incorporation of both β-hydroxyl and chlorine moieties in the tyrosine 6 residue of the teicoplanin peptide is gated by the specificity of the module 5/6 C domain.17 This C-domain demonstrates exquisite acceptor-site specificity for Cl-Bht, which is the completely modified residue incorporated in teicoplanin, and thus is able to pause the peptide assembly process to allow the trans-interacting enzymes to modify the PCP-bound Tyr residue.17 This NRPS system is therefore an excellent one to investigate the modification of aminoacyl-PCPs by trans interacting enzymes in vitro, although studies to date have relied upon the low selectivity of the module 6 A domain to use modified amino acids by proxy to probe the mechanism behind this selection process. Our results indicate that this system is indeed a versatile platform to investigate such biosynthetic processes and can be effectively used to explore hydroxylase and carrier protein selectivity in a facile manner.
Results and Discussion
CmlA Can Replace Tcp25 from Teicoplanin Biosynthesis in Vitro
Given that the module 5/6 C domain from the teicoplanin NRPS displays strict specificity for Tyr modifications, it is clear that the activated PCP-bound amino acid must be decorated prior to peptide bond formation. As such, this system provides an excellent reconstitution platform to investigate NRPS trans-acting enzyme activity and selectivity during NRPS-mediated peptide synthesis. Due to the fact that Tyr β-hydroxylation is the critical modification that gates C-domain specificity, we chose to focus on the reconstitution of nonheme diiron monooxygenase CmlA from the antibiotic chloramphenicol biosynthesis gene cluster.18−21 The native substrate for CmlA is a PCP-tethered l-para-aminophenylalanine (L-PAPA), with the enzyme demonstrating obligate selectivity for L-PAPA in the PCP-bound form. X-ray absorption studies have shown that both components (amino acid and PCP domain) are required for mediating structural changes of the diferrous enzyme, a requirement for the rapid addition of O2 and ensuing Cβ hydroxylation.21,22 Beyond these experiments, the scope of PCPs and amino acids that can be modified by CmlA has not yet been explored. Nonetheless, CmlA was chosen as a potential substitute for the native diiron β-hydroxylase from the teicoplanin biosynthesis gene cluster (Tcp25), as Tcp25 was unable to be expressed in soluble form using E. coli overexpression.23,24 Significant insight into the structure and mechanism of both nonheme diiron monooxygenases present in the chloramphenicol biosynthesis pathway—CmlA that is responsible for the β-hydroxylation of PCP-bound 4-aminophenylalanine and CmlI that is responsible for the six-electron oxidation aryl N-oxidation—has been performed.5,18−22,25−32 Given the level of identity shown between CmlA and the related enzymes from GPA biosynthesis (Tcp25/Dbv28/StaM: 34%),23,24,33,34 along with implicated roles of homologues in both bleomycin (orf3 (His): 39%)35,36 and lysobactin (orf78 (Phe, Leu and Asn): 34%) biosynthesis,37 this indicated a possibility that CmlA could serve to replace Tcp25 in teicoplanin peptide biosynthesis. One possible caveat was the low identity of the related PCP domains in chloramphenicol and teicoplanin biosynthesis (20%). However, a model of the PCP/CmlA complex indicates that the regions of highest conservation between PCPs that were modified by orthologous enzymes were likely clustered near the site of phosphopantetheine attachment and hence the possible interface of such a complex.19
To test CmlA compatibility with the teicoplanin biosynthesis system and the potential for this enzyme to modify compatible substrates on PCP domains from different biosynthetic NRPS assembly lines, we first utilized our previously described pentapeptide extension assay to examine the modification of Tyr-6 residues by CmlA during hexapeptide reconstitution (Figure 3). In these experiments, the synthetic pentapeptide was first loaded onto the PCP domain from Tcp11 module 5 (A–PCP–E–C architecture) using the promiscuous phosphopantetheinyl transferase Sfp (R4–4 mutant).38 In order to reconstitute hexapeptide biosynthesis in vitro, the peptidyl-PCP loaded Tcp11 module 5 and holo-PCP module 6 (A-PCP, holo-PCP prepared using CoA) were mixed with the desired test substrate for A6 (Tyr) along with ATP and MgCl2.17,39,40 Given that Tyr is not an effective substrate for the module 5/6 C domain and thus is not incorporated into the hexapeptide product beyond trace amounts, we next supplemented our peptide extension assay with an equimolar amount of the CmlA β-hydroxylase enzyme.21 To ensure optimal β-hydroxylation conditions in vitro, either phenazine methosulfate or methyl viologen was included as a redox mediator and NADH as a supply of reducing equivalents. The combination of methyl viologen/dithionite was also tested, although it was not able to effectively reconstitute CmlA activity under the conditions of this assay (Table 1).21,41 The complete reaction was then incubated overnight and was subsequently terminated by chemical cleavage of any PCP-bound peptides in their methyl amide form by the addition of methylamine. Gratifyingly, LC-MS analysis of the purified reconstitution reaction products confirmed pentapeptide extension and by inference the β-hydroxylation of the PCP6 bound Tyr residue by CmlA. The hexapeptide containing a Bht residue in position 6 of the peptide was detected by LCMS, and the incorporation of Bht at residue 6 was further confirmed by HRMS and MS/MS analysis of the peptide assay products. Given the expense of using CmlA as a single turnover enzyme, we also tested whether phenazine methosulfate/NADH could be used with CmlA in substoichiometric amounts to perform multiple turnovers. Indeed, this proved to be the case, and ∼10 turnovers per enzyme were routinely observed under the conditions of our assay. This compares well to those reported for other oxidative enzymes involved in either NRPS hydroxylation in the skyllamycin system (vide infra, 15–20 turnovers per cytochrome P450 enzyme)42 or the cross-linking enzymes from GPA biosynthesis (30–40 turnovers per cytochrome P450 enzyme under optimized conditions).43
Figure 3.

