

Abstract
5-(Adamantan-1-yl)-3-[(4-chlorobenzyl)sulfanyl]-4-methyl-4H-1,2,4-triazole (4) was identified as a potential 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitor and this paper describes the in-depth structural analysis thereof. Compound 4 was synthesized in a 92% yield and its 3D-structure confirmed by single-crystal X-ray diffraction. Hirshfeld surface analysis indicated that H…H, C-H…C, C-H…Cl and especially C-H…N hydrogen bond interactions are the primary contributors to the intermolecular stabilisation in the crystal. In order to explore the properties of 4, free from the influence of the crystal field, density functional theory (DFT) calculations were conducted. Results indicated that the DFT optimized geometry of 4 produced a conformer (4a) that is significantly different from the crystal structure. Further experiments confirmed that the crystal structure is not the absolute minimum conformation. This indicated that the crystal packing forces has significantly influenced the conformation thereof. Frontier molecular orbital energies and net atomic charges were also calculated to elucidate the electronic properties of 4a. These results provided insight into areas of the molecule that may present with the ability to form binding interactions at the 11β-HSD1 active site. Molecular docking experiments revealed important intermolecular interactions between 4a and 11β-HSD1. These results indicate that 4 may be considered for further drug design endeavors.
Subject terms: Computational chemistry, Medicinal chemistry
Introduction
The insertion of an adamantyl moiety into various bioactive molecules leads to compounds with relatively higher lipophilicity, which in turn results in modification of their bioavailability and modulates their therapeutic indices1–3. Several adamantane-based drugs are currently used as efficient antiviral4–7, anti-TB8,9 and anticancer10,11 agents. In addition, vildagliptin12 and saxagliptin13 are currently used as oral hypoglycemic adamantane-based agents for the treatment of type 2 diabetes acting via inhibition of dipeptidyl peptidase IV. In 2003, the adamantyl-1,2,4-triazoles I, II and III were discovered as potent inhibitors of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1)14,15. Currently a number of structurally related 1,2,4-triazole 11β-HSD1 inhibitors; IV16, V17, and VI18, are under clinical investigation (Fig. 1). The 11β-HSD1 enzyme is involved in the development of non-insulin-dependent diabetes, hyperglycemia, obesity, insulin resistance, hyperlipidemia, hypertension and other symptoms associated with excessive body cortisol19–22. Therefore, inhibitors of this enzyme could lead to the development of important therapeutic agents.
Figure 1.

The structures the adamanty l-1, 2, 4-triazoles (I–III) and related clinical 1, 2, 4-triazoles (IV–VI) 11β-HSD1 inhibitors.
In continuation of our on-going interest in the structural studies and biological application of adamantane derivatives23–27, we report herein the crystal structure, Hirshfeld surface analysis, density functional theoretical studies and electronic properties of the title adamantane-triazole hybrid derivative 4. In addition, compound 4 has a number of structural similarities to the 11β-HSD1 inhibitors shown in Fig. 1. Therefore, molecular docking experiments at the 11β-HSD1 active site were conducted in order to predict the potential 11β-HSD1 binding affinity and binding interactions of 4.
Results and Discussion
Chemical synthesis
The synthesis of compound 4 (Fig. 2) commenced from the reaction of adamantane-1-carbohydrazide 1 with methyl isothiocyanate yielding the corresponding thiosemicarbazide derivative 2, which was cyclized to its 1,2,4-triazole analogue 3. In the next step, 3 was reacted with 4-chlorobenzyl chloride in the presence of sodium methylate, in ethanol, at reflux temperature as previously described28. This method yielded the title compound 4 as a major product (65%) in addition to the undesirable N-chlorobenzyl analogue 5 as a minor product (22%). In an attempt to improve the yield of 4 and prevent the formation of 5, different reaction conditions were studied at the 4-chlorobenzyl chloride conjugation step. We found that the use of N,N-dimethylformamide (DMF) as solvent, anhydrous potassium carbonate as base, with the reaction conducted at room temperature for 6 hours, led to the formation of compound 4 with a yield of 92% and the formation of the N-chlorobenzyl analogue (5) was avoided (Fig. 2).
Figure 2.
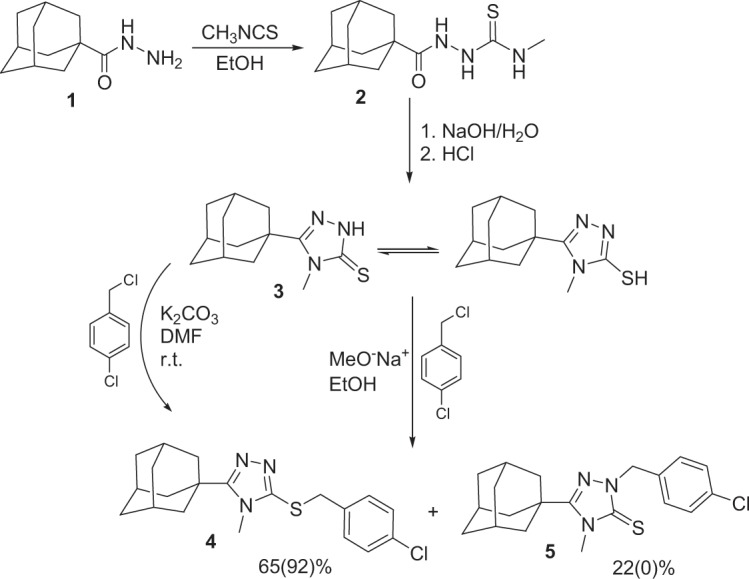
Synthetic approach for compound 4 (the percentage yields shown in parentheses are those obtained on carrying out the reaction in DMF with anhydrous potassium carbonate at room temperature).
