

Abstract
An important component of breast milk, calcium also appears as radiographically prominent microcalcifications in breast tissue that are often the earliest sign of malignancy. Ionic Ca2+ is a universal second messenger that controls a wide swathe of effector pathways integral to gene transcription, cell cycle control, differentiation, proliferation, cell migration, and apoptosis. Whereas prolonged elevation in resting Ca2+ levels drives proliferation to initiate and sustain tumor growth, depletion of calcium stores and attenuation of calcium influx pathways underlies tumor chemoresistance and evasion of apoptosis. This paradox of Ca2+ homeostasis highlights the challenge of targeting Ca2+ signaling pathways for breast cancer therapy. Furthermore, breast cancer is a heterogeneous disease classified into distinct subtypes based on tumor origin, stage of invasiveness and hormone receptor status. Classification is important for tailoring treatment, and in predicting clinical outcome or response to chemotherapy. There have been numerous reports of dysregulated expression, localization or activity of Ca2+ channels, regulators and pumps in breast cancer. An important aspect of these alterations is that they are specific to breast cancer subtype, as exemplified by a reciprocal switch in secretory pathway Ca2+-ATPase isoforms SPCA1 and SPCA2 depending on receptor status. In this review, we discuss the current knowledge of subtype specific changes in calcium channels and pumps, with a focus on functional insights that may inform new opportunities for breast cancer therapy.
Keywords: Calcium Signaling, Breast cancer subtypes, Calcium, SPCA2
Graphical Abstract
Breast Cancer: Statistics and Subtypes
Women born today will have about a 1 in 8 (12.5%) chance of developing breast cancer at some point during their lifetime [1, 2]. As the second most common cancer worldwide, after lung cancer, the incidence rate of breast cancer is increasing. In 2018, close to 2 million new cases were diagnosed worldwide and about 600,000 people died from this disease [2]. Although treatment of breast cancer has improved in recent decades, many patients still succumb to this disease. Moreover, during or after intensive treatment many breast cancer patients develop serious acute and chronic complications [3]. Therefore, there is an urgent need to improve patient outcomes by identifying new biomarker based targets and understanding mechanisms of resistance against current chemotherapeutic agents.
Breast cancer is a heterogeneous disease that can be subdivided based on origin, progression and molecular characteristics (Figure 1). This classification is important for tailoring treatment, predicting clinical outcome and response to chemotherapy. About 80% of breast cancers arise in the milk ducts (ductal carcinoma), while others originate in the lobules of milk producing glands (lobular carcinoma) [4]. The disease progresses in distinct stages beginning with a tumor localized in the breast (Stage 0), followed in succession through Stage I (tumor smaller than 2 cm), Stage II in which cancer has spread to the lymph nodes near the breast, and Stage III or “locally advanced breast cancer”, where the cancer has spread to the skin, chest wall, or internal mammary lymph nodes. Invasive breast cancer is described as stage IV, where the cancer has metastasized to other organs of the body, such as the lungs, distant lymph nodes, skin, bones, liver, or brain [5]. The 5-year overall survival rate for Stage I and II breast cancers is more than 95%, but drops to 72% for Stage III and 22% for Stage IV breast cancer [5]. In fact, 90% of breast cancer deaths are associated with metastasis. An estimated 150,000-plus women (and men) in the U.S. currently live with stage IV breast cancer that has metastasized [6]. This dismal statistic highlights the critical need to understand the cellular and molecular basis for tumor metastasis.
Figure 1.

Breast Cancer Subtypes are based on tumor origin, tumor stage and hormone receptor status. A. Tumors may originate in milk ducts or lobules, which are composed of a basal layer of myoepithelial cells surrounding a layer of luminal cells. B. Survival statistics for breast cancer are influenced by the stage of tumor progression and invasiveness. C. Breast cancers are subtyped at the molecular level depending on the presence or absence of receptors for estrogen (ER), progesterone (PR) or human epidermal growth factor (HER). Triple negative breast cancers (TNBC) lack all three of these receptors. Tumor aggressiveness increases from Luminal A to TNBC and is indicated by shading from orange to red.