Reconstitution of hexapeptide extension using separated modules (A–PCP–E–C architecture) from teicoplanin biosynthesis M5–M6 (Tcp11), different amino acid substrates for module 6, and the optional inclusion of the diiron monooxygenase enzyme CmlA from chloramphenicol biosynthesis. A, adenylation domain; C, condensation domain; PCP, peptidyl carrier protein domain; E, epimerization domain.
Table 1. Effect of Alternative Reductant/Mediator Combinations to Reconstitute CmlA Activity Using Tyr As a Substrate.
| reductant | redox coupler | conversion, %a |
|---|---|---|
| NADH | phenazine methosulfate | 56 ± 7 |
| NADH | methyl viologen | 61 ± 8 |
| Dithionite | methyl viologen | N/D |
| NADH | 24 ± 4 |
Triplicate experiments.
CmlA Has a Broad Substrate Specificity for Halogenated Tyrosine Residues
With an active Tyr β-hydroxylation system in hand, we were interested in investigating the substrate specificity of CmlA using modified tyrosine residues in the peptide reconstitution assay. Given that the A-domain from Tcp11 module 6 displays activation activity toward a wide range of modified tyrosine residues and the module 5/6 C-domain donor site preferentially accepts β-hydroxylated substrates, this assay served as a perfect vehicle to comprehensively explore CmlA substrate tolerance. Using this experimental design, seven synthetic halogenated Tyr substrates known to be activated by the teicoplanin A6 domain were tested (F-Tyr, Cl-Tyr, Br-Tyr, I-Tyr, 3,5-di-Cl-Tyr, 3,5-di-Br-Tyr, and 3,5-di-I-Tyr; Table 2).17 Five of these seven modified Tyr substrates tested led to the detection of corresponding hexapeptide products that included a halogenated 6-Bht residue (Figure 4). Only hexapeptide biosynthesis with 3,5-di-Br-Tyr and 3,5-di-I-Tyr was not detected in the LCMS analysis. Indeed, in the case of 3,5-di-I-Tyr, we determined that significant peptide formation did occur; however, the Tyr-6 residue detected in the peptide products in this case were in fact monoiodinated Tyr due to the decomposition of 3,5-di-I-Tyr. These findings demonstrate that single halogen atom alterations do not have a significant effect on CmlA activity and are readily tolerated as substrates. This matches the proposed biosynthesis pathway seen in teicoplanin biosynthesis as well as related GPAs, where chlorination of the tyrosine residue is anticipated to occur prior to β-hydroxylation.17
Table 2. Substrate Acceptance of CmlA Using a Coupled M5/M6 Teicoplanin Peptide Extension Assay.
| substrate | A-domain rate (vs Tyr), %a | hexapeptide yield, %a |
|---|---|---|
| Tyr | 100b | 56 ± 7 |
| F-Tyr | 77 ± 12b | 65 ± 6 |
| Cl-Tyr | 56 ± 13b | 55 ± 8 |
| Br-Tyr | 61 ± 7b | 37 ± 9 |
| I-Tyr | 59 ± 10b | 46 ± 6 |
| di-Cl-Tyr | 63 ± 2b | 37 ± 4 |
| di-Br-Tyr | 29 ± 6b | N/D |
| di-I-Tyr | 89 ± 17b | N/D (peptide from I-Tyr only) |
| Meta-Tyr | 31 ± 2 | 69 ± 7 |
| Homo-Tyr | 13 ± 10 (detection limit) | N/D |
| Phe | 68 ± 10 | 84 ± 4 |
| β-OH Phec | 25 ± 9 | 86 ± 6 |
Triplicate experiments.
Original data from Kaniusaite et al.17
Control testing acceptance of β-OH Phe by M6 A-domain; no CmlA present.
Figure 4.

LCMS analysis of the reconstitution of hexapeptide extension using the separated M5/M6 modules from teicoplanin biosynthesis and different substrates for module 6 in the presence of CmlA. Peptide biosynthesis and CmlA-mediated hydroxylation reconstituted using tyrosine (A), Cl-Tyr (B), Br-Tyr (C), I-Tyr (D), F-Tyr (E), m-Tyr (F), Phe (G), and 3,5-di-Cl-Tyr (H) as M6 substrates. Solid lines indicate methylamide peptides (PCP-bound); dashed lines indicate hydrolyzed peptides (pentapeptide, black line; hexapeptides: pink line (Tyr), orange line (Cl-Tyr), green line (Br-Tyr), red line (I-Tyr), blue line (F-Tyr), pale pink line (m-Tyr), pale blue line (Phe) or purple line (3,5-di-Cl-Tyr)).
The lack of activity on 3,5-di-Br-Tyr and 3,5-di-I-Tyr can be explained in these cases by the substrate being rejected by the module 5/6 C domain due to higher VDW radii for these halogen atoms. A model of CmlA with PPant-bound 4-aminophenylalanine also indicates that while one of the m positions of the aromatic ring is relatively exposed to solvent, the other approaches within 3 Å of the CmlA peptide backbone, and in particular residues that ligate the dinuclear iron center (Figure 5). This model, if correct, suggests a possible steric clash for substrates with large m-substituents at both positions of the aromatic ring.
Figure 5.