Crystal structure
Crystals of compound 4 suitable for single crystal X-ray analysis were grown via slow evaporation of an ethanolic solution at room temperature. The ORTEP diagram along with the atom-labelling scheme is given in Fig. 3. X-ray crystallographic analysis revealed that 4 crystallizes in the monoclinic space group P21/n, ß = 96.361(1)° and Z = 4 (full crystallographic data are presented in the materials and methods section. Selected bond lengths and bond angles of the adamantane ring, triazole-sulfanyl- and the chlorophenyl moiety are given in Table 1 and, in general, these parameters are comparable to other previously reported similar crystal structures24–27 (see the Supplementary material for a complete list of crystallographic parameters). The crystal packing arrangement shows the formation of pairs of dimers in an inversion related alternate head-to-tail molecular formation in the bc plane (Fig. 4a). Prominent non-classical C-H…N hydrogen-bonds are observed between the dimers and adjacent molecules (Table 2, Fig. 4c). Another non-prominent intramolecular C-H…N hydrogen bond is present between H14A of the adamantane moiety and N3 with an H…A distance of 2.652 Å and an angle of 123.57°. These hydrogen bonds add to the potential stability of the molecules within the crystal field and enables the formation, in addition to other short range contacts (Fig. 4b), of the three-dimensional network structure.
Figure 3.

ORTEP diagram of the asymmetric unit of compound 4 drawn at 50% ellipsoids for non-hydrogen atoms.
Table 1.
Selected bond lengths (Å) and angles (°) of the crystal structure of compound 4.
| Bond | Length (Å) | Bond | Angle (°) |
|---|---|---|---|
| C1-C2 | 1.373(3) | C2-C1-Cl1 | 119.39(15) |
| C4-C7 | 1.505(3) | C5-C4-C7 | 119.51(19) |
| C7-S1 | 1.832(2) | C4-C7-S1 | 107.11(13) |
| C8-S1 | 1.752(17) | N1-C8-S1 | 125.05(13) |
| C8-N1 | 1.370(2) | N2-C8-N1 | 110.83(14) |
| C9-N3 | 1.316(2) | C9-C11-C12 | 111.98(13) |
| C10-N1 | 1.464(2) | C20-C18-C17 | 109.56(15) |
| N2-N3 | 1.380(2) | C8-N1-C9 | 104.71(13) |
| C9-C11 | 1.505(2) | C9-N2-N3 | 106.74(14) |
| C11-C19 | 1.546(2) | C9-N3-N2 | 108.40(13) |
| C15-C20 | 1.534(3) | C8-S1-C7 | 102.14(8) |
Figure 4.

(a) Molecular crystal packing showing the head-to-tail dimers of compound 4 within the unit cell. (b) Illustration showing the complex series of short-range intermolecular interactions (cyan) within the crystal field. The molecule shown in magenta was used as the central point to illustrate all interactions present. (c) Schematic of the prominent non-classical C-H…N hydrogen-bonds (red) observed, as described in Table 2.
Table 2.
Prominent hydrogen bonds for 4.
| D-H…A | d (H…A)/Å | d (D…A)/Å | (D-H…A)/° |
|---|---|---|---|
| C5-H5…N21 | 2.41 | 3.353 (2) | 175.6 |
| C10-H10C…N3i | 2.59 | 3.215 (2) | 121.9 |
Symmetry codes: (i) –x + 1, −y + 1, −z + 1.
Hirshfeld surface analysis
The Hirshfeld surface and subsequent fingerprint plots were calculated to quantify the intermolecular contacts present within the crystal structure of compound 429,30. The respective acceptor and donor atoms showing strong C-H…N intermolecular hydrogen bonds (for C5—H5…N2, C10—H10C…N3 and C14-H14A…N3) are indicated as bright red spots on the Hirshfeld surface (Fig. 5a). This finding is substantiated by the calculated electrostatic potential (Fig. 5b) of the molecule that was used to generate the Hirshfeld surface. The negative potential (acceptor) is indicated as a red surface around the two nitrogen atoms (N2 and N3) and the blue surface area, indicating the positive potential (donor), is mapped in the proximity of the hydrogen atoms (H5, H10C and H14A).
Figure 5.

(a) The three-dimensional Hirshfeld surface showing the intermolecular interactions of 4 plotted over dnorm. (b) Electrostatic potential of 4 mapped using the 6-311 G(d,p) basis set at B3LYP level theory over a range ± 0.03 a.u. Dotted lines (green) signify prominent C-H…N hydrogen bonds.
Significant intermolecular interactions are mapped in Fig. 6. On the Hirshfeld surfaces the H…H interactions appear as the largest region (55.1%) of the fingerprint plot with a high concentration at de = di ~1.2 Å. It is of interest that the H…H bond between H2 and H17A is indicated as a red spot on the Hirshfeld surface indicating that this is a significant short range H…H contact. Two sharp spikes (de + di ~2.2 Å) on the fingerprint plot were observed for the N⋯H/H⋯N contacts, corresponding to the C—H…N interactions. These spikes are indicative of a strong hydrogen-bond interaction. The C…H/H…C contacts contribute to 10.3% of the Hirshfeld area. These contacts also appear as two spikes in the vicinity of de + di ~2.5 Å. Cl…H/Cl…H contacts (de + di ~3.6 Å), corresponding to C-H…Cl bond interactions, contribute to 12.8% of the surface area. This contact is however not visible as a red spot on the Hirshfeld surface. Most probably attributed to the fact that the sum of the van der Waals radii between the interacting C-H…Cl atoms is smaller than the interatomic distances between these atoms. All other contacts observed were found to contribute less than 4.3%. It is therefore clear that the H…H, C…H/H…C, Cl…H/Cl…H and especially N…H/H…N contacts, were the most significant contributors among the interacting atoms. This finding therefore indicates the significance of these contacts in the packing arrangement of the crystal structure. Based on these findings a detailed model was constructed showing the most prominent short range intermolecular contacts that are responsible for the packing arrangement and formation of the three-dimensional network structure of 4 (Fig. 7).