At the molecular level, breast cancers are categorized based on the presence of key hormone or growth factor receptors (Figure 1). Approximately 70% of breast cancers are positive for estrogen receptor (ER) and/or progesterone receptor (PR) [7]. Human epidermal growth factor 2 (HER2) positive cases account for 15–20% [8], and triple-negative tumors lacking all three receptors constitute up to 15% of total cases (Table 1) [9]. The receptor-positive breast cancers are further classified as Luminal A and B. Luminal A cancer is HER2-negative whereas Luminal B cancer may be HER2 positive or negative with higher expression of Ki67, a marker associated with aggressive tumor proliferation and growth [10]. Triple-negative breast cancer (TNBC) is more likely to recur than the other subtypes. The 5-year overall survival for TNBC stage I survival is 85% versus 94% to 99% for hormone receptor positive and HER2 positive breast cancers. Median overall survival for metastatic triple-negative breast cancer is approximately 1 year versus approximately 4–5 years for the other 2 subtypes (Table 1) [11].
Table 1.
Breast Cancer Prognosis Based on Receptor Subtype [11]
| Breast Cancer Subtype | Receptor Status | Breast Cancer Cases | Stage I 5-year survival | Metastatic Median Survival |
|---|---|---|---|---|
| Estrogen Receptor and Progesterone Receptor Positive | ER+/PR+ | 70% | ≥ 99 | 4–5 y |
| Human Epidermal Growth Factor 2 Receptor Positive | HER2+ | 15–20% | ≥ 94 | 5 y |
| Triple Receptor Negative Breast Cancer | TNBC | 15% | ≥ 85 | 10–13 months |
Hormone receptor-positive patients receive endocrine therapy, such as tamoxifen that competitively inhibits the binding of estrogen to ER, aromatase inhibitors (anastrozole, exemestane, and letrozole) that decrease circulating estrogen levels by inhibiting conversion of androgens to estrogen, and a minority of patients receive chemotherapy as well. HER2 positive patients are treated with HER2-specific monoclonal antibodies or small-molecule tyrosine kinase inhibitors combined with chemotherapy. TNBC patients receive only chemotherapy, as there is no targeted treatment [11].
Paradoxical Role of Calcium Signaling in Breast Cancer
A crucial component of breast milk, calcium plays an essential role in the mineralization of bones and teeth [12]. In breast tissue, the coordinated regulation of a functional module of Ca2+ transporters, sensors and buffers named CALTRANS, occurs during milk production, to efficiently move calcium from the blood, into milk [13]. In addition, ionized Ca2+ is a versatile, essential and ubiquitous second messenger that impacts all aspects of cell fate and function. Unlike other second messengers, elemental Ca2+ cannot be synthesized or degraded. Instead, a plethora of membrane-embedded Ca2+ transporting ion channels, ATPases (pumps), and carrier proteins rapidly (in the millisecond time scale) move Ca2+ across organellar or cell membranes for Ca2+ homeostasis. Resting cytosolic free Ca2+ is maintained at low levels (~50–100 nM) against a steep, ~10,000-fold gradient relative to extracellular free Ca2+ (1–2 mM). In non-excitable cells, a family of P-type Ca2+-ATPases, localized to the endoplasmic reticulum, Golgi, secretory vesicles and plasma membrane, pump Ca2+ out of the cytoplasm to maintain resting cytoplasmic Ca2+ levels. This allows elevations of cytoplasmic Ca2+ caused by the transient opening of Ca2+ channels in response to a host of intracellular and extracellular ligands, membrane voltage, mechanical forces or volume changes, to activate downstream effector pathways integral to gene transcription, cell cycle control, differentiation, proliferation, cell migration, and apoptosis (Figure 2). Each of these cellular processes is central to tumorigenesis. Thus, alterations in the precise timing, spatial distribution and amplitude of the Ca2+ signal leads to the establishment and maintenance of tumor malignancy [14–16].
Figure 2.

