

Abstract
Glial cell-line-derived neurotrophic factor (GDNF) is a potent neuroprotective agent in cellular and animal models of Parkinson’s disease (PD). However, CNS delivery of GDNF in clinical trials has proven challenging due to blood-brain barrier (BBB) impermeability, poor diffusion within brain tissue, and large brain size. We report that using non-toxic mobilization-enabled preconditioning, hematopoietic stem cell (HSC) transplantation-based macrophage-mediated gene delivery may provide a solution to overcome these obstacles. Syngeneic bone marrow HSCs were transduced ex vivo with a lentiviral vector expressing macrophage promoter-driven GDNF and transplanted into 14-week-old MitoPark mice exhibiting PD-like impairments. Transplant preconditioning with granulocyte colony-stimulating factor (G-CSF) and AMD3100 was used to vacate bone marrow stem cell niches. Chimerism reached ∼80% after seven transplantation cycles. Transgene-expressing macrophages infiltrated degenerating CNS regions of MitoPark mice (not wild-type littermate controls), resulting in increased GDNF levels in the midbrain. Macrophage GDNF delivery not only markedly improved motor and non-motor dysfunction, but also dramatically mitigated the loss of dopaminergic neurons in both substantia nigra and the ventral tegmental area and preserved axonal terminals in the striatum. Striatal dopamine levels were almost completely restored. Our data support further development of mobilization-enabled HSC transplantation (HSCT)-based macrophage-mediated GDNF gene delivery as a disease-modifying therapy for PD.
Keywords: Neurodegenerative diseases, Parkinson’s disease, Gene therapy, Neurotrophic factors, GDNF, Hematopoietic stem cell, Hematopoietic stem cell transplantation, Macrophage, Neuroprotective, Cell therapy
Graphical Abstract
Introduction
Parkinson’s disease (PD), a prevalent chronic neurodegenerative disease affecting millions of people,1, 2, 3 is epitomized by a progressive loss of dopaminergic neurons in the substantia nigra (SN) pars compacta (SNpc), which leads to a dopamine (DA) deficit in the primary projection site; the striatum. The consequent dysregulation of neural circuits, particularly the basal ganglia, causes impairment of both motor and non-motor functions.4, 5, 6 Current treatments provide only symptomatic relief, without arresting the progressive loss of DA neurons. Additionally, these treatments carry significant side effects, such as dyskinesia and motor fluctuations, and they eventually become ineffective.7 Therefore, an important unsolved challenge is to develop treatments capable of slowing or reversing the progressive neurodegeneration. Unfortunately, virtually all potential disease-modifying interventions have been unsuccessful to date.8
Glial cell-line-derived neurotrophic factor (GDNF) and GDNF family ligand neurturin (NRTN) are the most potent agents known to protect DA neurons affected in PD,9,10 although many neurotrophic factors, including platelet‐derived growth factor (PDGF), cerebral dopaminergic neurotrophic factor (CDNF), and insulin-like growth factor-1 (IGF-1) have been identified experimentally to be neuroprotective in PD models.11,12 Considering that the cause of nigral DA neuron loss in PD is still poorly understood, the use of GDNF as a therapeutic molecule is compelling since it can protect neurons from a variety of insults that cause damage through different mechanisms.13,14 However, when delivered peripherally, GDNF does not cross the blood-brain barrier (BBB), posing a substantial technical challenge for therapeutic application. Direct brain injection of either GDNF protein or NRTN-expressing vectors was tested in animal models rather successfully, but its therapeutic efficacy was not confirmed in clinical trials.15, 16, 17, 18 The lack of translatability from experimental settings to the clinic19 is likely due to insufficient diffusion of GDNF to areas of neurodegeneration, which are substantially larger in human brain.16, 17, 18 Along the same lines, impairment of axonal (retrograde) transport in PD patients may also limit effectiveness of neurotrophic factor delivery to the distal SN, where the protein product was intended to be transported to achieve a robust neurotrophic response.20,21
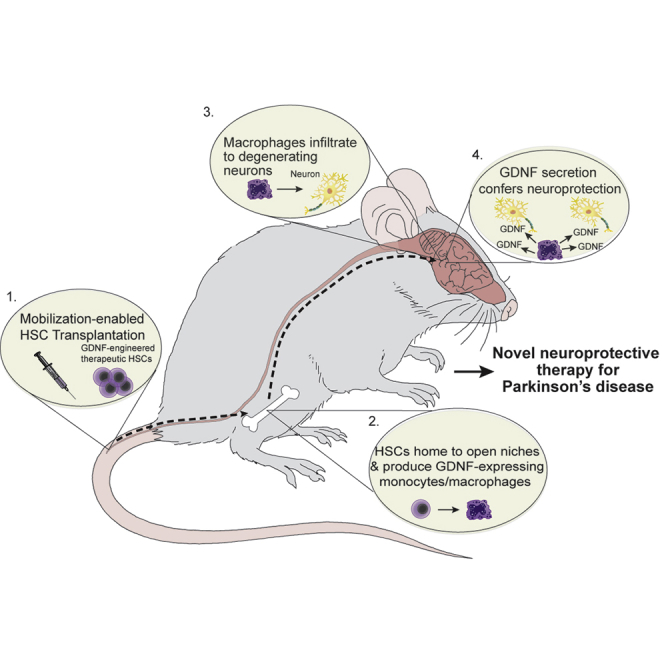
To overcome these limitations, we previously introduced an entirely novel strategy—hematopoietic stem cell (HSC) transplantation (HSCT)-based macrophage-mediated GDNF delivery.22 This approach capitalizes on the propensity of macrophages (a progeny of HSCs) to home to sites of neurodegeneration. Sustained therapeutic macrophage levels are maintained by HSCs modified with highly efficient lentiviral transduction. Bone marrow HSCs were harvested and transduced ex vivo with lentivirus expressing either the GDNF or NRTN gene driven by our highly active macrophage synthetic promoter (MSP) followed by transplantation into recipient mice. The neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was administered to the animals to induce PD-like dopaminergic neurodegeneration. Subsequently, HSC-derived macrophages infiltrated and homed to neurodegenerating sites within the brain. Macrophage-mediated GDNF or NRTN delivery significantly ameliorated MPTP-induced degeneration of tyrosine hydroxylase-positive (TH+) neurons of the SN and TH+ terminals in the striatum, stimulated axon regeneration, and ameliorated the decline in general ambulatory activity. However, a caveat of these initial studies was the use of whole-body irradiation for HSCT pre-conditioning, potentially compromising the integrity of the BBB and/or inducing neuroinflammation. Therefore, further studies were performed using a head-shielded irradiation procedure, clearly indicating that macrophage infiltration into SN required neurodegeneration and was not simply a consequence of BBB disruption. Moreover, we showed that GDNF expression/delivery was controllable using a doxycycline-regulated lentiviral vector. More recently, MitoPark mice were employed to recapitulate chronic/progressive neurodegeneration in PD and compensate for limitations of the acute neuronal injury in the MPTP model. MitoPark mice exhibit both PD-like motor and non-motor dysfunction. Using this genetic model, we validated our previous work from the toxin model and confirmed the effectiveness of our approach to mitigate PD-like neurodegeneration, motor deficits, and non-motor impairment.22, 23, 24, 25 However, conventional HSCT requires high-dose chemotherapy and/or irradiation,26 which can trigger both short-term and long-term adverse effects, which may limit clinical utility due to unfavorable benefit-to-harm ratio in PD patients.26 To solve this problem, we recently conceptualized and developed a novel non-toxic HSCT technology. Here, we report a combined study in the MitoPark mouse model of PD. The results of this study not only confirmed the dispensability of procedure-associated brain conditioning for macrophage CNS infiltration but also revealed that non-toxic HSCT-based macrophage-mediated delivery of GDNF effectively protected against dopaminergic neurodegeneration in MitoPark mice, leading to significant reversal of both motor and non-motor dysfunction, while remaining free of adverse effects.
Results
Non-toxic HSCT Was Achievable through Mobilization-Aided Conditioning
At physiologic steady state, the majority of HSCs reside within specialized bone marrow niches. However, 1%–5% leave the niche and enter circulation each day.27 Egress of HSCs is dramatically increased by administration of mobilizers such as granulocyte colony-stimulating factor (G-CSF),28 either alone or in combination with additional pharmacological agents, like AMD3100,29 a CXCR4 antagonist. Administration of mobilizers increases the population of HSCs circulating in the peripheral blood, which, consequently, is the basic mechanism underlying collection of peripheral blood donor stem cells in the clinic. Importantly, the increased HSC egress also creates temporary voids within the bone marrow niches. We hypothesized that donor cells infused during peak mobilization would mix in the peripheral blood with mobilized endogenous bone marrow cells, and by infusing large numbers of donor cells, they would compete with endogenous cells to repopulate mobilization-induced void in the niche, leading to favorable kinetics of donor cell engraftment. In humans, this process could be improved further by removing mobilized endogenous stem cells via apheresis immediately prior to donor cell infusion. Due to size limitations, apheresis cannot be re-configured to test the removal of endogenous cells in mice. Thus, we skewed the probability of donor cell engraftment by infusing superior numbers of donor cells into recipients. This concept is illustrated in Figure 1A and can be represented by a mathematical model (Figure 1B). To test our mobilization-based, non-cytotoxic conditioning regimen, we transplanted 10-week-old female wild-type C57BL/6J mice (n = 3) with 2.0 × 106 lineage-negative (HSCs-enriched) C57BL/6J GFP+ bone marrow cells via tail vein injections after receiving mobilization factors. This procedure was repeated every 2 weeks for a total of seven transplantation cycles. Donor cell engraftment was estimated by measuring recipient peripheral blood donor chimerism (GFP+) via flow cytometry at various time points after transplantation. Because only long-term HSCs (LT-HSCs) are capable of long-term self-renewal and continued contribution to hematopoiesis 4 months post-transplant,30 we collected peripheral blood samples at 1 or 4 months post-transplant to assess chimerism and long-term donor cell reconstitution. Figure 2 shows the percentage of GFP+ donor cells after each cycle of transplantation, demonstrating that donor chimerism increased with every transplantation cycle, stabilizing at ∼77% by 4 months post 7th cycle, in agreement with our mathematical model (Figure 1B, “a” was set at 0.25 and “d” was set at 0.80). These findings in females were confirmed in male C57BL/6J wild-type mice (n = 4) transplanted with C57BL/6J GFP+ HSCs for seven cycles as described. We detected minimal donor chimerism (1.24% ± 0.75% 1 month post-HSCT and 1.12% ± 0.23% 4 months post-HSCT) following a single HSCT cycle performed in non-mobilized control mice (Figures 2C and 2D), consistent with previous reports.31,32
Figure 1.