Figure 6.

Fingerprint plots of 4, showing the contributions of atoms within specific interacting pairs (blue areas). For each fingerprint map, the grey area is a representation of the whole plot. Surface maps next to each fingerprint plot indicate the applicable areas (indicated in blue) that are associated with the specific intermolecular contact(s).
Figure 7.
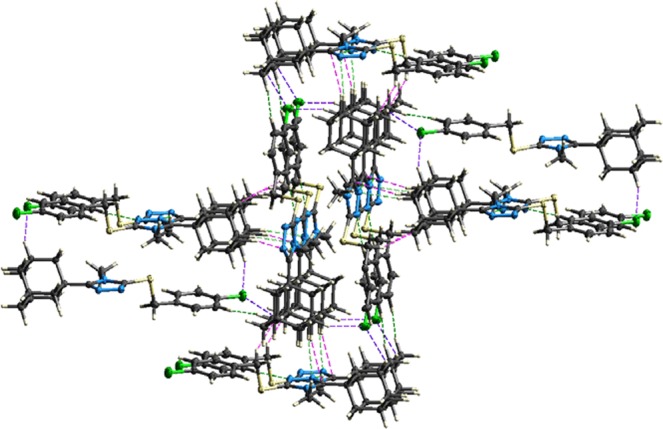
Partial packing diagram showing the most prominent intermolecular interactions present within the crystal structure of 4 based on the Hirshfeld surface analysis findings. Interaction color scheme: H…H = Red; CH…N = Green; C…H = Magenta; Cl…H = Blue.
Geometry optimization
Since we also wanted to investigate the potential 11β-HSD1 binding affinity of 4, we were interested in the molecular conformation thereof released from the crystalline field and without the influence of the intermolecular contacts. After determining the compound’s three-dimensional structure in crystalline state, the next step was to define the energy minima conformation thereof. To do this, the Gaussian09 program package was used to perform a density functional theory (DFT) geometry optimization according to the methods as previously described31–34.
The conformation of the molecule changed significantly in its DFT optimized state (4a) when compared to the crystal structure (Fig. 8). This is confirmed by comparing selected torsion angles of the molecule, obtained in the crystal structure analyses and after DFT optimization (Table 3). Table 3 shows that the DFT optimized N1-C8-S1-C7 dihedral angle is quantitatively closer to being flat. This could suggest that the sulfur π-π lone pair may be orthogonally oriented and possibly participates in a π-electron delocalization that is extended to the planar triazole ring leading to the observed DFT molecular minimum of 4a. To verify this point, the torsion around C8-S1 was fixed orthogonally and the DFT experiment was repeated in order to generate a conformer (4b) that is very close (RMSD = 0.221 Å) to the one observed in the crystal structure (see Supplementary information Figs. S2 and S3). Comparison of the energy differences between 4a (27.973 kcal mol−1) and 4b (47.495 kcal mol−1) indicates that 4a is a more stable system by as much as 19.5 kcal/mol. It is therefore confirmed that the experimental crystal structure is not the absolute minimum and that in the solid state the observed conformation is attributable to the crystalline packing forces. This phenomenon is similar to findings described in the literature35,36. The DFT optimized structure (4a) is therefore deemed the appropriate conformer for further structural investigation in order to determine the potential 11β-HSD1 binding affinity of 4.
Figure 8.
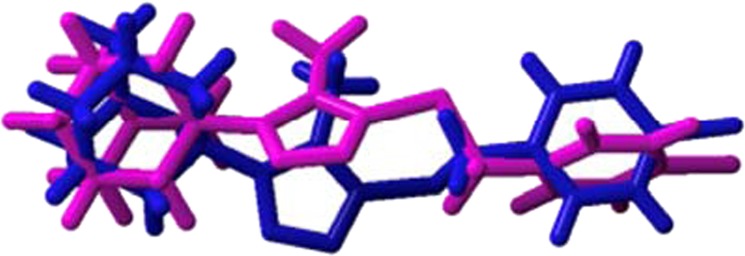
Atom-by-atom superimposition of the DFT optimized compound (4a, magenta) on the X-ray structure (blue) of the title compound (RMSD = 1.491 Å, performed using YASARA version 18.11.21 (YASARA Biosciences GmbH)).
Table 3.
Selected torsion angles (Å) of the crystal structure and the DFT optimized structure (4a).
| Crystal structure | Crystal structure | DFT optimized structurea |
|---|---|---|
| Cl1-C1-C2-C3 | 179.12 | −179.92 |
| C3-C4-C7-S1 | −85.68 | −87.33 |
| C4-C7-S1-C8 | 179.33 | 169.53 |
| N2-C8-N1-C10 | −177.78 | 177.56 |
| S1-C8-N1-C9 | 177.28 | 175.58 |
| S1-C8-N2-N3 | −177.37 | −174.80 |
| N1-C8-S1-C7 | 82.82 | −147.06 |
| N3-C9-C11-C12 | 126.1 | 98.62 |
| C11-C9-N1-C8 | 179.65 | −178.58 |
| N3-C9-N1-C10 | 177.85 | −177.99 |
| C9-C11-C12-C13 | −178.43 | −178.64 |
| C19-C11-C12-C13 | −59.54 | −59.24 |
aThe B3LYP/6-311++G(d,p) level of theory was used for optimization.