Ionic Ca2+ is a ubiquitous second messenger that impacts all aspects of cell fate and function. Ca2+ signaling pathways regulate key processes from inflammation to apoptosis that are involved in breast cancer tumorigenesis and resistance to chemotherapy.
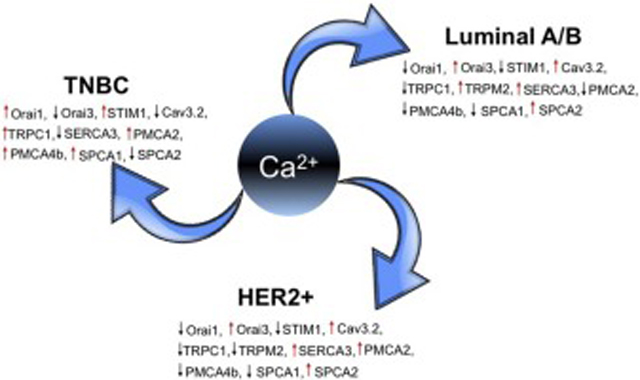
Prolonged elevation in resting Ca2+ levels drives the malignant potential for gene expression, migration, and proliferation to initiate and sustain tumor growth. Conversely, depletion of calcium stores and attenuation of calcium influx pathways underlie tumor chemoresistance and evasion of apoptosis [17, 18]. There have been numerous reports of dysregulated expression, localization or activity of Ca2+ channels, regulators and pumps in breast cancer. An important aspect of these alterations is that they are typically cancer-subtype and isoform-specific (Table 2), as exemplified by reciprocal changes in secretory pathway Ca2+-ATPase isoforms, SPCA1 and SPCA2, observed in receptor positive and negative breast cancers described ahead [19, 20]. Furthermore, dysregulated expression of Ca2+ channels or pumps may be associated with poor prognosis in one breast cancer subtype while being linked to improved patient survival in a different breast cancer subtype [21]. This paradoxical state of Ca2+ homeostasis explains why therapeutic targeting is not straightforward, and may fail or have undesirable effects unless the underlying molecular mechanisms are clearly understood.
Table 2.
Differential Expression of Calcium Channels, Pumps and Regulators in Breast Cancer Subtypes
| Channel/Pump/Regulator | Receptor Positive | Negative | Reference | ||
|---|---|---|---|---|---|
| Luminal A | Luminal B | Her2 | TNBC/basal | ||
| Orai1 | Low | Low | Low | High | [27, 28] |
| Orai3 | High | High | High | Low | [27, 28] |
| STIM1 | Low | Low | Low | High | [30–33] |
| Cav3.2 | High | High | High | Low | [35] |
| TRPC1 | Low | Low | Low | High | [36] |
| TRPM2 | High | Low | [37] | ||
| SERCA3 | High | High | High | Low | [39, 40] |
| PMCA2 | Low | Low | High | High | [45, 46] |
| PMCA4b | Low | Low | Low | High | [40] |
| SPCA1 | Low | Low | Low | High | [53] |
| SPCA2 | High | High | High | Low | [21, 53] |
Among the numerous Ca2+ influx pathways altered in malignancy are members of the transient receptor potential (TRP) family of Ca2+ permeable ion channels, voltage-gated Ca2+ channels, and components of the store operated calcium entry (SOCE) pathway Orai1 and STIM1. Activation of G protein-coupled receptors (GPCRs), leads to generation of inositol triphosphate (IP3) and subsequent stimulation of IP3 receptors (IP3Rs), which results in Ca2+ release from the endoplasmic reticulum stores. Active transport of Ca2+ out of the cytoplasm restores baseline Ca2+ levels and terminates second messenger signaling. The sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), mitochondrial uniporter (MCU), Golgi/secretory pathway Ca2+-ATPase (SPCA) and the H+/Ca2+ antiporter TMEM165 sequester cytosolic Ca2+ into intracellular organelles. Plasma membrane Ca2+-ATPases (PMCA) and Na+/Ca2+ exchanger (NCX) actively extrude Ca2+ from the cytosol into the extracellular space to restore resting calcium [14, 15]. Several recent publications have extensively catalogued the dysregulation in gene expression or activity of Ca2+ channels, pumps and transporters in relation to multiple cancer types [22–26]. Here, we focus on breast cancer with a specific effort to understand how subtype-selective changes in Ca2+ regulation contribute to tumor growth, metastasis and resistance.