Schematic Illustration and Theoretical Model of the Mobilization-Aided HSCT
(A) Schematic illustration. First column depicts HSCT without conditioning. Second column depicts HSCT following mobilization-based conditioning, allowing superior donor cell populations to out-compete endogenous cells. Third column depicts a theoretical HSCT procedure incorporating mobilization-based conditioning coupled with blood apheresis to remove endogenous HSCs prior to injecting donor HSCs, further enhancing donor cell competition for recently vacated niche space. Because murine models are too small to perform apheresis, this hypothetical procedure would be investigated in larger animal models. (B) Theoretical mathematical model. Assumptions are as follows: (1) the ratio of mobilized donor to recipient HSCs in circulation prior to injection of additional donor HSCs is equal to the ratio of donor to recipient HSCs in the bone marrow; (2) the total number of HSC niche spaces remains fixed; (3) each donor and recipient HSC in the blood has an equal probability of engrafting in a BM HSC niche space; and (4) remaining HSCs that are not engrafted after each cycle are cleared and are no longer present in circulation or available for engraftment during the next cycle of mobilization and transplant.
Figure 2.

Donor HSC Chimerism following Mobilization-Enabled HSCT
(A and B) Peripheral blood samples were collected from recipient C57BL/6J wild-type mice following each mobilization-enabled HSCT at (A) 1-month post-HSCT and (B) 4-months post-HSCT. Each bar represents mean ± SEM (n = 3/ bar). (C and D) Donor chimerism following a single cycle of HSCT in control mice without mobilization was (C) 1.24% ± 0.75% 1 month post-HSCT and (D) 1.12% ± 0.23% 4 months post-HSCT.
Flow cytometric analysis of peripheral blood samples 5 months after the final transplant revealed high GFP+ donor chimerism for both myeloid and lymphoid lineages in circulation (Figures 3A and 3B). Shortly thereafter, these mice were euthanized for bone marrow cell analysis (Figure 3C). Because bone marrow lineage-negative (Lin−), Sca1-positive, c-Kit-positive (LSK) cells are responsible for long-term competitive repopulation capacity of the hematopoietic system,33,34 we focused bone marrow analysis on GFP+ donor LSK+ cell populations. Flow cytometry revealed that approximately 6% of the lineage-negative bone marrow consisted of LSK cells (Figure 3D), agreeing with previous findings.35 Importantly, within the LSK population, ∼80% were donor-derived GFP+ cells, similar to the donor chimerism level of 77% observed in the peripheral blood.
Figure 3.

Mobilization-Aided HSCT Yielded High GFP+ Donor Chimerism
Peripheral blood and bone marrow were collected from four recipient male C57BL/6J wild-type mice at 5 months post G-CSF/AMD3100-aided syngeneic GFP+ HSCT. (A) Representative flow cytometry dot plots of peripheral blood cells immunostained with anti-B220, CD3e, CD11b, and Gr-1. (B) Percentage of donor GFP+ T cells (CD3e+), B cells (B220+), monocytes (CD11b+/Gr-1-), granulocytes (Gr-1+/CD11b-), and myeloid cells (CD11b+/Gr-1+) (excluded here) in the peripheral blood (n = 4). (C) Representative flow cytometry dot plots of bone marrow LSK (Lin-, Sca1+, c-Kit+) cells. The isolated lineage-negative (Lin-) cells were stained with anti-Sca1, -c-Kit antibodies for identification of LSK cells. (D) Percentage of GFP+ LSK cells engrafted in bone marrow of recipient mice (n = 4).
HSCT-Based GDNF Delivery Relieved Progressive Motor and Non-motor Dysfunction in MitoPark Mice
To assess the therapeutic effects of non-toxic HSCT-based, macrophage-mediated GDNF delivery on functional deficits in MitoPark mice, we initiated the transplant protocol at 14 weeks of age when MitoPark mice begin to exhibit spontaneous motor deficits and non-motor phenotypes. Thereafter, the mobilization-aided HSCT procedure was repeated once every 2 weeks on all experimental groups up to seven cycles, as described. Spontaneous horizontal and vertical locomotor activities were measured before and after each HSCT cycle. As shown in Figures 4A and 3B, at 14 weeks of age and prior to HSCT, the MitoPark mice had developed PD-like motor deficits with a 49.7% and 54.3% decline in horizontal and vertical activities, respectively (as a baseline) compared with normal control mice. The Mito-LT-MSP-GFP group displayed further progressive decline from baseline with age, as is typical of the MitoPark mouse model.36 During the 18-week treatment period (14 to 32 weeks of age), Mito-LT-MSP-GFP mice exhibited a further decline of 84.8% in horizontal activity and 91.6% in vertical activity (Figures 4A and 4B). In contrast, the Mito-LT-MSP-hGDNF-2A-GFP (expressing both hGDNF and GFP at equal levels to facilitate tracking) and Mito-LT-MSP-hGDNF groups showed increases in horizontal (36.2% and 46.3%) and vertical (45.2% and 54.9%) activities relative to baseline. The therapeutic effects of non-toxic HSCT-based GDNF delivery on the non-motor parameters, cognitive ability, and anhedonia, were determined by a novel-object recognition test (NOR) and the sucrose preference test (SPT), respectively. As shown in Figure 4C, all MitoPark mouse groups exhibited cognitive impairment before HSCT, as indicated by a 40.2% lower discrimination index compared with normal control mice. The Mito-LT-MSP-GFP group exhibited progressive impairment in cognitive function with an 88.4% decrease in discrimination index from baseline during the 18-week period, whereas Mito-LT-MSP-hGDNF-2A-GFP and Mito-LT-MSP-hGDNF groups revealed 65.4% and 66.8% improvement from the baseline, respectively, after seven cycles of HSCT-based GDNF therapy. Sucrose preference before HSCT was significantly lower in MitoPark mice compared to normal control mice, averaging 70.7%. The LT-MSP-GFP-transplanted MitoPark also showed progressive reduction of sucrose preference scores. Notably, sucrose preference in LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice was restored to 96.5% of normal control levels after multiple cycles of HSCT (Figure 4D). Together, these data indicate that the non-toxic HSCT-based macrophage-mediated GDNF delivery not only prevented further motor and non-motor functional decline but also led to a significant improvement in the deficits observed in MitoPark mice.
Figure 4.

Non-toxic HSCT-Based Macrophage-Mediated GDNF Delivery Mitigated Progressive Motor Deficits and Non-motor Dysfunction in MitoPark Mice
(A) Spontaneous horizontal locomotor activities. (B) Spontaneous vertical locomotor activities. Each point represents mean ± SEM of 6 × 10 min test recorded from 15 animals per treatment group (n = 15). Two-way ANOVA was performed, followed by Bonferroni post-test. *p < 0.05, **p < 0.01, ***p < 0.001, Mito-LT-MSP-GFP versus Mito-LT-MSP-hGDNF-2A-GFP and Mito-LT-MSP-hGDNF groups. (C) Novel-object recognition. (D) Sucrose preference. Each bar represents mean ± SEM from 15 animals per treatment group (n = 15). Two-way ANOVA was performed, followed by Bonferroni post-test. *p < 0.05, **p < 0.01, ***p < 0.001, Mito-LT-MSP-GFP versus Mito-LT-MSP-hGDNF-2A-GFP and Mito-LT-MSP-hGDNF groups.
HSCT-Based GDNF Delivery Reduced DA Neuronal Loss and Protected against Degeneration of Dopaminergic Terminals
Four weeks after the last HSCT cycle, brains were collected from all recipient mice. To assess the neuroprotective effects of HSCT-based GDNF delivery on the nigrostriatal dopaminergic system, we performed quantitative analyses of TH+ neurons in the SNpc and the ventral tegmental area (VTA) and density of TH+ terminals in the striatum. As shown by TH immunohistochemistry of midbrain sections in Figures 5A and 5B, LT-MSP-GFP-transplanted MitoPark mice suffered a significant 84% decrease in DA neurons compared to the LT-MSP-GFP-transplanted normal control mice. In contrast, LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice exhibited only 28% and 25% DA neuron loss, respectively. Similarly, a 66% loss of TH+ neurons was seen in the VTA of LT-MSP-GFP-transplanted MitoPark mice compared to LT-MSP-GFP-transplanted normal controls. In contrast, only 25% and 26% losses were observed in the LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice, respectively (Figures 5C and 5D). High intensity TH staining was observed in the striatum of LT-MSP-GFP-transplanted normal control mice, whereas a 66% decrease in TH stain intensity was seen in the LT-MSP-GFP-transplanted MitoPark mice, indicative of a dramatic loss of dopaminergic terminal fibers. In striking contrast, the LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice displayed preserved (79% and 67%, respectively) intensities of striatal TH+ terminals (Figures 5E and 5F), indicating that the degeneration of striatal dopaminergic terminals in MitoPark mice was also mitigated by this approach.
Figure 5.