Frontier molecular orbitals (FMO) and net atomic charges
Frontier orbital energy studies can provide valuable insights into biologically active compounds’ potential biological mechanisms37–40. Figure 9 shows the distribution and energy levels of the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) orbitals calculated for conformer 4a at the B3LYP/6-311 G level. The electron distribution is mainly scattered in HOMO over the triazole ring and the sulfur moiety, whereas the LUMO is mainly disseminated over the sulfur- and chlorophenyl moieties. This indicates that there is a transfer of charge between the triazole ring and the chlorophenyl moiety. The Eigen values of LUMO (−0.5151 eV) and HOMO (−9.1739 eV) and their large energy gap (8.6588 eV), provides further support that a charge transfer exist within 4a which could promote its bioactivity and ability to form biological interactions at the triazole- and/or chlorophenyl moiety. In addition, the low total energy and large energy gap of HOMO-LUMO suggest that the molecule has good stability and is in its lowest energy conformation.
Figure 9.
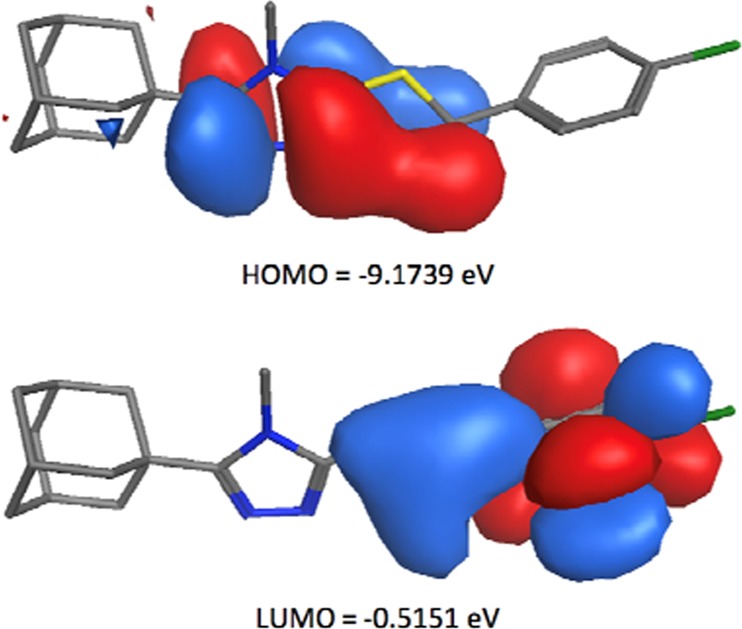
The HOMO and LUMO FMOs of conformer 4a calculated using B3LYP/6-311++G (d,p).
To study the electronic properties of 4a, the net atomic charges of the DFT optimized structure were calculated using Avogadro 1.2.0 (Fig. 10). The three N-atoms in the triazole ring have the highest electronegativity and the lowest net atomic charge values (N1 = −0.29 e, N2 = −0.12 e and N3 = −0.13). It is therefore expected that the triazole moiety as a whole or the individual N-atoms contained within the structure will be the most favored to interact through hydrogen bond interactions and/or other intermolecular forces with closely positioned amino acids within the 11β-HSD1 active site. In addition, the sulfur and chlorine moieties also have relatively low net atomic charges (S = −0.071 e and Cl = −0.082 e) and may also be involved in productive intermolecular interactions or may aid the ability of functional moieties they are conjugated to, such as the phenyl moiety, to form binding interactions with the enzyme.
Figure 10.
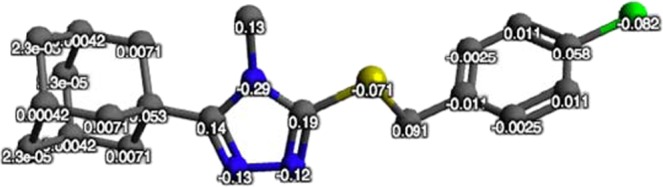
The net atomic charges (e) of the DFT optimized structure of compound 4.
Molecular docking studies
The Molecular Operating Environment (MOE) 2018 suite was used for the molecular docking experiments of compound 441. The crystal structure (Protein Data Bank I.D.: 4C7J) of human 11β-HSD1 co-crystallized with the 11β-HSD1 inhibitor 4-cyclopropyl-N-(trans-5-hydroxy-2-adamantyl)-2-(2-hydroxyethoxy)-thiazole-5-carboxamide (4YQ)40 was used for all the docking experiments. 4YQ has a potent 11β-HSD1 IC50 value of 9.9 nM and is structurally related to compound 4 of this study42. This enzyme and the co-crystallized ligand is therefore deemed appropriate to explore and predict the potential 11β-HSD1 inhibitory activity of compound 4. The DFT optimized conformer (4a) was used for the docking experiments.