Differential Expression of Calcium Channels in Breast Cancer Subtypes
Isoform-specific changes in the expression and function of calcium channels correlate with hormone receptor status and differ significantly between breast cancer subtypes, summarized in Table 2. Of the three isoforms of the store-operated Orai Ca2+ channel family, Orai1 was highly expressed in basal, receptor-negative subtypes whereas Orai3 levels were higher in ER-positive breast cancer cell lines compared to ER-negative cell lines, as observed in cell lines and tumor samples mined from The Cancer Genome Atlas (TCGA) database. Silencing of Orai3 resulted in anti-proliferative effects on ER-positive cell line MCF-7 in vitro and in vivo xenograft models in immune-deficient mice, but no effect was reported on the anchorage-independent growth in TNBC cell line MDA-MB-231 consistent with a subtype specific function of Orai3. Further, silencing of estrogen receptor α (ERα) in MCF-7 cells reduced Orai3 but did not affect Orai1 [27, 28]. These studies establish that the mammalian-specific Orai3 isoform is regulated by ERα upon stimulation with 17β-estradiol in hormone-positive tumors and could be targeted for therapy in ER+ breast cancers. In contrast, hormone receptor negative, or TNBC cell lines utilize the canonical pathway of store operated Ca2+ entry through the Orai1 isoform [29]. Activation of Orai channels by two related Ca2+ sensor proteins, STIM1 and STIM2, occurs in response to depletion of endoplasmic reticulum Ca2+ stores. Overall, TNBC cell lines showed higher STIM1 overexpression compared to ER+ breast cancer cell lines and patients [30, 31]. Further, basal breast cancers with high STIM1/low STIM2 were associated with poorer survival [32]. In summary, SOCE in the ER+ MCF-7 cell line is mediated by STIM1, STIM2, and Orai3, whereas, in the TNBC line MDA-MB-231 SOCE is mediated by STIM1 and Orai1 [33]. Although the mechanistic implications of these differences are currently unclear, emerging evidence suggests that sex hormones could differentially regulate expression of Orai isoforms for sex-specific functional outcomes that relate to cancers of the breast [27] and prostate [34].
Remodeling Ca2+ signaling pathways may be associated with chemoresistance. For example, while patients with HER2+ breast cancers are candidates for treatment with the monoclonal antibody trastuzumab (Herceptin), some patients fail to respond or develop resistance. Monteith and co-workers examined trastuzumab sensitive and resistant HER2+ cell lines for expression of a panel of 44 Ca2+ pumps, channels and regulators and identified higher levels of voltage gated Ca2+ channel Cav3.2 in two of three resistant lines. Luminal A and B subtypes tumors and cell lines also showed the highest expression level of Cav3.2, while the basal subtype expressed significantly lower levels of Cav3.2 compared to the other subtypes [35]. Although no evidence was found for a direct role for Cav3.2 in driving trastuzumab resistance, the authors suggest that elevated levels of this ion channel could serve as a biomarker for overall survival in HER2 positive breast cancer patients undergoing monoclonal antibody therapy.