Non-toxic HSCT-Based GDNF Delivery Protected against Loss of DA Neurons in the SN and VTA, as well as Degeneration of Dopaminergic Terminals in the Striatum of MitoPark Mice
Coronal midbrain and forebrain sections from randomly selected brain samples of LT-MSP-hGDNF-2A-GFP-, LT-MSP-hGDNF-, and LT-MSP-GFP-transplanted MitoPark mice and LT-MSP-GFP-transplanted non-MitoPark normal control mice after seven cycles of non-toxic HSCT were stained with TH immunohistochemistry. (A) Representative images of TH-immunoreactive DA neurons (stained in brown) in SN (original magnification, 100×). SNpc, substantia nigra pars compacta; SNpr, substantia nigra pars reticulata. (B) The number of DA neurons in SNpc was counted from each of five sections (n = 5) per group using NIH ImageJ software by two blinded investigators following the same criteria. Each bar represents mean ± SEM. One-way ANOVA was performed, followed by Tukey’s post-test. **p < 0.01, ***p < 0.001 versus LT-MSP-GFP-transplanted MitoPark mice. (C) TH-immunoreactive DA neurons in VTA (original magnification, 100×). (D) The number of DA neurons in VTA was counted from each of five sections (n = 5) per group. Each bar represents mean ± SEM. One-way ANOVA was performed, followed by Tukey’s post-test. **p < 0.01, ***p < 0.001 versus LT-MSP-GFP-transplanted MitoPark mice. (E) Representative images of TH-immunoreactive dopaminergic terminals in the striatum (original magnification, 20×). (F) Optical densities of striatal TH+ terminals were measured from digitized images of the striatum using NIH ImageJ software by two blinded investigators following the same criteria. Each bar represents mean ± SEM from six animals per treatment group (n = 6). One-way ANOVA was performed, followed by Tukey’s post-test. *p < 0.05, ***p < 0.001 versus LT-MSP-GFP-transplanted MitoPark mice.
Gene-Modified Macrophages Selectively Infiltrated the Area of Neurodegeneration
To investigate whether bone-marrow-derived gene-modified macrophages infiltrated the sites of DA neuron degeneration, we examined midbrain sections stained with TH and Iba1 (microglia/macrophage marker) by fluorescent immunohistochemistry. The normal control mice showed abundant TH-stained DA neurons (in red) in the SN without GFP+ macrophage (in green) infiltration (Figure 6A). In contrast, the LT-MSP-GFP-transplanted MitoPark mice exhibited severe loss of DA neurons, which was accompanied by a marked infiltration of GFP-expressing macrophages into the SN. Similarly, midbrain sections from the LT-hGDNF-2A-GFP-transplanted MitoPark mice exhibited marked infiltration of GFP+/hGDNF-expressing macrophages into the SN (Figure 6A). Furthermore, we quantified the proportion of HSCT-derived gene-modified macrophages among the total population of Iba1-positive cells. In the SN of LT-MSP-GFP-MitoPark mice, 52.3% of microglia were of donor origin, whereas only 7.8% of microglia were of donor origin in the LT-MSP-GFP-transplanted normal control mice (Figures 6B and 6C). Similar levels of bone-marrow-derived gene-modified macrophage infiltration into sites of DA neuron degeneration were observed in the VTA (Figures 6D and 6E), which plays an important role in cognition and motivation, among other processes.37,38 In addition to studying the SN and VTA, we investigated whether the bone marrow-derived gene-modified macrophages infiltrated other regions of the mid-brain. GFP-expressing cells were rarely found in these regions of the brain (Figure S1), suggesting selective homing of HSC-derived macrophages to sites of prominent neurodegeneration in the SN and VTA of MitoPark mice.
Figure 6.

Gene-Modified Macrophages/Microglia Selectively Infiltrated the Area of DA Neuron Degeneration in the SN and VTA
Coronal midbrain sections were from randomly selected brain samples of LT-MSP-hGDNF-2A-GFP- and LT-MSP-GFP-transplanted MitoPark mice and LT-MSP-GFP-transplanted non-MitoPark normal control mice after seven cycles of non-toxic HSCT. (A) Representative images of TH fluorescent immunohistochemistry. TH-immunoreactive DA neurons (stained in red) and infiltrated gene-modified GFP-expressing macrophages/microglia (in green) in SN (original magnification, 100×). (B) Representative images of DA neurons and macrophages/microglia co-immunostained with TH and Iba1 in SN (original magnification, 100×). TH-immunoreactive DA neurons (stained in blue), Iba1+ macrophages/microglia (stained in red) and infiltrated GFP-expressing macrophages/microglia (in green). (C) The number of Iba1+ and GFP+ macrophages/microglia in SNpc were counted from the sections of five individuals per group (n = 5) using NIH ImageJ software by two blinded investigators following the same criteria. Each bar represents mean ± SEM. One-way ANOVA was performed, followed by Tukey’s post-test. **p < 0.01, ***p < 0.001 versus LT-MSP-GFP transplanted normal control mice. (D) Representative images of DA neurons and macrophages/microglia co-immunostained with TH and Iba1 in VTA (original magnification, 100×). TH-immunoreactive DA neurons (stained in blue), Iba1+ macrophages/microglia (stained in red), and infiltrated GFP-expressing macrophages/microglia (in green). (E) The number of Iba1+ and GFP+ macrophages/microglia in VTA were counted from the sections of five individuals per group (n = 5) using NIH ImageJ software by two blinded investigators following the same criteria. Each bar represents mean ± SEM. One-way ANOVA was performed, followed by Tukey’s post-test. *p < 0.05, ***p < 0.001 versus LT-MSP-GFP-transplanted normal control mice.
Disease-associated disruption of the BBB has been proposed as necessary for bone-marrow-derived macrophages to infiltrate to sites of neural injury in conditions such as PD. BBB breakdown has been confirmed in amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), epilepsy, and multiple sclerosis (MS); however, whether BBB breakdown occurs in PD remains controversial.39 Controlled, regulated trafficking of peripheral immune cells into the CNS has been reported in the absence of overt disruption of the BBB.40,41 We used the Evans blue (EB) dye extravasation technique to evaluate BBB permeability in our mouse model. EB did not permeate the BBB of saline treated wild-type mice (negative control), whereas BBB disruption by high-dose peripheral lipopolysaccharide (LPS) allowed a significant increase in EB penetration into the brain (positive control). MitoPark mice did not exhibit increased permeability compared with wild-type littermates, suggesting that the BBB is not compromised in the MitoPark mouse model39,42 (Figure S2). Similarly, permeability of the BBB to low molecular weight (∼10 kDa) fluorescein isothiocyanate (FITC)-dextran was increased by peripheral LPS administration, but not by MitoPark genotype (data not shown). It is possible that other unidentified non-structural changes of BBB42 physiology may occur to contribute to macrophage infiltration, as bone marrow-derived macrophages were observed in the brain parenchyma of these mice (Figure 6).
HSCT-Based GDNF Delivery Protected against Loss of Striatal DA and Its Metabolites
At the termination of the experiment, we compared GDNF levels in the plasma, SN, and striatum between the different treatment groups. Compared with control groups (LT-MSP-GFP-transplanted MitoPark and normal control mice), we saw a significant increase in plasma GDNF levels in the LT-MSP-hGDNF-2A-GFP and LT-MSP-hGDNF MitoPark mice (Figure 7A). Importantly, GDNF levels were increased by ∼7-fold and ∼4.5-fold in the SN and striatum, respectively, of these mice (Figures 7B and 7C) compared to control groups. Striatal levels of DA and its major metabolites, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), in LT-MSP-GFP-transplanted MitoPark mice were reduced by ∼90% compared to LT-MSP-GFP-transplanted normal control. In contrast, LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice exhibited restorations of striatal DA, DOPAC, and HVA, reaching approximately 84%, 86%, and 83% of levels measured in normal control mice (Figures 7D–7F). Taken together, these data indicate that the HSCT-based therapeutic approach can deliver GDNF effectively to the brain regions most affected during PD progression, leading to recovery of striatal DA levels. Restoration of DA levels is very likely responsible for the improved motor activities and mitigation of non-motor deficits observed in MitoPark mice receiving HSCT.
Figure 7.

HSCT-Based GDNF Delivery Restored Striatal Levels of DA and Its Metabolites in MitoPark Mice
The GDNF levels in the plasma, SN, and striatum were measured by ELISA and the striatal DA and its metabolites were measured by HPLC. Each bar represents mean ± SEM. One-way ANOVA was performed, followed by Tukey’s post-test. (A) Plasma GDNF levels (n = 6), ***p < 0.001 versus LT-MSP-GFP-transplanted MitoPark and normal control mice. (B) SN GDNF content (n = 6), ***p < 0.001 versus LT-MSP-GFP-transplanted MitoPark and normal control mice. (C) Striatum GDNF content (n = 6), ***p < 0.001 versus LT-MSP-GFP-transplanted MitoPark and normal control mice. (D) Striatal DA (n = 5), ***p < 0.001 versus LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice and LT-MSP-GFP-transplanted normal control mice. (E) Striatal DOPAC (n = 5), **p < 0.01 versus LT-MSP-GFP-transplanted MitoPark mice. ***p < 0.001 versus LT-MSP-hGDNF-2A-GFP-transplanted MitoPark mice and LT-MSP-GFP-transplanted-normal control mice. (F) Striatal HVA (n = 5), ***p < 0.001 versus LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice and LT-MSP-GFP-transplanted normal control mice.