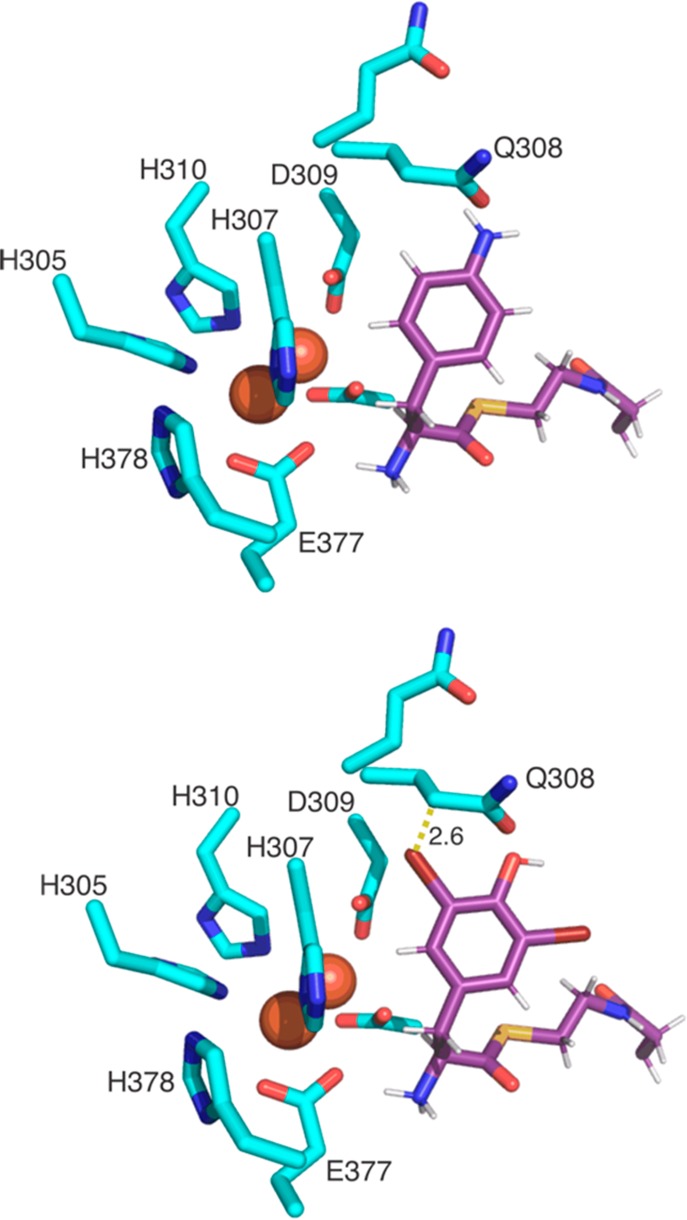
CmlA active site model showing the diiron center and coordinating residues with a model of the pantetheine arm bearing either the native CmlA substrate 4-amino-Phe (upper) or 3,5-di-Br-Tyr that is not accepted as a substrate. The side chains of CmlA residues are shown in cyan sticks. The Fe atoms are shown as red spheres, and the modeled 4-amino-Phe-Pant/3,5-di-Cl-Tyr-Pant residues are shown in purple sticks.
CmlA Activity Extends beyond Modified Tyrosine Substrates
Next, we tested the ability of the coupled peptide extension/CmlA hydroxylation assay with substrates that diverged from tyrosine. Initially, we tested meta-Tyr and homo-Tyr as substrates for both A-domain and C-domain acceptance, both with and without CmlA present. These experiments show that meta-Tyr is a good substrate for both the M6 A-domain and CmlA, with the hydroxylated amino acid then also well accepted by the M5 C-domain. In this case, the pentapeptide extension to hexapeptide using meta-Tyr in the reaction without CmlA showed that this substrate was more effectively processed by the M5 C-domain, even without the β-hydroxyl group, than Tyr, possibly due to the different position of the phenol moiety in this substrate. Homo-Tyr was very poorly accepted as an A-domain substrate, and in the absence of CmlA, a relatively small peptide product peak was observed, albeit all hydrolyzed from the NRPS. Curiously, inclusion of CmlA led to a lack of this peptide product in this case.