Ca2+ signaling appears to play a key role in the epithelial to mesenchymal transition, resulting in more invasive and therapy resistant phenotype. Silencing TRPC1 channels in the basal, receptor negative cell line MDA-MB-468 blocked the ability of a certain cell-permeant Ca2+ chelator (EGTA-AM), but not another (BAPTA-AM), to elicit expression of EMT marker vimentin, suggesting distinct pathways of Ca2+ influx that occur during EMT. TRPC1 levels are significantly higher in TNBC subtypes compared to Luminal A, Luminal B, and HER2+ [36]. There are several other examples of Ca2+ channels that are differentially regulated in breast cancer subtypes resulting in dramatically different outcomes. For example, poor patient outcome was associated with high expression of TRPM2 in the Luminal B subtype, whereas, low expression of TRPM2 mRNA correlated with poor outcome in HER2+ breast cancer patients [37]. Therefore, TRPM2 is a potential biomarker for subtype-dependent outcomes and likely has distinct, subtype-specific role in cancer evolution that remains to be elucidated. The prognostic implications of TRP channel isoforms in patient outcomes and as a biomarker of specific breast cancer subtypes have been reviewed [38].
Differential Expression of Calcium Pumps in Breast Cancer Subtypes
Remodeling of Ca2+ pump expression has been observed to accompany neoplastic progression. The non-muscle Ca2+-ATPase of the endoplasmic reticulum, SERCA3, is highly expressed in normal, lobular epithelial cells and is associated with the fully differentiated acinar epithelium. Very early in pre-cancerous lesions, SERCA3 expression is found to be markedly decreased [39]. In invasive carcinomas, SERCA3 is strongly and inversely correlated with tumor grade and is significantly lower in TNBC tumor samples compared to hormone receptor positive tumors [39, 40]. On the other hand, SERCA3 transcript was prominently elevated upon transition of luminal and receptor-positive breast cancer cells from epithelial to the less differentiated and more metastatic mesenchymal state in vitro in response to TGF-β [41]. Papp and colleagues speculate that loss of SERCA3 may lead to a loss of an IP3-mobilizable sub-compartment of the endoplasmic reticulum, which could alter the ability to respond to external stimuli that work through the IP3 second messenger.
The plasma membrane Ca2+-ATPases acts in concert with other Ca2+ pumps to maintain cellular Ca2+ homeostasis. Of the four human isoforms, PMCA2 is reversibly induced in lactating epithelial mammary gland under normal physiological conditions, but is abnormally overexpressed in breast cancer cells [42, 43]. Interestingly, a study of 85 human breast tumors in comparison with 69 adjacent non-tumor tissue revealed associations between specific splice variants of PMCA2 and clinical variables, including receptor status, tumor size, metastasis and stage [44]. Splice variants at the N- and C-termini of PMCA2 are known to alter apical to basal localization of the pump and affinity for the regulatory protein calmodulin suggesting that pump activity is tailored for specific outcomes in cancer cells [44]. High levels of PMCA2 were found to co-express strongly with HER2 and correlate with poor survival prognosis. Wysolmerski and colleagues demonstrated that PMCA2 interacts with HER2 within specific actin-rich plasma membrane domains where pump activity maintains local cytoplasmic Ca2+ concentrations at low levels to block HER2 endocytosis and maintain oncogenic signaling. PMCA2 is part of a multiprotein scaffolding complex that includes NHERF1, Hsp90, as well as HER2, EGFR and HER3, required for surface retention of receptors [45]. Thus, both ATPase dependent Ca2+ pumping activity and scaffolding activity of PMCA2 are important to the tumor cell function. However, a pump-inactive PMCA2 mutant (T692K) capable of normal trafficking increased HER2 levels at the plasma membrane but did not support downstream oncogenic pathways including tumor cell proliferation and inhibition of apoptosis demonstrating that Ca2+ homeostasis is critical in HER2 signaling [45]. Peters et al. reported that basal cancers have highest PMCA2 expression compared to other subtypes, and silencing of PMCA2 in TNBC cell line decreased proliferation and sensitized cells to doxorubicin. These findings show that calcium pumps may play a role in drug resistance to cancer therapy [46].
In contrast to PMCA2, PMCA4b expression is elevated in TNBC lines whereas ER/PR+ cell lines showed a relatively very low expression of this isoform [40]. HDAC inhibitors which have been FDA-approved for treatment of T-cell lymphomas [47] are currently in breast cancer clinical trials [48] and found to increase the expression of PMCA4b in ER/PR+ cell lines. However, this effect was less pronounced in the TNBC cells [40]. These results highlight the fact that chemotherapeutic drugs may also differentially regulate components of the calcium toolkit.