HSCT-Based GDNF Delivery Reduced Fragmentation of Golgi Apparatus in the DA Neurons
In addition to neuronal loss and impaired neurotransmitter release, previous studies have reported subcellular abnormalities in both cellular and mouse models of PD.43,44 Overexpression of either wild-type or PD-related mutant α-synuclein disrupts Golgi structures in striatal neurons.45,46 To investigate whether impairment of subcellular organelles occurs in MitoPark DA neurons and whether our GDNF delivery exerts a therapeutic effect, we examined the structure of the Golgi apparatus in nigral DA neurons by fluorescent immunohistochemistry using the cis-Golgi matrix protein marker GM130 (Figure 8). GM130 is responsible for connecting distinct Golgi compartments in soma and dendritic branch points.44,47,48 In LT-MSP-GFP-transplanted normal control mice, GM130 staining revealed integrated thick tubular structures primarily stacked at one side of DA neurons, whereas the GM130-positive tubular structures in the LT-MSP-GFP-transplanted MitoPark mice appeared thin and fragmented. Relative to the LT-MSP-GFP-transplanted MitoPark mice, GM130-stained Golgi apparatus in the LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice showed more robust tubular structures and less fragmentation (Figures 8A and 8B), suggesting that the Golgi apparatus in nigral DA neurons of MitoPark mice was protected by our GDNF therapy.
Figure 8.

HSCT-Based GDNF Delivery Protected against Fragmentation of Golgi Apparatus in the DA Neurons of MitoPark Mice
Coronal midbrain sections were from randomly selected brain samples of LT-MSP-GFP-transplanted normal control mice and MitoPark mice, and LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF-transplanted MitoPark mice after seven cycles of non-toxic HSCT. (A) The sections were co-immunostained with TH (red) for DA neurons and GM130 (green) for Golgi apparatus. Asterisks label TH+ neurons. Arrowheads point to non-TH+ cells. (B) Morphometric analysis using NIH ImageJ software of Golgi apparatus in the different treatment groups. Percentage of GM130 staining area over TH+ area of neurons (n ≥ 20) were presented as mean ± SEM ***p < 0.001.
Non-toxic HSCT-Based GDNF Gene Therapy Was Not Associated with Adverse Effects
We did not observe any adverse effects in our previous conventional HSCT-based macrophage-mediated GDNF gene therapy studies. In this study, we monitored mouse body weight and examined H&E-stained tissue sections of the heart, lung, liver, spleen, kidney, ovary, uterus, and pancreas of mice from each group. Body weight was recorded every 2 weeks as a proxy for general health and vitality. The body weight of the LT-MSP-GFP-transplanted normal control mice remained stable throughout the experimental period, as expected. MitoPark mice exhibited progressive loss of body weight with age, which was significantly mitigated in both the LT-MSP-hGDNF-2A-GFP- and LT-MSP-hGDNF groups (Figure S3). Pathological examination of the kidneys and ovaries from GDNF-treated mice demonstrated normal tissue morphology comparable with that of normal control mice. GDNF-treated mice exhibited neither pancreatic acinar-to-ductular metaplasia nor heart abnormalities, such as multifocal degeneration with interstitial fibrosis (Figure S4).
Discussion
The major unmet need for patients diagnosed with PD is the development of neuroprotective, disease-modifying therapy. Here, we demonstrated one such potential therapy in the MitoPark mouse model. Non-toxic HSCT-based macrophage-mediated GDNF delivery dramatically ameliorated the loss of TH+ dopaminergic neurons, thereby leading to remarkable mitigation of motor and non-motor Parkinsonian symptoms.
Although GDNF-based therapy has strong disease-modifying potential for the treatment of PD, CNS delivery has proven challenging, as peripheral GDNF does not cross the BBB. Macrophages represent a cellular delivery platform that offers targeted GDNF delivery to sites of neurodegeneration. Although an earlier report showed that no donor macrophage infiltration was detected in the CNS of normal HSC recipient mice preconditioned with brain-shielded irradiation,49 we have reported that donor-derived macrophages are capable of infiltrating the CNS of MitoPark mice, even when the brain was shielded from irradiation during traditional myeloablative conditioning.24 Consistent with previous findings, robust donor macrophage infiltration occurred in the brain of MitoPark mice when mobilization-based non-toxic pre-conditioning was used instead of irradiation or chemotherapy, which demonstrates that PD-like pathogenesis was sufficient to elicit macrophage infiltration into brain. These results are consistent with reports that left-heart-ventricle-infused macrophages are capable of homing to areas of neurodegeneration in mouse models of both PD and AD.50,51 Nevertheless, macrophage infiltration has not been detected in all models of CNS injury, such as facial nerve axotomy.49 Taken together, peripheral macrophage passage through the BBB and recruitment to sites of injury within the brain is likely disease-dependent and pathogenesis-specific.41 Critically, whether macrophage infiltration to areas of DA neuron degeneration occurs in PD patients remains unknown. Encouragingly, however, clinical trials of lentiviral HSC gene therapy for X-linked adrenoleukodystrophy (ALD)52,53 and metachromatic leukodystrophy (MLD)54 have been successful, in which macrophages/microglia are believed to be the effector cells delivering therapeutic protein to the CNS. These examples provide evidence in favor of macrophage infiltration in neurodegenerative diseases. Prospectively, a similar approach may be used to deliver specialized neurotrophic factors to treat other types of neurodegenerative diseases. For instance, macrophage-mediated delivery of BDNF to the degenerating brain of Alzheimer’s patients may be a promising strategy to treat this devastating disease. Throughout these endeavors, a microglia-specific promoter is highly desired and a recent study has pointed out new perspectives on this strategy.55
It is worthwhile to note that not only are HSC-derived GDNF-expressing macrophages capable of homing to PD-like brain in mouse models, but systemically infused macrophages can also do so as well. Zhao et al. showed that GDNF-transfected macrophages infused via the left ventricle homed to the inflamed brain and produced potent neuroprotective effects in a 6-hydroxydopamine (6-OHDA) -induced mouse model of PD.50 In that model, due to the acute nature of the 6-OHDA toxicity, only short-term (21 days) neuroprotection was observed. Considering the chronic nature of human PD, the relatively short lifespan of monocytes, and the dynamic and homeostatic properties of macrophage homing to the CNS,41 approaches utilizing lineage-committed monocytes may prove challenging. Periodic administration of large numbers of genetically modified cells would be required to sustain long-term GDNF delivery. In contrast, our HSCT-based approach would ensure a continuous source of enhanced macrophages capable of long-term GDNF delivery. Although sustained GDNF delivery to the affected DA neurons may be key to therapeutic success,56 long-term GDNF overexpression can lead ultimately to decreases in DA levels via downregulation of TH expression.57 However, in that study the levels of GDNF in SN (2.5–4.5 ng/mg) were 50-fold higher than those reached in our approach (∼0.06 ng/mg).57 The lowest tissue GDNF concentrations that led to DA downregulation were ∼0.4 ng/mg,57 which is 5-fold higher than the highest concentration we observed in the present study, suggesting that our approach could provide safe brain GDNF levels to PD subjects. Together, these key features make our HSC-derived GDNF-expressing macrophages an attractive cellular vehicle to deliver sustained therapeutic dosing and trophic support directly to the neurons most in need. Of note, systemic administration of GDNF-expressing macrophages of regenerative M2 phenotype may provide short-term benefits, along with the advantage of fading expression if adverse reactions are encountered.50
Genetic lysosomal or peroxisomal disorders such as MLD and ALD are rare but serious genetic conditions that typically manifest at a young age, justifying conventional bone marrow HSCT treatments in spite of its associated risks. However, the application of conventional chemotherapy/irradiation-based HSCT may not be justified for most PD patients, given their older age profile and disease chronicity. In this report, we provide proof-of-concept evidence in a mouse model for a non-toxic, non-ablative HSCT by manipulating the mobilization and homing mechanisms of HSC physiology. In patients, removal of mobilized endogenous stem cells from the peripheral blood via apheresis immediately before donor cell infusion should allow for even higher engraftment efficiency than in our mouse models, by decreasing competition for niche occupation (as illustrated in Figure 1). Recent data questioned the conventional belief that essentially all bone marrow niches are occupied by HSCs in unconditioned mice and argued that the bone marrow contains more habitable niches for HSCs. Nonetheless, these constitutive niches may provide lower accessibility to transplanted HSCs than newly vacated niches. Moreover, we detected only about 1.2% chimerism following HSCT in non-mobilized mice. Because HSCT is one of the fastest growing hospital procedures, maximizing efficiency while minimizing adverse effects is crucial for expanding its application. Based on the literature, current clinical practice, and our animal studies, this novel HSCT procedure, utilizing well-established drugs, is anticipated to have a favorable safety profile in the clinical setting, although further studies are required to more thoroughly investigate its tolerability.
Mobilization-based HSCT was explored previously with limited success.31,58 The level of donor chimerism was far lower (9.1%) than observed in our current study (∼40%) after three rounds of transplantation. In the previous study, PeP3b mice were used as recipients with C57BL/6J as donors.31 According to the literature and our unpublished data, PeP3b mice were found to be poor mobilizers compared with C57BL/6J mice, and therefore may be a poor model for the study of mobilization-aided HSCT. In humans, G-CSF-based mobilization regimens have a 70%–95% success rate among healthy donors and patients.59 The response rates to AMD3100 are even higher, and the combination of G-CSF and AMD3100 reliably results in fewer mobilization failures.60 Moreover, new mobilizers and optimized procedures are continuously being developed. Taken together, the evidence suggests that effective mobilization and transplantation should be achievable in most patients.
To provide proof of concept for our approach, we chose seven cycles of transplantation to achieve high levels of chimerism. However, fewer cycles may be sufficient for a measurable therapeutic effect on PD, since the level of chimerism per se is less relevant than is delivery of therapeutic levels of GDNF to the DA neurons. In the SNpc of chronic Parkinsonian macaques, microglial cell processes and cell bodies establish contacts with dopaminergic neurites and cell bodies.61 Intimate contacts of microglial cells with surrounding neurons were found—one microglial cell can touch several neurons, and a neuron can be reached by several microglia.62 When these macrophages/microglial cells are genetically engineered to secrete GDNF protein, one can envision them (even only a small percentage of the pool) effectively delivering this factor to the nerve cells that require trophic support the most. Moreover, because these studies were performed in mouse models, small body size precluded the use of apheresis to remove competing endogenous HSCs. In larger species, including humans, it is likely that fewer cycles of transplantation would be required to achieve sufficient engraftment of therapeutic HSCs. Moving forward, timely evaluation of therapeutic effects after each cycle of transplant may be conducted to guide decisions on the optimum number of cycles and level of chimerism.