The binding mode of the co-crystallized ligand, 4YQ, indicated the formation of six hydrogen bond contacts through the interaction of the carbonyl, sulfur and hydroxyl moieties with residues Ser170, Leu215 and Asp259 (Figs. 11 and 12). These interactions are responsible for the potent inhibition of 11β-HSD1 by 4YQ (human 11β-HSD1 IC50 = 9.9 nM)42. The binding affinity of the co-crystallized ligand, 4YQ, was calculate as −12.511 kcal/mol. Using the protocol as described in the experimental section, the DFT optimized minimum energy conformer (4a) was docked. The results showed that 4a was able to access and bind to the 11β-HSD1 active site, with an orientation similar to 4YQ (Figs. 11 and 12). Two H-π interactions between the adamantane and Tyr183 and the triazole and Leu126 were observed. In addition, a hydrogen bond interaction was observed between the halogen function and Asp259. These results were as expected based on the FMO and net atomic charge findings as discussed in the previous sections. The predicted binding affinity of 4a was found to be −11.953 kcal/mol. These interactions, together with the predicted binding affinity, which is similar to the binding affinity of 4YQ, indicate that 4 should show some significant 11β-HSD1 inhibitory activity within a similar range as 4YQ. However, biological tests need to be done in order to validate the computational predictions.
Figure 11.

The binding mode, orientation and predicted binding affinity (kcal/mol) of 4YQ (magenta) and 4a (green) within the 11β-HSD1 active site.
Figure 12.

Binding interactions of the co-crystallized ligand, 4YQ (left) and conformer 4a (right), within the 11β-HSD1 active site.
Conclusion
5-(Adamantan-1-yl)-3-[(4-chlorobenzyl)sulfanyl]-4-methyl-4H-1,2,4-triazole (4) was synthesised in a 94% yield using an optimized synthesis method. Single crystal X-ray diffraction analysis was used to confirm the three-dimensional conformation of 4 and showed that the molecules form pairs of dimers in an inversion related alternate head-to-tail molecular formation in the bc plane. Hirshfeld surface analysis revealed the important contacts within the crystal structure that led to the molecular packing and observed conformation of 4. Using DFT calculations, the optimised molecular structure (4a) free from the influence of the crystal field, FMO and electronic properties were deduced. Results indicated that the conformation of the crystal structure was significantly influenced by the crystal packing forces and confirmed that 4a is the optimal conformer for docking analysis. Docking experiments showed that 4a forms important binding interactions with amino acids within the active site of the 11β-HSD1 enzyme and presented with a binding affinity similar to a known potent inhibitor. These results have therefore provided valuable structural information of compound 4 that may be used in the further development of this compound or derivatives thereof as potential therapeutic agents.
Materials and Methods
Synthesis of 5-(adamantan-1-yl)-3-[(4-chlorobenzyl)sulfanyl]-4-methyl-4H-1,2,4-triazole (4)
The synthesis method followed is similar to previously described by our group28 with some minor optimization steps. 4-Chlorobenzyl chloride (322 mg, 2 mmol) and anhydrous potassium carbonate (276 mg, 2 mmol) were added to a solution of 5-(adamantan-1-yl)-4-methyl-4H-1,2,4-triazole-3-thiol 3 (499 mg, 2 mmol) in DMF (8 mL), and the mixture was stirred at room temperature for 6 hours. Water (20 mL) was then gradually added to the reaction mixture with stirring for 30 minutes. The precipitated crude product was filtered, washed with water (20 mL), dried and crystallized from aqueous ethanol to yield 688 mg (92%) compound 4 (C20H24ClN3S) as colorless prism crystals. M.p.: 175–177 °C. Single crystals suitable for X-ray analysis were obtained by slow evaporation of an ethanolic solution at room temperature. 1H NMR (DMSO-d6, 700.17 MHz): δ 1.72–1.77 (m, 6 H, adamantane-H), 2.0 (s, 6 H, adamantane-H), 2.04 (s, 3 H, adamantane-H), 3.47 (s, 3 H, CH3), 4.26 (s, 2 H, benzylic CH2), 7.26 (d, 2 H, Ar-H, J = 8.3 Hz), 7.35 (d, 2 H, Ar-H, J = 8.3 Hz). 13C NMR (DMSO-d6, 176.08 MHz): δ 32.67 (CH3), 27.99, 34.94, 37.11, 40.49 (adamantane-C), 39.35 (benzylic-CH2), 128.84, 131.21, 132.50, 137.03 (Ar-C), 150.34, 131.26 (Triazole-C).
X-ray crystallography
Single-crystal X-ray diffraction data was collected at 160(1) K on a Rigaku OD XtaLAB Synergy, Dualflex, Pilatus 200 K diffractometer using a single wavelength X-ray source (Cu Kα radiation: λ = 1.54184 Å)43 from a micro-focus sealed X-ray tube and an Oxford liquid-nitrogen Cryostream cooler. The selected suitable single crystal was mounted using polybutene oil on a flexible loop fixed on a goniometer head and immediately transferred to the diffractometer. Pre-experiment, data collection, data reduction and analytical absorption correction44,45 were performed with the program suite CrysAlisPro44 using Olex246. The structure was solved with the SHELXT47 small molecule structure solution program and refined with the SHELXL 2018/3 program package48 by full-matrix least-squares minimization on F2. PLATON49 was used to check the result of the X-ray analysis. The crystal data and refinement parameters are shown in Table 4.
Table 4.