The secretory pathway Ca2+-ATPases (SPCA) sequester Ca2+ and Mn2+ from the cytoplasm into the Golgi and post-Golgi vesicles, where they are critical for protein folding, glycosylation, sorting and quality control. There are two isoforms, SPCA1 and SPCA2, and they share ~65% sequence similarity. Differences include a significantly longer N-terminus and higher affinity for Ca2+ transport in SPCA1. Whereas SPCA1 is the essential housekeeping isoform, ubiquitously expressed in mammalian tissues, SPCA2 expression is confined to highly absorptive and secretory epithelia, which includes mammary, testis, salivary glands, intestinal tract, and lung. During lactation, there is increased demand for calcium transport into the milk. In response, SPCA2 expression is massively induced immediately prior to parturition whereas SPCA1 expression is moderately elevated during the mid-phase of lactation [49]. Further, both SPCA isoforms have been shown to interact with Orai1 channels and elicit store-independent Ca2+ entry (SICE), which has been proposed to mediate efficient basolateral Ca2+ influx into mammary epithelia to support transcytosis of Ca2+ from the blood to milk [50, 51].
Both SPCA isoforms have been implicated in early stages of breast cancer oncogenesis, contributing to the formation of microcalcifications, which are radiographically dense deposits of mineralized calcium in the soft tissue of breast. Microcalcifications are the earliest evidence of malignant Ca2+ dysregulation observed in mammograms, serving as outward manifestations of the underlying molecular changes that drive carcinogenesis. Hydroxyapatite is the main component of malignant microcalcification and implicated to stimulate tumor growth [52]. Both SPCA1 and SPCA2 are prominently elevated in breast cancer subtypes associated with microcalcifications, such as ductal carcinoma in situ. Using an in vitro model of human breast cancer cells, Dang et al. showed that osteogenic conditions that elicit formation of microcalcifications induce expression of SPCA pumps. Formation of hydroxyapatite crystals was enhanced by ectopic expression of SPCA pumps and significantly attenuated by SPCA knockdown [20]. These findings suggest that dysregulated expression of SPCA pumps may initiate mineralization in breast tissue by driving Ca2+ transport into the lumen of the secretory pathway.
Isoform Switching of SPCA Pumps in Breast Cancer Subtypes: Functional Insights
Hierarchical clustering analysis of gene expression in breast tumors revealed a significant difference in the expression of the two SPCA isoforms: whereas SPCA1 clustered with mesenchymal signature genes, SPCA2 was co-expressed with epithelial signature genes [53]. Similarly, analysis of SPCA gene expression in breast cancer cell lines revealed up regulation of SPCA2 and, conversely, down regulation of SPCA1 in epithelial-like breast cancer cell lines. Furthermore, upon growth to confluency, non-tumorigenic mammary cells increased gene expression of SPCA2 and epithelial markers, and reciprocally decreased SPCA1 and mesenchymal gene markers [53]. These observations suggest distinct roles of the secretory pathway Ca2+-ATPases in cell differentiation in both normal and cancerous cells that may reveal new vulnerabilities to target in cancer therapy.