Gene therapy is quickly becoming an established form of medical care with manageable risk.63 In 2017, the FDA approved the first three gene-therapy products for use in the United States.63 Given the rapid evolution of this field, it is foreseeable that gene therapy will be a mainstay treatment for many diseases. Although the risk of integration-induced carcinogenesis, which occurred in early clinical trials of retroviral HSC gene therapy,64 cannot be entirely discounted, lentiviral HSC gene therapy has enjoyed a relatively safe history thus far. Moreover, the latest genome-editing technologies, CRISPR/Cas9 in particular, provide powerful tools for addressing the possible risk of random integration-related tumorigenesis.65,66 Using these technologies, therapeutic GDNF cassettes could be placed in a safe harbor (such as AAVS1) of the genome to enhance the safety profile of the therapy for clinical trials and beyond.
There are some limitations to this study. PD pathogenesis in humans likely involves a combination of genetic and environmental factors. It is thus unlikely there will ever be a single ideal animal model for PD.67 Available models have contributed greatly to our understanding of the pathophysiology68 and have led to effective treatment for the motor symptoms. However, when we turn attention to neuroprotection, conventional animal models often lose their ability to predict clinical effects.69 The MitoPark mouse, which carries a genetic disruption of the mitochondrial transcription factor A (Tfam) gene in DA neurons,70 was the first animal model to show the slowly progressive but robust degeneration of DA neurons and other essential features seen in PD, including impairment of non-motor functions. Although no PD patient has yet been found with mutations in Tfam, many of the genes affected in the rare familial forms of PD are involved in the regulation of mitochondrial function.71 Nonetheless, it is not yet clear whether MitoPark mice provide a predictive model for clinical neuroprotection; additional animal models would ideally be tested to verify this GDNF delivery approach for PD.
Lastly, GDNF family receptor alpha (GFRα) receptors are widespread throughout the body, raising possible concern regarding off-target effects of GDNF therapies. Allodynia, constipation, dizziness, muscle spasms, and weight loss were observed in patients who received intracerebroventricular (i.c.v.) injections of GDNF protein.72 Outside of the CNS, GDNF acts as a kidney morphogen during embryonic development and regulates the differentiation of spermatogonia in the testis.73 Mice with targeted overexpression of GDNF in undifferentiated spermatogonia tend to develop malignant testicular tumors.74 Moreover, monoclonal antibody-GDNF fusion was reported to associate with proliferative pancreatic and myocardiocyte lesions.75 Although our present and previous studies did not observe any of these potential adverse effects during the relatively short experimental period (∼5 months), long-term study would be required to address these concerns. Because MitoPark mice have a limited lifespan, wild-type C57BL/6J mice are used in our ongoing long-term investigations. The long-term study will also address two major safety issues surrounding GDNF administration: antibody development and cerebellar pathology.16
In conclusion, we have demonstrated a neuroprotective/disease-modifying therapy for PD in the MitoPark mouse model by a combination of novel non-toxic HSCT and macrophage-mediated GDNF delivery to the degenerating neurons. We believe the combination of desirable characteristics offered by our approach represents a major technical/therapeutic advance by offering sustained GDNF delivery directly to the degenerating DA neuron microenvironment. Because macrophage CNS recruitment is unlikely to be affected by brain size, our approach should more reliably translate from animal studies to human clinical trials. Considering the dire need for effective disease-modifying therapy, further pre-clinical investigations, as well as early trials in PD patients, appear to be warranted.
Materials and Methods
Mice
Colony founders (GFP-positive, C57BL/6J mice) from the Jackson Laboratory (Bar Harbor, ME, USA) were housed and bred at the UTHSCSA Laboratory Animal Resources facility. MitoPark mice were described previously.24,36 Mice were randomly allocated to experimental groups and group size was estimated by power analysis based on our previous behavioral test data to achieve 95% power with α less than 0.05. All mice were group-housed in same-sex, standard shoebox cages within a ventilated caging system and provided with ad libitum access to food and water. The room was maintained at 26°C on a 12-hr light/12-hr dark cycle. All animal care and use procedures were in accordance with the Guide for the Care and Use of Laboratory Animals, 8th edition (National Research Council).
Non-toxic HSCT Regimen
8-week old, female C57BL/6J mice were divided into seven groups (n = 3 per group). Mice from each group received G-CSF (Neupogen, Filgrastim, Amgen, Thousand Oaks, CA, USA) intra-peritoneal injections (125 μg/kg) every 12 hr for 4 consecutive days to mobilize resident HSCs prior to transplantation. AMD3100 (Mozobil, Genzyme, Cambridge, MA, USA) was administered (5 mg/kg) through subcutaneous injection 14 hr after the last dose of G-CSF and 1 hr prior to HSC transplantation. 8-week old GFP+ lineage-negative donor cells (2.0 × 106) were transplanted into each G-CSF/AMD3100-treated mouse via tail vein injection. This procedure was repeated once every 2 weeks for a number of cycles corresponding to individual group numbers (i.e., group 1 received one transplant, group 2 received two transplant cycles, and so on, with group 7 receiving seven transplant cycles). HSC transplant efficacy was assessed by determination of percentage of GFP-positive cells in peripheral blood by flow cytometry at 1 and 4 months after the last transplantation cycle. Data represent mean ± SD from three animals per different cycle of HSCT group (n = 3).
Construction and Validation of Lentiviral Vectors
Construction of LT-MSP-GFP and LT-MSP-hGDNF (human GDNF) vectors was described previously.76 The LT-MSP-hGDNF-2A-GFP vector was designed for expressing both hGDNF and GFP at equal levels, allowing the monitoring of the infiltration of transplanted hGDNF-expressing macrophages in mouse brain by tracking their GFP positivity (Figure S5). The vector was assembled using fragments of GFP-T2A and 2A-hGDNF with an 8.4 kb AgeI-BsrGI fragment of the Lenti-MSP-GFP backbone using NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs, Ipswich, MA, USA). The GFP-T2A fragment was amplified by PCR (forward primer: 5′-CAGGGAGCACTGGAGGCCACCCAGTATGGTGAGCAAGGGCGAGGA-3′, reverse primer: 5′-CACGACATCCCATAACTTCATAGGGCCGGGATTCTCCTCCA-3′) and EGFP-2A-luciferase DNA as a template. The 2A-hGDNF fragment was amplified by PCR (forward primer: 5′-CCTATGAAGTTATGGGATGTCGTGGC-3′, reverse primer: 5′-CTAGTAGGATCTGAGTCCGGACTTTCAGATACATCCACACCTTTTAGCG-3′) and tdTomato-2A-hGDNF DNA as a template. To validate the lentiviral vectors, we individually transfected plasmids LT-MSP-hGDNF-2A-GFP and LT-MSP-GFP into 293T cells followed by determination of GFP expression. The plasmids LT-MSP-GFP, LT-MSP-hGDNF-2A-GFP, and LT-MSP-hGDNF were also separately transfected into the murine macrophage cell line RAW 264.7 (ATCC, Rockville, MD, USA). The culture media containing GDNF produced by RAW 264.7 cells were collected at 48 hr post-transfection, and GDNF concentrations were measured by ELISA. The cytotoxicity of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridinium ion (MPP+) and cell viability were measured as previously described.77,78 Briefly, human SH-SY5Y neuroblastoma cells (ATCC, Manassas, VA, USA) were cultured at 0.5 × 105 cells per well in 96-well plates and treated with LT-MSP-hGDNF-2A-GFP, LT-MSP-hGDNF, or LT-MSP-GFP medium produced from transfected RAW 264.7 cells for 24 hr, along with parallel control groups of SH-SY5Y cells cultured without vectors. All wells of SH-SY5Y cells were then treated with 300 μM MPP+ (Sigma, St. Louis, MO, USA) for 24 hr and cell viability was determined by colorimetric MTT (tetrazolium) assay.
Preparation of Lentivirus
Lentiviruses were prepared as described previously.76 Briefly, plasmid LT-MSP-hGDNF-2A-GFP, LT-MSP-hGDNF, or LT-MSP-GFP was co-transfected with packaging plasmids pMDLg/pRRE, pRSV/Rev, and pMD2G into 293T cells. The culture media containing lentiviral particles were collected at 24 and 48 hr post-transfection, filtered through 0.45 μm pore size filters, and then concentrated 1,000-fold by two rounds of ultracentrifugation at 56,000 × g for 2 hr and then 72,000 × g for 1.5 hr. The viral pellets were resuspended in StemPro-34 SFM medium (Invitrogen, Carlsbad, CA, USA) and stored at −80°C until use.