To further investigate CmlA substrate specificity, we also tested phenylalanine (Phe) as a potential substrate. Surprisingly, β-hydroxylated Phe was detected at position 6 in the final reconstituted hexapeptide product in assays when CmlA was present (Figure 4). A negative control (without CmlA) clearly showed no peptide extension (i.e., Phe alone was not accepted by the C-domain), once again highlighting the importance of substrate β-hydroxylation in teicoplanin precursor biosynthesis. In contrast, when the analogous experiment was repeated using a kistamicin biosynthesis reconstitution system,17 where the comparable C-domain does not accept modified tyrosine substrates, the hexapeptide product using Phe was only detected under conditions where CmlA was omitted. The absence of pentapeptide extension to hexapeptide in the presence of CmlA suggests that β-hydroxy Phe is not able to be accepted by the kistamicin C domain in this case, supporting previous results obtained with a mutated M6 that utilized a modified A-domain to accept larger substrates.17
PCP-Specificity of CmlA Appears Restricted to Biosynthetically Related Systems
Next, we assayed whether CmlA was compatible either with alternative PCP domains from GPA biosynthesis (kistamicin module 6)44 or with the PCP domains from other NRPS systems in which a similar hydroxylation occurs (Figure 6). To test this, we designed hybrid Tcp11 module 6 A-PCP constructs, in which the natural carrier protein was replaced with one of two analogous domains from skyllamycin biosynthesis machinery.42,45 The rational for choosing these PCP domains is that these domains display different interacting properties with the hydroxylase (in this case a cytochrome P450)46 in skyllamycin biosynthesis. Furthermore, the P450 in the skyllamycin system shows acceptance of only some PCP-bound amino acid substrates, with PCP7-bound amino acids accepted as substrates and PCP10-bound amino acids not accepted.42 Hence, we wanted to see if these PCP acceptance preferences were at all maintained when using CmlA. Furthermore, the skyllamycin and teicoplanin systems also share activity of the hydroxylase enzymes against PCP-bound amino acids in the main NRPS machinery, as opposed to specialized offline modules. Both hybrids were successfully cloned, expressed, and isolated as monomeric species. Initial experiments with Bht to test A- and C-domain acceptance in these hybrid constructs were successful in both cases, although the majority of peptide was now found to be hydrolyzed at the end of the assay in contrast to the native system (Figure 6). Inclusion of CmlA in the assay together with Tyr as an A-domain substrate was not able to reconstitute hexapeptide formation, which indicates that these PCP domains are not able to functionally replace the favorable interaction between CmlA and the teicoplanin PCP. In the case of the kistamicin PCP, CmlA activity was detected, and hexapeptide formation was detected, even though there is no hydroxylation of the tyrosine residue at this position during kistamicin biosynthesis. While the specificity that governs these PCP interactions is not clear without the structure of a complex, what is clear from these results is that there appears to be conservation of interactions between biosynthetic systems employing different diiron hydroxylase enzymes and their respective carrier protein targets. In the case of the skyllamycin P450/PCP interactions, positive interactions rely on an unusual exposed tryptophan on the surface of the PCP binding site of the P450,47 while the lack of interaction shown by PCP10 is believed to be governed by a rearrangement of the helices in the PCP due to the loss of a phenylalanine residue mediating interactions between α2 and α3 of the PCP.47 Such relatively small changes in PCP domains are known to be able to cause significant variation in the interaction of these domains with their partner proteins—however, what is intriguing in the case of CmlA is the ability of this enzyme to target PCP domains that are of very low sequence identity to the native chloramphenicol PCP domain, yet with selectivity toward PCP domains that are also modified by comparable diiron monooxygenase enzymes (or related to them, as is the case with kistamicin). As shown here, this offers an alternative to homologues that cannot be easily accessed for in vitro experiments, which can often severely limit biosynthetic studies in such cases.
Figure 6.

Investigating CmlA specificity for alternate PCP domains. (A) Sequence alignments of PCP domains from chloramphenicol biosynthesis with PCP domains accepted as CmlA substrates (from the teicoplanin and kistamicin NRPS module 6) and those not accepted by CmlA (from the skyllamycin NRPS modules 7 and 10). Predicated PCP secondary structure is shown, with the degree of homology highlighted in yellow (similar), orange (highly similar), and pink (identical). The post-translationally modified serine residue (green asterisk) and residues implicated in the PCP-acceptance in skyllamycin biosynthesis (blue squares) are indicated. A structural model of PCP6tei demonstrates the location of similar residues on the structure of such a PCP domain (colors as previously indicated). (B–G) LCMS analysis of the reconstitution of hexapeptide extension using separated M5 modules from teicoplanin biosynthesis with hybrid module 6 constructs combining the A-domain from teicoplanin biosynthesis together with PCP domains from the NRPS machinery from kistamicin biosynthesis (module 6, B/E) and skyllamycin biosynthesis (module 7, C/F; module 10, D/G). Peptide biosynthesis was reconstituted with either Bht (upper panels) or Tyr + CmlA (lower panels). Solid lines indicate methylamide peptides (PCP-bound), and dashed lines indicate hydrolyzed peptides (pentapeptide, black line; Bht-containing hexapeptide, pink line).
Conclusions
Within NRPS-mediated peptide biosynthesis, the exciting prospects for tailored redesign of biosynthesis pathways to produce desired secondary metabolites will remain elusive until greater understanding of the mechanisms controlling the exquisite selectivity of these machineries is reached.1 In this regard, trans-interacting enzymes such as hydroxylases and halogenases show particular promise, given the ease with which such enzymes can be added or removed from in vivo biosynthesis pathways.7 However, it is becoming clear that several alternate mechanisms within NRPS biosynthesis exist to control the actions of these enzymes, making the ability to explore such interactions in vitro a key priority. Even within the incorporation of β-hydroxyl groups during peptide biosynthesis, significant diversity exists in terms of the timing of hydroxylation, enzyme selectivity, and the interactions that control substrate binding.5,6 In this regard, the GPAs are excellent examples, for even within this closely related family of structures, two distinct pathways to generate β-hydroxylated amino acid residues exist.9 In teicoplanin biosynthesis, control of hydroxylation on the main assembly line appears to be governed by the selectivity of the upstream peptide bonding forming domain,17 while in vancomycin-related systems the offline amino acid hydroxylation is controlled through specific PCP binding by the hydroxylase.10,12,48 A recent report concerning the hydroxylation of amino acids during siderophore biosynthesis has even implicated a role of specific catalytically inactive C-domains as recruitment domains for hydroxylase enzymes (termed interface or I-domains) in a similar manner to the oxidative cross-linking cascade from GPA biosynthesis,49 showing again the potential scope for diversity of function within even relatively simple biosynthetic processes. Thus, our ability to explore hydroxylase activity and PCP selectivity in vitro using a peptide extension assay is an important step in understanding the controlling mechanisms behind such processes. In doing so, we were able to show that the CmlA enzyme from chloramphenicol biosynthesis can replace the equivalent enzyme from teicoplanin biosynthesis in spite of the low sequence homology of both the hydroxylase and PCP domains involved. Furthermore, the ability of CmlA to hydroxylate a wide variety of amino acid structures related to tyrosine bodes well for the ability to couple such an enzyme with assembly lines displaying altered A-domain or halogenase specificities. With the presence of such diiron monooxygenase enzymes in NRPS systems appearing to be a relatively common occurrence—even when the products of the clusters they are present in are not fully resolved—the possible interchangeability of these enzymes and their PCP substrates coupled with an understanding of C-domain mediated gating offers tantalizing prospects for future NRPS reengineering efforts.