Single crystal X-ray crystallographic data of 4.
| Data | Compound 4 |
|---|---|
| Formula | C20H24ClN3S |
| Formula weight | 373.93 |
| Temperature (K) | 160 |
| Crystal system, Space group | Monoclinic, P21/n |
| a, b, c (Å) | 6.61380(10), 13.3456(2), 20.9061(4) |
| ß (°) | 96.361(1) |
| V (Å3) | 1833.92(5) |
| Z | 4 |
| Radiation type | CuKα (λ = 1.54184) |
| μ (mm−1) | 2.954 |
| No. of reflections | 13268 |
| No. of unique reflections/obs. reflections | 3708/3107 |
| No. of parameters | 227 |
| No. of restraints | 0 |
| Δρmax, Δρmin (e Å−3) | +0.39, −0.39 |
| Tmin, Tmax | 0.706, 0.875 |
| Rint | 0.0353 |
| Crystal size (mm) | 0.18 × 0.08 × 0.05 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.0379, 0.0865, 1.020 |
| CCDC number | 1918378 |
A CIF file containing complete information of the studied structure was deposited with CCDC, deposition number 1918378 and is freely available upon request from the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (Fax: + 44-1223-336033; e-mail: deposit@ccdc.cam.ac.uk) or from the following website: www.ccdc.cam.ac.uk/data_request/cif.
Hirshfeld surface analysis
The Hirshfeld surfaces and 2D fingerprint plots were generated using Crystal Explorer 3.150. The X-ray single-crystal crystallographic information file of 4 was used as input file. The TONTO application within Crystal Explorer was used to calculate the electrostatic potential surface map using B3LYP/6-311G(d,p) ± 0.03 a.u.
Quantum chemical calculations
A crystal unit within the crystal structure of the 4 was selected as the starting structure for the DFT calculations. Structural optimization was performed using the Gaussian09 program package31, in the same manner as previously reported by our group26. Energy minima calculations were carried out using the Becke’s three parameters Lee-Yang-Parr exchange correlation functional (B3LYP). This calculation combines Becke’s hybrid exchange functional32 with Lee, Yang and Parr’s gradient-correlation functional using the base set of 6-311 G++(d,p)33. Previous studies have shown a good correlation between gas phase calculations and crystal structures, therefore no solvent corrections were made34. The geometry was optimized and the DFT optimized molecule (4a) was generated. Vibration analysis showed no negative Eigen values indicating that the optimized structure (4a) represents a minimum on the potential energy surface. A complete list of coordinates, bond lengths and bond angles for both the experimental crystal structure and 4a are given in the Supplementary materials (Tables S1–S3 and Fig. S1). For the optimized structure, the HOMO, LUMO and atomic net charges were drawn using Avogadro 1.2.051,52.
Molecular docking studies
The molecular docking studies were performed using the human 11β-HSD1 structure (Protein Data Bank I.D.: 4C7J). The docking method employed is similar to that reported by our group for studies on protease- and neuronal nitric oxide synthase enzymes26,53. The Molecular Operating Environment (MOE) 2018 software suite41 was used for docking studies with the following protocol. (1) The enzyme protein structure was checked for missing atoms, bonds and contacts. (2) Removal of water molecules, 3D protonation and energy minimization was carried out with parameters, force field: MMFF94X + solvation, gradient: 0.05, chiral constraint and current geometry. This minimized structure was used as enzyme for docking analysis. (3) The DFT B3LYP/6-311++G (d,p) optimized structure 4a was saved as a pdb file and imported into the MOE database. (4) Conformer 4a was subsequently docked within the human 11β-HSD1 active site using the MOE Dock application. The active site was selected based on the proximity of the co-crystallized ligand, 4YQ, with the help of the MOE Site Finder tool. The docking algorithm, which was chosen for these experiments, was based on induced fit docking to allow for flexible interactions of the test ligand with the protein. (5) The best binding pose of the title compound was visually inspected and the interactions with the binding pocket residues were analyzed. The selected parameters that were used to calculate the score and interaction of the ligand molecule with the 11β-HSD1 enzyme were; Rescoring function: London dG, Placement: Triangle matcher, Retain: 30, Refinement: Force field, Rescoring 2: London dG. The build-in scoring function of MOE, S-score, was used to predict the binding affinity (kcal/mol) of 4a with the enzyme protein active site after docking.
Supplementary information
Acknowledgements
This work was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Research Group Program (Grant No. RGP-1438-0010) and the University of the Western Cape. The Centre for High Performance Computing at the Council for Scientific and Industrial Research (South Africa) is acknowledged for granting access to Gaussian09 software.
Author contributions
L.H. Al-Wahaibi., N.H. Al-Shaalan and A.A. El-Emam designed the study, synthesized and characterized the title compound and prepared the single crystals. J. Joubert conducted and interpreted the DFT, FMO, Hirshfeld analysis and docking studies. O. Blacque performed the X-ray data collection and the resolution of the crystal structure. All authors contributed in the preparation of the manuscript, discussed the contents and approved the submission.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-56331-z.