Lentiviral Transduction and Hematopoietic Stem Cell Transplantation
14-week-old recipient MitoPark mice exhibiting motor and non-motor PD-like features and normal control littermates were randomly divided into four groups of 15 animals each. Group 1 (Mito-LT-hGDNF-2A-GFP) mice were transplanted with HSCs that had been transduced with LT-hGDNF-2A-GFP vectors expressing both hGDNF- and GFP cDNAs. Group 2 (Mito-LT-MSP-hGDNF) mice were transplanted with HSCs that had been transduced with LT-MSP-hGDNF vector expressing hGDNF cDNA. Group 3 (Mito-LT-MSP-GFP) and group 4 (normal control-LT-MSP-GFP) mice were transplanted with HSCs that had been transduced with LT-MSP-GFP vector expressing GFP cDNA. Bone marrow cells were harvested from the femurs, tibias, humeri, and hip bones of 12-week-old syngeneic non-MitoPark donor mice by flushing the bones with Iscove’s Modified Dulbecco’s Medium (IMDM) containing 0.5% heparin. After red blood cell lysis, lineage-negative (Lin−) cells were isolated using the Lineage Cell Depletion kit (Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer’s protocol. The enriched bone-marrow-derived HSCs (Lin−) were cultured overnight in StemSpan SFEM II medium (STEMCELL Technologies, Cambridge, MA, USA) supplemented with 50 ng/mL of murine TPO (thrombopoietin), 100 ng/mL of murine SCF (stem cell factor), and 100 ng/mL murine Flt3-Ligand (PeproTech, Rocky Hill, NJ, USA), as previously described.79 The next day, the harvested cells were centrifuged and resuspended in 800 μL of concentrated viral supernatant supplemented with the aforementioned growth factors. Infections were performed on RetroNectin (Takara, Otsu, Japan) coated plates for 6 hr in the presence of 8 μg/mL of protamine sulfate (Sigma, St. Louis, MO, USA). Following infection, the cells were harvested and washed with StemSpan SFEM II medium, and then 2 × 106 lentiviral transduced cells were transplanted into recipient mice through tail vein injection. This HSCT procedure was repeated once every 2 weeks on each animal group for seven cycles.
Behavioral Tests
The mice were acclimated to the testing room for a minimum of 1 hr prior to behavioral tests. All testing was performed before and after HSCT for each animal by technicians blinded to the mouse group information. Horizontal and vertical locomotor activities were measured using the Photobeam Activity System (San Diego, CA, USA) according to the manufacturer’s protocol. The mice were individually placed in a clear polycarbonate testing cage (18 × 29 × 12 cm) with approximately 0.5 cm of corncob bedding lining the floor. The horizontal and vertical locomotor activities were recorded for 60 min and assessed in 10-min time bins. Novel-object recognition (NOR) task was tested in a training/testing open field (40 × 40 × 38 cm) as previously described. The time spent by each mouse investigating the familiar or novel objects was scored from video records by two blinded observers. A discrimination index was calculated using the following formula: (time exploring novel object/time spent exploring novel + time spent exploring familiar) × 100. Sucrose preference was measured as previously described.80,81 Briefly, the mice were individually housed in a clean plastic cage with approximately 2 cm of corncob bedding lining the floor and allowed ad libitum access to drink from two identical volumetric bottles containing either water or 1% sucrose solution. The positions of the two bottles were switched daily and the bottles were weighed to measure the water or sucrose solution consumption every day for 4 days. Sucrose preference was calculated using the following formula: % sucrose preference = (sucrose solution intake / water intake + sucrose solution intake) × 100.
Immunohistochemistry
Mice were anesthetized at the termination of experiment and perfused transcardially with 20 mL of PBS, pH 7.4 followed by an equal volume of 4% paraformaldehyde solution. Brains were dissected out and fixed in the same fixative overnight at 4°C. The brains were cryo-protected in sequential 10% (2 hr), 20% (2 hr), and 30% sucrose in PBS solution overnight. The brains were embedded in Tissue-Tek OCT compound (Sakura Finetek USA, Torrance, CA, USA) and processed for cryosectioning at 30 μm thickness in the coronal plane. Four series of slides containing every fourth section were prepared for SN, whereas six series of every sixth section were prepared for striatum. The brain sections were treated with 1% BSA in PBS containing 0.3% Triton X-100 for 30 min and then incubated with rabbit anti-TH antibody (Millipore, Billerica, MA, USA) at 1:1,000 dilution overnight at 4°C. The sections were rinsed with PBS and incubated with biotinylated goat anti-rabbit secondary antibody (1:100) for 1 hr, followed by avidin-biotin peroxidase complex (ABC Elite Kit, Vector Laboratories, Burlingame, CA, USA) at room temperature for 1 hr. The chromogen was either 3-amino-9-ethyl carbazole (AEC Chromogen Kit, Sigma) or 3,3′-diaminobenzidine tetrahydrochloride (Liquid DAB Substrate Kit, Invitrogen, Carisbad, CA, USA). DAB-stained midbrain sections were counterstained with cresyl violet and used for DA neuron count. For immunofluorescent staining, the sections were first incubated with either anti-TH or anti-Iba1 (1:1,000, Wako Diagnostics, Richmond, VA, USA) or anti-GM130 (BD Biosciences, San Jose, CA, USA), followed by Alexa Fluor 568 (1:100, Invitrogen) or either Alexa Fluor 350 (1: 100, ThermoFisher) or Alexa Fluor 488 (1:100, Molecular Probes) as secondary antibodies. For quantification of microglia infiltrated into SN and VTA, the midbrain sections from normal control-LT-MSP-GFP, Mito-LT-MSP-GFP, or Mito-LT-hGDNF-2A-GFP mice were double immunostained with sheep anti-TH antibody (ThermoFisher, Rockford, IL, USA) and rabbit anti-Iba1 antibody at 1:1,000 dilution, followed by Alexa Fluor 350 and Alexa Fluor 568-conjugated secondary antibodies. The images were captured with a fluorescent microscope (Nikon Eclipse TE2000-U, Nikon Instruments, Melville, NY, USA).
Optical Density
Optical densities (ODs) of the TH-stained fibers in the striatum were measured from digitized images of every sixth section using NIH ImageJ software (NIH, Bethesda, MD, USA) by two blinded investigators following the same criteria. The measurements were taken from dorsolateral aspects of the striatum, which receives the majority of innervation from SNpc DA neurons. The relative ODs of TH+ fibers in the striatum were calculated by subtracting the background OD from the measured OD of the dorsolateral aspects of the striatum.
ELISA
Plasma was prepared from peripheral blood collected in EDTA-treated tubes. Midbrain tissue containing SN and forebrain tissue containing striatum were dissected out in 1.0 mm thick coronal slices using a mouse brain slicer matrix (Zivic Instruments, Pittsburgh, PA, USA) according to the mouse brain atlas. The brain tissue slices were collected in cryovials, quickly frozen in liquid nitrogen and stored at −80°C until analysis. The frozen tissue samples were homogenized in tissue lysis buffer containing 137 mM NaCl, 20 mM Tris (pH 8.0), 1% NP-40, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 1 μg/mL leupeptin, and 0.5 mM sodium vanadate at a tissue concentration of 30 mg/mL and centrifuged at 20,000 × g for 10 min at 4°C. The GDNF concentrations in plasma, SN, and striatum were measured using a commercially available GDNF Emax ImmunoAssay System (Promega, Madison, WI, USA), according to the manufacturer’s protocol. The assay detects both human and rodent GDNF with pg/mL level sensitivity.
Measurement of DA Content in the Striatum by High-Performance Liquid Chromatography
A 0.5 mm thick slice of coronal forebrain containing striatum was dissected out using a mouse brain slicer matrix (Zivic Instruments, Pittsburgh, PA, USA) and stored at −80°C until analysis. The concentrations of DA, DOPAC, and HVA were determined by high performance liquid chromatography (HPLC) combined with electrochemical detection. The frozen tissues were sonicated in 0.2 mol/L perchloric acid (20% wt/vol) containing 100 ng/mL 3,4-dihydroxybenzylamine (internal standard) and centrifuged at 15,000 × g for 7 minutes. The supernatant was filtered using a 0.2 μm Nylon-66 filter and a 25 μL aliquot of the filtrate, equivalent to 2.5 mg of tissue, was injected directly into the HPLC/electrochemical system. The mobile phase consisted of 0.07 mol/L potassium phosphate, pH 3.0, 8% methanol, and 1.02 mmol/L 1-heptane sulfonic acid. The concentrations of DA, DOPAC, and HVA were calculated using a curve generated with authentic standards.
Flow Cytometry
Peripheral blood was collected from LT-hGDNF-2A-GFP- and LT-MSP-GFP-transplanted MitoPark mice and LT-MSP-GFP-transplanted non-MitoPark normal control mice. After erythrocytes were lysed with Red Blood Lysing Buffer (Sigma, St. Louis, MO, USA), the GFP-expressing (GFP+) cells were determined by flow cytometry (BD FACSCablibur System, BD Bioscience, San Jose, CA, USA). The peripheral blood samples collected from wild-type C57BL/6J and GFP transgenic mice were used as negative and positive controls, respectively.
Statistical Analysis
The statistical analysis of the data was performed using GraphPad Prism 6.02 (GraphPad Software, LaJolla, CA, USA). Multiple group comparisons were analyzed by two-way ANOVA, followed by post hoc analyses using Bonferroni post-test or one-way ANOVA, followed by Tukey’s post-test. Differences among treatment groups were considered statistically significant at p < 0.05.
Study Approval
The procedures for all animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of The University of Texas Health San Antonio.
Author Contributions
C.C., J.C.O., and S.L. designed research and analyzed data. Y.L., Z.H., and E.M. provided critical scientific and technical insights. C.C. carried out all of the experimental work with the help of M.J.G., G.G., and A.B. S.L. and J.C.O. directed the project. C.C., M.J.G., and S.L. wrote the manuscript with the help of Y.L., J.C.O., and R.A.C.
Conflicts of Interest
S.L., R.A.C., C.C., and M.J.G. have filed a provisional patent application. The authors have declared no additional conflicts of interest.
Acknowledgments
We thank Dr. Nils-Göran Larsson (Max Planck Institute for Biology of Aging, Cologne, Germany) for providing breeders of the MitoPark mice, Ms. Karla Moncada Gorena for excellent technical support with flow cytometry analysis, and Dr. Martha A. Hanes for professional pathological review. This study was supported by a Merit Review grant from the Department of Veterans Affairs Biomedical Laboratory Research & Development(2I01BX000737), the William and Ella Owens Medical Research Foundation, and the Perry & Ruby Stevens Parkinson’s Disease Center of Excellence.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2019.11.013.