Experimental Methods
Tcp11 Module 6 A-PCP Hybrid Cloning
Constructs were cloned into a pET-GB1–1d vector17 using an In-Fusion HD Cloning kit (Clontech). PCR primers were designed that share 15 bases of homology with adjacent DNA fragments. Next, these primers (Supporting Information Table 1) were used to amplify both the insert(s) and vector DNA. The plasmid DNA encoding the genes of interest were used as the template sequence for PCR. DNA fragments were analyzed on 1.5–0.8% agarose gel in a TAE buffer and the desired DNA subsequently gel-extracted using the GeneJET Gel Extraction kit (Thermo Fisher Scientific). Extracted DNA fragments were combined in the in-fusion cloning reaction as per the manufacturer’s instructions. A total of 2.5 μL of the reaction mixture was used for transformation of NEB 10-beta competent E. coli cells following standard procedures.
Protein Expression and Purification
Tcp11 module 5 (A-PCP-E-C architecture) and Tcp11 module 6 (A-PCP architecture) were expressed and purified as described previously.17 CmlA was also expressed and purified as described previously.21
Activity Assays
PCP loading and A-domain activity assays were performed as has been reported previously.17In vitro reconstitution of nonribosomal peptide synthesis was also performed as described previously, using both halogenated amino acids and peptidyl-CoA substrates that were synthesized based on previous reports.17 To reconstitute substrate β-hydroxylation, assays additionally were supplemented with 10–100 mol % of CmlA (molar ratio to module 6 A-PCP (8 μM)) and 10 μM of redox cycler (phenazine methosulfate or methyl viologen). Reactions were started with the addition of 1 mM NADH.17
HRMS and MS2 Measurements
HRMS and MS2 measurements of the products of peptide extension were performed as described previously.17 Samples were analyzed using an Orbitrap QExactive Plus (MS1 at 35K resolution, MS2 at 17.5K res; Thermo Scientific), except for meta-Bht that was analyzed using an Orbitrap QExactive HF (MS1 @ 60K res, MS2 @ 30K res; Thermo Scientific), all at 27% normalized collision energy (nce). Hexapeptide products were confirmed by HRMS (Supporting Information Table 2), and the incorporation of the variable residues at position 6 of the peptide were confirmed by monitoring the amino acid specific ammonium ions of the peptides in MS2 measurements from the hydrolyzed peptides. Conformation of the presence of a β-hydroxyl moiety in these C-terminal residues was further confirmed by monitoring for these amino acid specific ammonium ions having also eliminated a water molecule (Supporting Information Table 3).
CmlA Docking Studies
A homology model of Tcp11 PCP6 was generated using Phyre.50 Docking to CmlA was done using the previously generated model of CmlA (PDB: 4JO0) with the PCP domain of CmlP.19 The docked model of L-PAPA-PPant from that same study, generated using Sybyl-X 2.0 Surflex-Dock Suite,51 was used to visualize the interaction of CmlA with halogenated tyrosine analogs.
Structural Model of Teicoplanin PCP6 Domain
The PCP model was generated by Swiss-Model52 in alignment mode using the structure of the teicoplanin PCP7 domain (2MR8).53
Acknowledgments
Acknowledgment is given to E. Marschall (Monash) for synthesis of halogenated amino acids and J. Tailhades (Monash) for synthesis of the peptidyl CoA substrate; G. Stier (BZH-Heidelberg) for fusion protein vectors; J. Yin (University of Chicago) for the R4-4 Sfp expression plasmid; J. Lipscomb (University of Minnesota) for the CmlA expression plasmid; and E. Stegmann and N. Ziemert (University of Tübingen) for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.9b00862.
Primers and construct sequences, HRMS, and MS2 data (PDF)
Author Contributions
M.J.C. designed the study. M.K. cloned constructs, expressed proteins, and performed all activity assays. R.J.A.G. and R.B.S. performed and analyzed HRMS and MS2 experiments. T.M.M. generated the CmlA substrate model. M.J.C. wrote the manuscript and prepared the figures with input from all coauthors.
This work was supported by Monash University, EMBL Australia, and the National Health and Medical Research Council (APP1140619 (to M.J.C.)) and further supported under the Australian Research Council’s Discovery Projects funding scheme (project number DP190101272 to M.J.C.) and National Institutes of Health Grant GM135315 (to T.M.M.).
The authors declare no competing financial interest.