References
- 1.Wanka L, Iqbal K, Schreiner PR. The lipophilic bullet hits the targets: Medicinal chemistry of adamantane derivatives. Chem. Rev. 2013;113:3516–3604. doi: 10.1021/cr100264t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J, Obando D, Liao V, Lifa T, Codd R. The many faces of the adamantyl group in drug design. Eur. J. Med. Chem. 2011;46:1949–1963. doi: 10.1016/j.ejmech.2011.01.047. [DOI] [PubMed] [Google Scholar]
- 3.Lamoureux G, Artavia G. Use of the adamantane structure in medicinal chemistry. Curr. Med. Chem. 2010;17:2967–2978. doi: 10.2174/092986710792065027. [DOI] [PubMed] [Google Scholar]
- 4.Davies WL, et al. Antiviral activity of 1-adamantamine (amantadine) Science. 1964;144:862–863. doi: 10.1126/science.144.3620.862. [DOI] [PubMed] [Google Scholar]
- 5.Wendel HA, Snyder MT, Pell S. Trial of amantadine in epidemic influenza. Clin. Pharmacol. Therap. 1966;7:38–43. doi: 10.1002/cpt19667138. [DOI] [PubMed] [Google Scholar]
- 6.Schwab RS, England AC, Jr., Poskanzer DC, Young RR. Amantadine in the treatment of Parkinson’s disease. J. Am. Med. Assoc. 1969;208:1168–1170. doi: 10.1001/jama.1969.03160070046011. [DOI] [PubMed] [Google Scholar]
- 7.Rosenthal KS, Sokol MS, Ingram RL, Subramanian R, Fort RC. Tromantadine: Inhibitor of early and late events in herpes simplex virus replication. Antimicrob. Agents Chemother. 1982;22:1031–1036. doi: 10.1128/AAC.22.6.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia L, et al. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Brit. J. Pharmacol. 2005;144:80–87. doi: 10.1038/sj.bjp.0705984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Protopopova M, et al. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005;56:968–974. doi: 10.1093/jac/dki319. [DOI] [PubMed] [Google Scholar]
- 10.Sun SY, Yue P, Chen X, Hong WK, Lotan R. The synthetic retinoid CD437 selectively induces apoptosis in human lung cancer cells while sparing normal human lung epithelial cells. Cancer Res. 2002;62:2430–2436. [PubMed] [Google Scholar]
- 11.Britten CD, et al. A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017;23:4642–4650. doi: 10.1158/1078-0432.CCR-16-2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villhauer EB, et al. 1-(3-Hydroxy-1-adamantyl)aminoacetyl-2-cyano-(S)-pyrrolidine: a potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J. Med. Chem. 2003;2003(46):2774–2789. doi: 10.1021/jm030091l. [DOI] [PubMed] [Google Scholar]
- 13.Augeri DJ, et al. Discovery and preclinical profile of saxagliptin (BMS-477118): A highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005;48:5025–5037. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- 14.Balkovec, J. M. et al. Preparation of 1,2,4-triazole derivatives as 11β-hydroxysteroid dehydrogenase 1 inhibitors useful for treatment of diabetes, obesity and dyslipidemia. WO 20030814 (2003); Chem. Abstr. 139, 180065 (2003).
- 15.Olson S, et al. Adamantyl triazoles as selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1. Bioorg. Med. Chem. Lett. 2005;15:4359–4362. doi: 10.1016/j.bmcl.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 16.Cott, J. S. & Choormun, J. 11β-Hydroxysteroid dehydrogenase Type 1 (11β-HSD-1) inhibitors in development. In: New therapeutic strategies for type-2 diabetes, small molecule approaches (Jones, R.N., Ed.), RSC, Cambridge, UK, Chapter 5, 109–133 (2012).
- 17.Zhu Y, et al. Phenylcyclobutyl triazoles as selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1. Bioorg. Med. Chem. Lett. 2008;18:3412–3416. doi: 10.1016/j.bmcl.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 18.Tu H, et al. Distinctive molecular inhibition mechanisms for selective inhibitors of human 11β-hydroxysteroid dehydrogenase type 1. Bioorg. Med. Chem. 2008;16:8922–8931. doi: 10.1016/j.bmc.2008.08.065. [DOI] [PubMed] [Google Scholar]
- 19.Joharapurkar A, Dhanesha N, Shah G, Kharul R, Jain M. 11β-Hydroxysteroid dehydrogenase type 1: potential therapeutic target for metabolic syndrome. Pharmacol. Rep. 2012;64:1055–1065. doi: 10.1016/S1734-1140(12)70903-9. [DOI] [PubMed] [Google Scholar]
- 20.Bujalska IJ, et al. A novel selective 11β-hydroxysteroid dehydrogenase type 1 inhibitor prevents human adipogenesis. J. Endocrinol. 2008;197:297–307. doi: 10.1677/JOE-08-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart PM, Tomlinson JW. Selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1 for patients with metabolic syndrome. Diabetes. 2009;58:14–15. doi: 10.2337/db08-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gathercole LL, Stewart PM. Targeting the pre-receptor metabolism of cortisol as a novel therapy in obesity and diabetes. J. Steroid Biochem. Mol. Biol. 2010;122:21–27. doi: 10.1016/j.jsbmb.2010.03.060. [DOI] [PubMed] [Google Scholar]
- 23.El-Emam AA, Al-Tamimi A-MS, Al-Omar MA, Alrashood KA, Habib EE. Synthesis and antimicrobial activity of novel 5-(1-adamantyl)-2-aminomethyl-4-substituted-1,2,4-triazoline-3-thiones. Eur. J. Med. Chem. 2013;68:96–102. doi: 10.1016/j.ejmech.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 24.Al-Abdullah ES, et al. Antimicrobial and hypoglycemic activities of novel N-Mannich bases derived from 5-(1-adamantyl)-4-substituted-1,2,4-triazoline-3-thiones. Int. J. Mol. Sci. 2014;15:22995–23010. doi: 10.3390/ijms151222995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Al-Wahaibi LH, et al. Theoretical investigations of two adamantane derivatives: A combined X-ray, DFT, QTAIM analysis and molecular docking. J. Mol. Struct. 2018;1159:233–245. doi: 10.1016/j.molstruc.2018.01.064. [DOI] [Google Scholar]
- 26.Joubert J. Synthesis, crystal structure, DFT studies, docking studies, and fluorescent properties of 2-(adamantan-1-yl)-2H-isoindole-1-carbonitrile. Crystals. 2019;9:24. doi: 10.3390/cryst9010024. [DOI] [Google Scholar]
- 27.Al-Wahaibi LH, Alsfouk A, El-Emam AA, Blacque O. Crystal structures and Hirshfeld surface analysis of 2-(adamantan-1-yl)-5-(4-fluorophenyl)-1,3,4-oxadiazole and 2-(adamantan-1-yl)-5-(4-chlorophenyl)-1,3,4-oxadiazole. Acta Crystallog. Cryst. Commun. 2019;E75:611–615. doi: 10.1107/S2056989019004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Emam AA, Ibrahim TM. Synthesis and anti-inflammatory and analgesic activity of some 3-(1-adamantyl)-4-substituted-5-mercapto-1,2,4-triazoles. Arzneim.-Forsch./Drug Res. 1992;41:1260–1264. [PubMed] [Google Scholar]
- 29.Spackman MA, Byrom PG. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997;267:215–220. doi: 10.1016/S0009-2614(97)00100-0. [DOI] [Google Scholar]
- 30.Spackman MA, Jayatilaka D. Hirshfeld surface analysis. CrystEngComm. 2009;11:19–32. doi: 10.1039/B818330A. [DOI] [Google Scholar]
- 31.Frisch, M. J. et al. Gaussian 09, Gaussian, Inc: Wallingford, CT, USA (2009).