Supplemental Information
References
- 1.Emborg M.E. Evaluation of animal models of Parkinson’s disease for neuroprotective strategies. J. Neurosci. Methods. 2004;139:121–143. doi: 10.1016/j.jneumeth.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Wood-Kaczmar A., Gandhi S., Wood N.W. Understanding the molecular causes of Parkinson’s disease. Trends Mol. Med. 2006;12:521–528. doi: 10.1016/j.molmed.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Obeso J.A., Stamelou M., Goetz C.G., Poewe W., Lang A.E., Weintraub D., Burn D., Halliday G.M., Bezard E., Przedborski S. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017;32:1264–1310. doi: 10.1002/mds.27115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalia L.V., Lang A.E. Parkinson’s disease. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 5.Stacy M., Bowron A., Guttman M., Hauser R., Hughes K., Larsen J.P., LeWitt P., Oertel W., Quinn N., Sethi K., Stocchi F. Identification of motor and nonmotor wearing-off in Parkinson’s disease: comparison of a patient questionnaire versus a clinician assessment. Mov. Disord. 2005;20:726–733. doi: 10.1002/mds.20383. [DOI] [PubMed] [Google Scholar]
- 6.Shetty A.S., Bhatia K.P., Lang A.E. Dystonia and Parkinson’s disease: What is the relationship? Neurobiol. Dis. 2019;132:104462. doi: 10.1016/j.nbd.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Ahlskog J.E., Muenter M.D. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. 2001;16:448–458. doi: 10.1002/mds.1090. [DOI] [PubMed] [Google Scholar]
- 8.Park A., Stacy M. Disease-Modifying Drugs in Parkinson’s Disease. Drugs. 2015;75:2065–2071. doi: 10.1007/s40265-015-0497-4. [DOI] [PubMed] [Google Scholar]
- 9.Lin L.F., Doherty D.H., Lile J.D., Bektesh S., Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 10.Tomac A., Lindqvist E., Lin L.F., Ogren S.O., Young D., Hoffer B.J., Olson L. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. 1995;373:335–339. doi: 10.1038/373335a0. [DOI] [PubMed] [Google Scholar]
- 11.Paul G., Sullivan A.M. Trophic factors for Parkinson’s disease: Where are we and where do we go from here? Eur. J. Neurosci. 2019;49:440–452. doi: 10.1111/ejn.14102. [DOI] [PubMed] [Google Scholar]
- 12.Bianchi V.E., Locatelli V., Rizzi L. Neurotrophic and Neuroregenerative Effects of GH/IGF1. Int. J. Mol. Sci. 2017;18:2441. doi: 10.3390/ijms18112441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi-Lundberg D.L., Lin Q., Chang Y.N., Chiang Y.L., Hay C.M., Mohajeri H., Davidson B.L., Bohn M.C. Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science. 1997;275:838–841. doi: 10.1126/science.275.5301.838. [DOI] [PubMed] [Google Scholar]
- 14.Espay A.J., Brundin P., Lang A.E. Precision medicine for disease modification in Parkinson disease. Nat. Rev. Neurol. 2017;13:119–126. doi: 10.1038/nrneurol.2016.196. [DOI] [PubMed] [Google Scholar]
- 15.Brundin P. GDNF treatment in Parkinson’s disease: time for controlled clinical trials? Brain. 2002;125:2149–2151. doi: 10.1093/brain/awf217. [DOI] [PubMed] [Google Scholar]
- 16.Sherer T.B., Fiske B.K., Svendsen C.N., Lang A.E., Langston J.W. Crossroads in GDNF therapy for Parkinson’s disease. Mov. Disord. 2006;21:136–141. doi: 10.1002/mds.20861. [DOI] [PubMed] [Google Scholar]
- 17.Salvatore M.F., Ai Y., Fischer B., Zhang A.M., Grondin R.C., Zhang Z., Gerhardt G.A., Gash D.M. Point source concentration of GDNF may explain failure of phase II clinical trial. Exp. Neurol. 2006;202:497–505. doi: 10.1016/j.expneurol.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 18.Bartus R.T., Weinberg M.S., Samulski R.J. Parkinson’s disease gene therapy: success by design meets failure by efficacy. Mol. Ther. 2014;22:487–497. doi: 10.1038/mt.2013.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behrstock S., Ebert A., McHugh J., Vosberg S., Moore J., Schneider B. Human neural progenitors deliver glial cell line-derived neurotrophic factor to parkinsonian rodents and aged primates. Gene Ther. 2005;13:379–388. doi: 10.1038/sj.gt.3302679. [DOI] [PubMed] [Google Scholar]
- 20.Bartus R.T., Herzog C.D., Chu Y., Wilson A., Brown L., Siffert J., Johnson E.M., Jr., Olanow C.W., Mufson E.J., Kordower J.H. Bioactivity of AAV2-neurturin gene therapy (CERE-120): differences between Parkinson’s disease and nonhuman primate brains. Mov. Disord. 2011;26:27–36. doi: 10.1002/mds.23442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bartus R.T., Baumann T.L., Siffert J., Herzog C.D., Alterman R., Boulis N., Turner D.A., Stacy M., Lang A.E., Lozano A.M., Olanow C.W. Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology. 2013;80:1698–1701. doi: 10.1212/WNL.0b013e3182904faa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biju K., Zhou Q., Li G., Imam S.Z., Roberts J.L., Morgan W.W., Clark R.A., Li S. Macrophage-mediated GDNF delivery protects against dopaminergic neurodegeneration: a therapeutic strategy for Parkinson’s disease. Mol. Ther. 2010;18:1536–1544. doi: 10.1038/mt.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biju K.C., Santacruz R.A., Chen C., Zhou Q., Yao J., Rohrabaugh S.L., Clark R.A., Roberts J.L., Phillips K.A., Imam S.Z., Li S. Bone marrow-derived microglia-based neurturin delivery protects against dopaminergic neurodegeneration in a mouse model of Parkinson’s disease. Neurosci. Lett. 2013;535:24–29. doi: 10.1016/j.neulet.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C., Li X., Ge G., Liu J., Biju K.C., Laing S.D., Qian Y., Ballard C., He Z., Masliah E. GDNF-expressing macrophages mitigate loss of dopamine neurons and improve Parkinsonian symptoms in MitoPark mice. Sci. Rep. 2018;8:5460. doi: 10.1038/s41598-018-23795-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge G., Chen C., Guderyon M.J., Liu J., He Z., Yu Y., Clark R.A., Li S. Regulatable Lentiviral Hematopoietic Stem Cell Gene Therapy in a Mouse Model of Parkinson’s Disease. Stem Cells Dev. 2018;27:995–1005. doi: 10.1089/scd.2018.0030. [DOI] [PubMed] [Google Scholar]
- 26.Copelan E.A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharya D., Czechowicz A., Ooi A.G.L., Rossi D.J., Bryder D., Weissman I.L. Niche recycling through division-independent egress of hematopoietic stem cells. J. Exp. Med. 2009;206:2837–2850. doi: 10.1084/jem.20090778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pessach I., Shimoni A., Nagler A. Granulocyte-colony stimulating factor for hematopoietic stem cell donation from healthy female donors during pregnancy and lactation: what do we know? Hum. Reprod. Update. 2013;19:259–267. doi: 10.1093/humupd/dms053. [DOI] [PubMed] [Google Scholar]
- 29.Pusic I., DiPersio J.F. Update on clinical experience with AMD3100, an SDF-1/CXCL12-CXCR4 inhibitor, in mobilization of hematopoietic stem and progenitor cells. Curr. Opin. Hematol. 2010;17:319–326. doi: 10.1097/MOH.0b013e328338b7d5. [DOI] [PubMed] [Google Scholar]
- 30.Dykstra B., Kent D., Bowie M., McCaffrey L., Hamilton M., Lyons K., Lee S.J., Brinkman R., Eaves C. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell. 2007;1:218–229. doi: 10.1016/j.stem.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 31.Chen J., Larochelle A., Fricker S., Bridger G., Dunbar C.E., Abkowitz J.L. Mobilization as a preparative regimen for hematopoietic stem cell transplantation. Blood. 2006;107:3764–3771. doi: 10.1182/blood-2005-09-3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamminga L.M., van Os R., Ausema A., Noach E.J.K., Weersing E., Dontje B., Vellenga E., de Haan G. Impaired hematopoietic stem cell functioning after serial transplantation and during normal aging. Stem Cells. 2005;23:82–92. doi: 10.1634/stemcells.2004-0066. [DOI] [PubMed] [Google Scholar]
- 33.Randall T.D., Weissman I.L. Characterization of a population of cells in the bone marrow that phenotypically mimics hematopoietic stem cells: resting stem cells or mystery population? Stem Cells. 1998;16:38–48. doi: 10.1002/stem.160038. [DOI] [PubMed] [Google Scholar]