Supplementary Material
References
- Süssmuth R. D.; Mainz A. (2017) Nonribosomal Peptide Synthesis—Principles and Prospects. Angew. Chem., Int. Ed. 56, 3770–3821. 10.1002/anie.201609079. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; O’Brien R. V.; Khosla C. (2013) Nonproteinogenic Amino Acid Building Blocks for Nonribosomal Peptide and Hybrid Polyketide Scaffolds. Angew. Chem., Int. Ed. 52, 7098–7124. 10.1002/anie.201208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschke M.; Haslinger K.; Brieke C.; Reinstein J.; Cryle M. (2016) Regulation of the P450 oxygenation cascade involved in glycopeptide antibiotic biosynthesis. J. Am. Chem. Soc. 138, 6746–6753. 10.1021/jacs.6b00307. [DOI] [PubMed] [Google Scholar]
- Haslinger K.; Peschke M.; Brieke C.; Maximowitsch E.; Cryle M. J. (2015) X-domain of peptide synthetases recruits oxygenases crucial for glycopeptide biosynthesis. Nature 521, 105–109. 10.1038/nature14141. [DOI] [PubMed] [Google Scholar]
- Komor A. J.; Jasniewski A. J.; Que L.; Lipscomb J. D. (2018) Diiron monooxygenases in natural product biosynthesis. Nat. Prod. Rep. 35, 646–659. 10.1039/C7NP00061H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Thomas M. G.; O’Connor S. E.; Hubbard B. K.; Burkart M. D.; Walsh C. T. (2001) Aminoacyl-S-Enzyme Intermediates in β-Hydroxylations and α,β-Desaturations of Amino Acids in Peptide Antibiotics. Biochemistry 40, 11651–11659. 10.1021/bi0115434. [DOI] [PubMed] [Google Scholar]
- Winn M.; Fyans J. K.; Zhuo Y.; Micklefield J. (2016) Recent advances in engineering nonribosomal peptide assembly lines. Nat. Prod. Rep. 33, 317–347. 10.1039/C5NP00099H. [DOI] [PubMed] [Google Scholar]
- Kittilä T.; Kittel C.; Tailhades J.; Butz D.; Schoppet M.; Büttner A.; Goode R. J. A.; Schittenhelm R. B.; van Pee K.-H.; Süssmuth R. D.; Wohlleben W.; Cryle M. J.; Stegmann E. (2017) Halogenation of glycopeptide antibiotics occurs at the amino acid level during non-ribosomal peptide synthesis. Chem. Sci. 8, 5992–6004. 10.1039/C7SC00460E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim G.; Thaker M. N.; Koteva K.; Wright G. (2014) Glycopeptide antibiotic biosynthesis. J. Antibiot. 67, 31–41. 10.1038/ja.2013.117. [DOI] [PubMed] [Google Scholar]
- Puk O.; Bischoff D.; Kittel C.; Pelzer S.; Weist S.; Stegmann E.; Süssmuth R. D.; Wohlleben W. (2004) Biosynthesis of Chloro-β-Hydroxytyrosine, a Nonproteinogenic Amino Acid of the Peptidic Backbone of Glycopeptide Antibiotics. J. Bacteriol. 186, 6093–6100. 10.1128/JB.186.18.6093-6100.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulyani S.; Egel E.; Kittel C.; Turkanovic S.; Wohlleben W.; Süssmuth R. D.; van Pée K.-H. (2010) The Thioesterase Bhp is Involved in the Formation of β-Hydroxytyrosine during Balhimycin Biosynthesis in Amycolatopsis balhimycina. ChemBioChem 11, 266–271. 10.1002/cbic.200900600. [DOI] [PubMed] [Google Scholar]
- Cryle M. J.; Meinhart A.; Schlichting I. (2010) Structural Characterization of OxyD, a Cytochrome P450 Involved in b-Hydroxytyrosine Formation in Vancomycin Biosynthesis. J. Biol. Chem. 285, 24562–24574. 10.1074/jbc.M110.131904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise C. E.; Makris T. M. (2017) Recruitment and Regulation of the Non-ribosomal Peptide Synthetase Modifying Cytochrome P450 Involved in Nikkomycin Biosynthesis. ACS Chem. Biol. 12, 1316–1326. 10.1021/acschembio.7b00081. [DOI] [PubMed] [Google Scholar]
- Chen H.; Hubbard B. K.; O’Connor S. E.; Walsh C. T. (2002) Formation of β-Hydroxy Histidine in the Biosynthesis of Nikkomycin Antibiotics. Chem. Biol. 9, 103–112. 10.1016/S1074-5521(02)00090-X. [DOI] [PubMed] [Google Scholar]
- Chen H.; Walsh C. T. (2001) Coumarin formation in novobiocin biosynthesis: β-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI. Chem. Biol. 8, 301–312. 10.1016/S1074-5521(01)00009-6. [DOI] [PubMed] [Google Scholar]
- Stinchi S.; Carrano L.; Lazzarini A.; Feroggio M.; Grigoletto A.; Sosio M.; Donadio S. (2006) A derivative of the glycopeptide A40926 produced by inactivation of the β-hydroxylase gene in Nonomuraea sp. ATCC39727. FEMS Microbiol. Lett. 256, 229–235. 10.1111/j.1574-6968.2006.00120.x. [DOI] [PubMed] [Google Scholar]