- 32.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- 33.Lee C, Yang W, Parr RG. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 34.Honarparvar B, Govender T, Maguire GE, Soliman ME, Kruger HG. Integrated approach to structure-based enzymatic drug design: Molecular modeling, spectroscopy, and experimental bioactivity. Chem. Rev. 2013;114:493–537. doi: 10.1021/cr300314q. [DOI] [PubMed] [Google Scholar]
- 35.Grirrane A, et al. First example of a tetra-carboxylate bridged dimanganese species. Chem. Commun. 2003;4:512–513. doi: 10.1039/b211886f. [DOI] [PubMed] [Google Scholar]
- 36.Canossa S, et al. Hierarchy of Supramolecular Arrangements and Building Blocks: Inverted Paradigm of Crystal Engineering in the Unprecedented Metal Coordination of Methylene Blue. Inorg. Chem. 2017;56:3512–3516. doi: 10.1021/acs.inorgchem.6b02980. [DOI] [PubMed] [Google Scholar]
- 37.Clare BW. Frontier orbital energies in quantitative structure-activity relationships: A comparison of quantum chemical methods. Theor. Chim. Acta. 1994;87:415–430. doi: 10.1007/BF01127805. [DOI] [Google Scholar]
- 38.Clare BW. The relationship of charge transfer complexes to frontier orbital energies in QSAR. J. Mol. Struct. Theochem. 1995;331:63–78. doi: 10.1016/0166-1280(94)03783-H. [DOI] [Google Scholar]
- 39.Clare BW. Charge transfer complexes and frontier orbital energies in QSAR: A congeneric series of electron acceptors. J. Mol. Struct. Theochem. 1995;337:139–150. doi: 10.1016/0166-1280(95)04135-S. [DOI] [Google Scholar]
- 40.Heaton C, Miller A, Powell R. Predicting the reactivity of fluorinated compounds with copper using semi-empirical calculations. J. Fluor. Chem. 2001;107:1–3. doi: 10.1016/S0022-1139(00)00324-9. [DOI] [Google Scholar]
- 41.Molecular Operating Environment (MOE), Version 2018.01. http://www.chemcomp.com.
- 42.Goldberg FW, et al. Optimization of brain penetrant 11β-hydroxysteroid dehydrogenase type I inhibitors and in vivo testing in diet-induced obese mice. J. Med. Chem. 2014;57:970–986. doi: 10.1021/jm4016729. [DOI] [PubMed] [Google Scholar]
- 43.Dyadkin V, Pattison P, Dmitriev V, Chernyshov D. A new multipurpose diffractometer PILATUS@SNBL. J. Synchrotron Radiat. 2016;23:825–829. doi: 10.1107/S1600577516002411. [DOI] [PubMed] [Google Scholar]
- 44.Clark RC, Reid JS. The analytical calculation of absorption in multifaceted crystals. Acta Cryst. 1995;A51:887–897. doi: 10.1107/S0108767395007367. [DOI] [Google Scholar]
- 45.CrysAlisPro (version 1.171.40.39a), Rigaku Oxford Diffraction (2018).
- 46.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009;42:339–341. doi: 10.1107/S0021889808042726. [DOI] [Google Scholar]
- 47.Sheldrick GM. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 2015;A71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheldrick GM. Crystal structure refinement with SHELXL. Acta Cryst. 2015;C71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spek AL. Structure validation in chemical crystallography. Acta Cryst. 2009;D65:148–155. doi: 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolff, S., Grimwood, D., McKinnon, J., Jayatilaka, D. & Spackman, M. CrystalExplorer 2.0, University of Western Australia, Perth, Australia (2007).
- 51.Avogadro: an open-source molecular builder and visualization tool. Version 1.2.0. http://avogadro.cc/.
- 52.Hanwell MD, et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joubert J, Foxen EB, Malan SF. Microwave optimized synthesis of N-(adamantan-1-yl)-4-[(adamantan-1-yl)-sulfamoyl]benzamide and its derivatives for anti-dengue virus activity. Molecules. 2018;23:1678. doi: 10.3390/molecules23071678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.