- 34.Kumar R., Fossati V., Israel M., Snoeck H.-W. Lin-Sca1+kit- bone marrow cells contain early lymphoid-committed precursors that are distinct from common lymphoid progenitors. J. Immunol. 2008;181:7507–7513. doi: 10.4049/jimmunol.181.11.7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang H., Richter M., Psatha N., Li C., Kim J., Liu J., Ehrhardt A., Nilsson S.K., Cao B., Palmer D. A Combined In Vivo HSC Transduction/Selection Approach Results in Efficient and Stable Gene Expression in Peripheral Blood Cells in Mice. Mol. Ther. Methods Clin. Dev. 2017;8:52–64. doi: 10.1016/j.omtm.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X., Redus L., Chen C., Martinez P.A., Strong R., Li S., O’Connor J.C. Cognitive dysfunction precedes the onset of motor symptoms in the MitoPark mouse model of Parkinson’s disease. PLoS ONE. 2013;8:e71341. doi: 10.1371/journal.pone.0071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez A.M., Weintraub D., Claassen D.O. Impulse Control Disorders and Related Complications of Parkinson’s Disease Therapy. Semin. Neurol. 2017;37:186–192. doi: 10.1055/s-0037-1601887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holstege G., Georgiadis J.R., Paans A.M.J., Meiners L.C., van der Graaf F.H.C.E., Reinders A.A.T.S. Brain activation during human male ejaculation. J. Neurosci. 2003;23:9185–9193. doi: 10.1523/JNEUROSCI.23-27-09185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zlokovic B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Wohleb E.S., McKim D.B., Sheridan J.F., Godbout J.P. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front. Neurosci. 2015;8:447. doi: 10.3389/fnins.2014.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lund H., Pieber M., Parsa R., Han J., Grommisch D., Ewing E., Kular L., Needhamsen M., Espinosa A., Nilsson E. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat. Commun. 2018;9:4845. doi: 10.1038/s41467-018-07295-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varatharaj A., Galea I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017;60:1–12. doi: 10.1016/j.bbi.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 43.Auluck P.K., Caraveo G., Lindquist S. α-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- 44.Lin X., Parisiadou L., Sgobio C., Liu G., Yu J., Sun L., Shim H., Gu X.L., Luo J., Long C.X. Conditional expression of Parkinson’s disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J. Neurosci. 2012;32:9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gosavi N., Lee H.-J., Lee J.S., Patel S., Lee S.-J. Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 2002;277:48984–48992. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- 46.Lin X., Parisiadou L., Gu X.-L., Wang L., Shim H., Sun L., Xie C., Long C.X., Yang W.J., Ding J. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant α-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou W., Chang J., Wang X., Savelieff M.G., Zhao Y., Ke S., Ye B. GM130 is required for compartmental organization of dendritic golgi outposts. Curr. Biol. 2014;24:1227–1233. doi: 10.1016/j.cub.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakamura N., Rabouille C., Watson R., Nilsson T., Hui N., Slusarewicz P., Kreis T.E., Warren G. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mildner A., Schmidt H., Nitsche M., Merkler D., Hanisch U.K., Mack M., Heikenwalder M., Brück W., Priller J., Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 50.Zhao Y., Haney M.J., Gupta R., Bohnsack J.P., He Z., Kabanov A.V., Batrakova E.V. GDNF-transfected macrophages produce potent neuroprotective effects in Parkinson’s disease mouse model. PLoS ONE. 2014;9:e106867. doi: 10.1371/journal.pone.0106867. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Lebson L., Nash K., Kamath S., Herber D., Carty N., Lee D.C., Li Q., Szekeres K., Jinwal U., Koren J. Trafficking CD11b-positive blood cells deliver therapeutic genes to the brain of amyloid-depositing transgenic mice. J. Neurosci. 2010;30:9651–9658. doi: 10.1523/JNEUROSCI.0329-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cartier N., Aubourg P. Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X-linked adrenoleukodystrophy. Brain Pathol. 2010;20:857–862. doi: 10.1111/j.1750-3639.2010.00394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eichler F., Duncan C., Musolino P.L., Orchard P.J., De Oliveira S., Thrasher A.J., Armant M., Dansereau C., Lund T.C., Miller W.P. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017;377:1630–1638. doi: 10.1056/NEJMoa1700554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 55.Shemer A., Grozovski J., Tay T.L., Tao J., Volaski A., Süß P., Ardura-Fabregat A., Gross-Vered M., Kim J.S., David E. Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat. Commun. 2018;9:5206. doi: 10.1038/s41467-018-07548-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montero C.N., Hefti F. Rescue of lesioned septal cholinergic neurons by nerve growth factor: specificity and requirement for chronic treatment. J. Neurosci. 1988;8:2986–2999. doi: 10.1523/JNEUROSCI.08-08-02986.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Georgievska B., Kirik D., Björklund A. Overexpression of glial cell line-derived neurotrophic factor using a lentiviral vector induces time- and dose-dependent downregulation of tyrosine hydroxylase in the intact nigrostriatal dopamine system. J. Neurosci. 2004;24:6437–6445. doi: 10.1523/JNEUROSCI.1122-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson S., van Os R., Sheridan T., Mauch P. Reduction of marrow hematopoietic progenitor and stem cell content is not sufficient for enhanced syngeneic engraftment. Stem Cells. 2000;18:93–101. doi: 10.1634/stemcells.18-2-93. [DOI] [PubMed] [Google Scholar]
- 59.To L.B., Levesque J.P., Herbert K.E. How I treat patients who mobilize hematopoietic stem cells poorly. Blood. 2011;118:4530–4540. doi: 10.1182/blood-2011-06-318220. [DOI] [PubMed] [Google Scholar]
- 60.Mohty M., Duarte R.F., Croockewit S., Hübel K., Kvalheim G., Russell N. The role of plerixafor in optimizing peripheral blood stem cell mobilization for autologous stem cell transplantation. Leukemia. 2011;25:1–6. doi: 10.1038/leu.2010.224. [DOI] [PubMed] [Google Scholar]
- 61.Barcia C., Ros C.M., Ros-Bernal F., Gómez A., Annese V., Carrillo-de Sauvage M.A., Yuste J.E., Campuzano C.M., de Pablos V., Fernández-Villalba E., Herrero M.T. Persistent phagocytic characteristics of microglia in the substantia nigra of long-term Parkinsonian macaques. J. Neuroimmunol. 2013;261:60–66. doi: 10.1016/j.jneuroim.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 62.Mueller K.L., Hines P.J., Travis J. Neuroimmunology. Science. 2016;353:760–761. doi: 10.1126/science.353.6301.760. [DOI] [PubMed] [Google Scholar]
- 63.Collins F.S., Gottlieb S. The Next Phase of Human Gene-Therapy Oversight. N. Engl. J. Med. 2018;379:1393–1395. doi: 10.1056/NEJMp1810628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 65.Corrigan-Curay J., O’Reilly M., Kohn D.B., Cannon P.M., Bao G., Bushman F.D., Carroll D., Cathomen T., Joung J.K., Roth D. Genome editing technologies: defining a path to clinic. Mol. Ther. 2015;23:796–806. doi: 10.1038/mt.2015.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perrin S. Preclinical research: Make mouse studies work. Nature. 2014;507:423–425. doi: 10.1038/507423a. [DOI] [PubMed] [Google Scholar]
- 68.Jenner P. Functional models of Parkinson’s disease: a valuable tool in the development of novel therapies. Ann. Neurol. 2008;64(Suppl 2):S16–S29. doi: 10.1002/ana.21489. [DOI] [PubMed] [Google Scholar]
- 69.Olanow C.W., Kieburtz K., Schapira A.H. Why have we failed to achieve neuroprotection in Parkinson’s disease? Ann. Neurol. 2008;64(Suppl 2):S101–S110. doi: 10.1002/ana.21461. [DOI] [PubMed] [Google Scholar]
- 70.Ekstrand M.I., Terzioglu M., Galter D., Zhu S., Hofstetter C., Lindqvist E., Thams S., Bergstrand A., Hansson F.S., Trifunovic A. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shen J., Cookson M.R. Mitochondria and dopamine: new insights into recessive parkinsonism. Neuron. 2004;43:301–304. doi: 10.1016/j.neuron.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 72.Nutt J.G., Burchiel K.J., Comella C.L., Jankovic J., Lang A.E., Laws E.R., Jr., Lozano A.M., Penn R.D., Simpson R.K., Jr., Stacy M., Wooten G.F., ICV GDNF Study Group. Implanted intracerebroventricular. Glial cell line-derived neurotrophic factor Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60:69–73. doi: 10.1212/wnl.60.1.69. [DOI] [PubMed] [Google Scholar]
- 73.Sariola H., Saarma M. Novel functions and signalling pathways for GDNF. J. Cell Sci. 2003;116:3855–3862. doi: 10.1242/jcs.00786. [DOI] [PubMed] [Google Scholar]
- 74.Meng X., de Rooij D.G., Westerdahl K., Saarma M., Sariola H. Promotion of seminomatous tumors by targeted overexpression of glial cell line-derived neurotrophic factor in mouse testis. Cancer Res. 2001;61:3267–3271. [PubMed] [Google Scholar]
- 75.Ohshima-Hosoyama S., Simmons H.A., Goecks N., Joers V., Swanson C.R., Bondarenko V., Velotta R., Brunner K., Wood L.D., Hruban R.H., Emborg M.E. A monoclonal antibody-GDNF fusion protein is not neuroprotective and is associated with proliferative pancreatic lesions in parkinsonian monkeys. PLoS ONE. 2012;7:e39036. doi: 10.1371/journal.pone.0039036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.He W., Qiang M., Ma W., Valente A.J., Quinones M.P., Wang W., Reddick R.L., Xiao Q., Ahuja S.S., Clark R.A. Development of a synthetic promoter for macrophage gene therapy. Hum. Gene Ther. 2006;17:949–959. doi: 10.1089/hum.2006.17.949. [DOI] [PubMed] [Google Scholar]
- 77.Presgraves S.P., Ahmed T., Borwege S., Joyce J.N. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox. Res. 2004;5:579–598. doi: 10.1007/BF03033178. [DOI] [PubMed] [Google Scholar]
- 78.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 79.Liu X., Zheng H., Yu W.M., Cooper T.M., Bunting K.D., Qu C.K. Maintenance of mouse hematopoietic stem cells ex vivo by reprogramming cellular metabolism. Blood. 2015;125:1562–1565. doi: 10.1182/blood-2014-04-568949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salazar A., Gonzalez-Rivera B.L., Redus L., Parrott J.M., O’Connor J.C. Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm. Behav. 2012;62:202–209. doi: 10.1016/j.yhbeh.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parrott J.M., Redus L., Santana-Coelho D., Morales J., Gao X., O’Connor J.C. Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Transl. Psychiatry. 2016;6:e918. doi: 10.1038/tp.2016.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.