- Kaniusaite M.; Tailhades J.; Marschall E. A.; Goode R. J. A.; Schittenhelm R. B.; Cryle M. J. (2019) A proof-reading mechanism for non-proteinogenic amino acid incorporation into glycopeptide antibiotics. Chem. Sci. 10, 9466–9482. 10.1039/C9SC03678D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasniewski A. J.; Knoot C. J.; Lipscomb J. D.; Que L. (2016) A Carboxylate Shift Regulates Dioxygen Activation by the Diiron Nonheme β-Hydroxylase CmlA upon Binding of a Substrate-Loaded Nonribosomal Peptide Synthetase. Biochemistry 55, 5818–5831. 10.1021/acs.biochem.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris T. M.; Knoot C. J.; Wilmot C. M.; Lipscomb J. D. (2013) Structure of a Dinuclear Iron Cluster-Containing β-Hydroxylase Active in Antibiotic Biosynthesis. Biochemistry 52, 6662–6671. 10.1021/bi400845b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu V. V.; Makris T. M.; Lipscomb J. D.; Que L. (2011) Active-Site Structure of a β-Hydroxylase in Antibiotic Biosynthesis. J. Am. Chem. Soc. 133, 6938–6941. 10.1021/ja201822v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris T. M.; Chakrabarti M.; Münck E.; Lipscomb J. D. (2010) A family of diiron monooxygenases catalyzing amino acid beta-hydroxylation in antibiotic biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 107, 15391–15396. 10.1073/pnas.1007953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasniewski A. J.; Komor A. J.; Lipscomb J. D.; Que L. (2017) Unprecedented (μ-1,1-Peroxo)diferric Structure for the Ambiphilic Orange Peroxo Intermediate of the Nonheme N-Oxygenase CmlI. J. Am. Chem. Soc. 139, 10472–10485. 10.1021/jacs.7b05389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.-L.; Huang F.; Haydock S. F.; Mironenko T.; Leadlay P. F.; Spencer J. B. (2004) Biosynthetic Gene Cluster of the Glycopeptide Antibiotic Teicoplanin: Characterization of Two Glycosyltransferases and the Key Acyltransferase. Chem. Biol. 11, 107–119. 10.1016/S1074-5521(04)00002-X. [DOI] [PubMed] [Google Scholar]
- Sosio M.; Stinchi S.; Beltrametti F.; Lazzarini A.; Donadio S. (2003) The Gene Cluster for the Biosynthesis of the Glycopeptide Antibiotic A40926 by Nonomuraea Species. Chem. Biol. 10, 541–549. 10.1016/S1074-5521(03)00120-0. [DOI] [PubMed] [Google Scholar]
- Komor A. J.; Rivard B. S.; Fan R.; Guo Y.; Que L.; Lipscomb J. D. (2017) CmlI N-Oxygenase Catalyzes the Final Three Steps in Chloramphenicol Biosynthesis without Dissociation of Intermediates. Biochemistry 56, 4940–4950. 10.1021/acs.biochem.7b00695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor A. J.; Rivard B. S.; Fan R.; Guo Y.; Que L.; Lipscomb J. D. (2016) Mechanism for Six-Electron Aryl-N-Oxygenation by the Non-Heme Diiron Enzyme CmlI. J. Am. Chem. Soc. 138, 7411–7421. 10.1021/jacs.6b03341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoot C. J.; Kovaleva E. G.; Lipscomb J. D. (2016) Crystal structure of CmlI, the arylamine oxygenase from the chloramphenicol biosynthetic pathway. JBIC, J. Biol. Inorg. Chem. 21, 589–603. 10.1007/s00775-016-1363-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris T. M.; Vu V. V.; Meier K. K.; Komor A. J.; Rivard B. S.; Münck E.; Que L.; Lipscomb J. D. (2015) An Unusual Peroxo Intermediate of the Arylamine Oxygenase of the Chloramphenicol Biosynthetic Pathway. J. Am. Chem. Soc. 137, 1608–1617. 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Martínez L. T.; Borsetto C.; Gomez-Escribano J. P.; Bibb M. J.; Al-Bassam M. M.; Chandra G.; Bibb M. J. (2014) New Insights into Chloramphenicol Biosynthesis in Streptomyces venezuelae ATCC 10712. Antimicrob. Agents Chemother. 58, 7441–7450. 10.1128/AAC.04272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Chanco E.; Zhao H. (2012) CmlI is an N-oxygenase in the biosynthesis of chloramphenicol. Tetrahedron 68, 7651–7654. 10.1016/j.tet.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholec M.; Sello J. K.; Walsh C. T.; Thomas M. G. (2007) Formation of an aminoacyl-S-enzyme intermediate is a key step in the biosynthesis of chloramphenicol. Org. Biomol. Chem. 5, 1692–1694. 10.1039/b703356g. [DOI] [PubMed] [Google Scholar]
- He J.; Magarvey N.; Piraee M.; Vining L. C. (2001) The gene cluster for chloramphenicol biosynthesis in Streptomyces venezuelae ISP5230 includes novel shikimate pathway homologues and a monomodular non-ribosomal peptide synthetase gene. Microbiology 147, 2817–2829. 10.1099/00221287-147-10-2817. [DOI] [PubMed] [Google Scholar]
- Sosio M.; Kloosterman H.; Bianchi A.; de Vreugd P.; Dijkhuizen L.; Donadio S. (2004) Organization of the teicoplanin gene cluster in Actinoplanes teichomyceticus. Microbiology 150, 95–102. 10.1099/mic.0.26507-0. [DOI] [PubMed] [Google Scholar]
- Pootoolal J.; Thomas M. G.; Marshall C. G.; Neu J. M.; Hubbard B. K.; Walsh C. T.; Wright G. D. (2002) Assembling the glycopeptide antibiotic scaffold: The biosynthesis of from Streptomyces toyocaensis NRRL15009. Proc. Natl. Acad. Sci. U. S. A. 99, 8962–8967. 10.1073/pnas.102285099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B.; Du L.; Sanchez C.; Edwards D. J.; Chen M.; Murrell J. M. (2002) Cloning and Characterization of the Bleomycin Biosynthetic Gene Cluster from Streptomyces verticillus ATCC15003. J. Nat. Prod. 65, 422–431. 10.1021/np010550q. [DOI] [PubMed] [Google Scholar]
- Du L.; Sánchez C.; Chen M.; Edwards D. J.; Shen B. (2000) The biosynthetic gene cluster for the antitumor drug bleomycin from Streptomyces verticillus ATCC15003 supporting functional interactions between nonribosomal peptide synthetases and a polyketide synthase. Chem. Biol. 7, 623–642. 10.1016/S1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- Hou J.; Robbel L.; Marahiel M. A. (2011) Identification and Characterization of the Lysobactin Biosynthetic Gene Cluster Reveals Mechanistic Insights into an Unusual Termination Module Architecture. Chem. Biol. 18, 655–664. 10.1016/j.chembiol.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Sunbul M.; Marshall N. J.; Zou Y.; Zhang K.; Yin J. (2009) Catalytic Turnover-Based Phage Selection for Engineering the Substrate Specificity of Sfp Phosphopantetheinyl Transferase. J. Mol. Biol. 387, 883–898. 10.1016/j.jmb.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Tailhades J.; Schoppet M.; Greule A.; Peschke M.; Brieke C.; Cryle M. J. (2018) A route to diastereomerically pure phenylglycine thioester peptides: crucial intermediates for investigating glycopeptide antibiotic biosynthesis. Chem. Commun. 54, 2146–2149. 10.1039/C7CC09409D. [DOI] [PubMed] [Google Scholar]
- Brieke C.; Cryle M. J. (2014) A Facile Fmoc Solid Phase Synthesis Strategy To Access Epimerization-Prone Biosynthetic Intermediates of Glycopeptide Antibiotics. Org. Lett. 16, 2454–2457. 10.1021/ol500840f. [DOI] [PubMed] [Google Scholar]
- Chanco E.; Choi Y. S.; Sun N.; Vu M.; Zhao H. (2014) Characterization of the N-oxygenase AurF from Streptomyces thioletus. Bioorg. Med. Chem. 22, 5569–5577. 10.1016/j.bmc.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Uhlmann S.; Süssmuth R. D.; Cryle M. J. (2013) Cytochrome P450sky Interacts Directly with the Nonribosomal Peptide Synthetase to Generate Three Amino Acid Precursors in Skyllamycin Biosynthesis. ACS Chem. Biol. 8, 2586–2596. 10.1021/cb400555e. [DOI] [PubMed] [Google Scholar]
- Tailhades J.; Zhao Y.; Schoppet M.; Greule A.; Goode R. J. A.; Schittenhelm R. B.; De Voss J. J.; Cryle M. J. (2019) An enzymatic cascade to evaluate the tricyclization of glycopeptide antibiotic precursor peptides as a prequel to biosynthetic re-design. Org. Lett. 21, 8635–8640. 10.1021/acs.orglett.9b03245. [DOI] [PubMed] [Google Scholar]
- Greule A.; Izoré T.; Iftime D.; Tailhades J.; Schoppet M.; Zhao Y.; Peschke M.; Ahmed I.; Kulik A.; Adamek M.; Goode R. J. A.; Schittenhelm R. B.; Kaczmarski J. A.; Jackson C. J.; Ziemert N.; Krenske E. H.; De Voss J. J.; Stegmann E.; Cryle M. J. (2019) Kistamicin biosynthesis reveals the biosynthetic requirements for production of highly crosslinked glycopeptide antibiotics. Nat. Commun. 10, 2613. 10.1038/s41467-019-10384-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohle S.; Appelt C.; Roux M.; Fiedler H.-P.; Süssmuth R. D. (2011) Biosynthetic Gene Cluster of the Non-ribosomally Synthesized Cyclodepsipeptide Skyllamycin: Deciphering Unprecedented Ways of Unusual Hydroxylation Reactions. J. Am. Chem. Soc. 133, 6194–6205. 10.1021/ja108971p. [DOI] [PubMed] [Google Scholar]
- Greule A.; Stok J. E.; De Voss J. J.; Cryle M. J. (2018) Unrivalled diversity: the many roles and reactions of bacterial cytochromes P450 in secondary metabolism. Nat. Prod. Rep. 35, 757–791. 10.1039/C7NP00063D. [DOI] [PubMed] [Google Scholar]
- Haslinger K.; Brieke C.; Uhlmann S.; Sieverling L.; Süssmuth R. D.; Cryle M. J. (2014) The Structure of a Transient Complex of a Nonribosomal Peptide Synthetase and a Cytochrome P450 Monooxygenase. Angew. Chem., Int. Ed. 53, 8518. 10.1002/anie.201404977. [DOI] [PubMed] [Google Scholar]
- Stegmann E.; Frasch H.-J.; Wohlleben W. (2010) Glycopeptide biosynthesis in the context of basic cellular functions. Curr. Opin. Microbiol. 13, 595–602. 10.1016/j.mib.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Reitz Z. L.; Hardy C. D.; Suk J.; Bouvet J.; Butler A. (2019) Genomic analysis of siderophore β-hydroxylases reveals divergent stereocontrol and expands the condensation domain family. Proc. Natl. Acad. Sci. U. S. A. 116, 19805. 10.1073/pnas.1903161116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. A.; Mezulis S.; Yates C. M.; Wass M. N.; Sternberg M. J. E. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sybyl-X 2.0; Tripos International: St. Louis, MO. [Google Scholar]
- Waterhouse A.; Bertoni M.; Bienert S.; Studer G.; Tauriello G.; Gumienny R.; Heer F. T.; de Beer T. A P.; Rempfer C.; Bordoli L.; Lepore R.; Schwede T. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslinger K.; Redfield C.; Cryle M. J. (2015) Structure of the terminal PCP domain of the non-ribosomal peptide synthetase in teicoplanin biosynthesis. Proteins: Struct., Funct., Genet. 83, 711–721. 10.1002/prot.24758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.