SPCA1 and SPCA2 also have distinct expression profiles that correlate with receptor status and tumor cell origin. Whereas SPCA1 is elevated in tumors derived from basal cells (“basal-like”) [19], SPCA2 is highly expressed in luminal and receptor positive tumor subtypes, including HER2+ breast cancers [20]. There is some evidence that elevated expression of the housekeeping isoform SPCA1 is relevant to basal subtypes of breast tumors and hormone receptor-negative, basal-like breast cancer cell lines, MDA-MB-231 and Hs578T [19, 53]. Silencing of SPCA1 in MDA-MB-231 reduced tumor cell proliferation although cell viability was not affected [19]. The expression of IGF1R (insulin-like growth factor 1 receptor) has been shown to correlate with poor prognosis in breast cancer, and knockdown of SPCA1 halted the proteolytic processing of the pro-IGF1R protein in MDA-MB-231 cells [19]. As a result, levels of the functional IGF1Rβ were reduced by ~80% with corresponding increase in the TGN-associated unprocessed intermediate, likely due to the depletion of Golgi Ca2+ that is required for activation of the proprotein convertase furin. Small, but significant changes in cytoplasmic Ca2+ signaling were also observed in response to protease activated receptor (PAR) signaling in basal-like MDA-MB-231 cells upon silencing of SPCA1. More studies are warranted to replicate these findings in a wider array of breast cancer cell lines. It will also be of interest to determine if SPCA1 regulates processing and expression of other key oncogenic receptors in basal cancers and modulates their downstream signaling pathways. These findings may explain why high SPCA1 levels are associated with poor patient survival in the combined aggregate of breast cancer patients. However, this difference was not statistically significance upon stratifying tumors based on receptor type, including luminal, HER2+ and TNBC.
Feng et al., reported that SPCA2 transcript was highly up regulated in cell lines derived from luminal breast cancers, compared to nonmalignant mammary epithelial cells. SPCA2 plays a prominent role in promoting tumor growth in receptor positive and luminal breast cancers through activation of the RAS-RAF-MEK-ERK axis, a well-known driver of cell proliferation, differentiation, growth, and apoptosis. Thus, knockdown of SPCA2 in the MCF-7 cell line decreased cell proliferation and anchorage-independent colony formation in soft agar, concomitantly decreasing phosphorylation of ERK and levels of the cell cycle regulator cyclin D1. The mechanistic basis for these observations was revealed by analysis of cytoplasmic calcium levels: unexpectedly, SPCA2 was shown to elicit constitutive, store-independent Ca2+ entry in ER+/PR+ MCF7 cells by interacting with, and activating the Orai1 Ca2+ channel. The elevation in basal Ca2+ levels was sufficient to activate the Ca2+/calmodulin-responsive phosphatase calcineurin, resulting in dephosphorylation and nuclear translocation of the transcription factor NFAT. Furthermore, the RAS/ERK pathway is activated by Ca2+ signaling to drive cell cycle progression and cell proliferation [54]. Curiously, Orai1 activation by SPCA2 does not require active Ca2+ pumping activity, and appears to have evolved as an independent, moonlighting function encoded within the SPCA N- and C-termini. In this context, an alternative transcription start site in the SPCA2 gene has been discovered that encodes a membrane-embedded C-terminal fragment of SPCA2 lacking ATPase-depending pumping activity [55]. This fragment is capable activating Ca2+ influx although it has not been investigated in breast cancer cells [56]. These results could explain how inappropriate expression of SPCA2 or its splice variants in non-lactation conditions drives Ca2+-mediated oncogenesis in receptor positive breast cancers. The translational significance of these findings arise from in vivo observation that tumor incidence in mice implanted with MCF7 breast cancer cells was significantly attenuated by SPCA2 knockdown. Consistent with these experimental findings, high SPCA2 expression was associated with poor survival prognosis not only for the aggregate of all breast cancer subtypes, but also specifically for luminal and HER2+ positive breast cancers.
Highlighting the subtype-specific roles of Ca2+ pumps, low SPCA2 expression is characteristic of triple receptor negative breast cancers [53], where it is associated with poor patient survival prognosis. These observations are explained by the recent finding that loss of SPCA2 expression closely parallels loss of the Ca2+ binding cell adhesion protein E-cadherin, and reciprocal increase of N-cadherin and other mesenchymal genes. These changes are characteristic of epithelial-mesenchymal transition, a key step in cancer metastasis. Mesenchymal cells are multipotent stromal cells that can differentiate into a variety of cell types. The characteristic features of mesenchymal cells include enhanced migratory effect, invasiveness, elevated resistance to apoptosis and greatly increased production of ECM components. The most critical epithelial marker that is lost during EMT is the Ca2+ binding cell adhesion protein E-cadherin [57]. SPCA2 is strongly co-expressed with E-cadherin, pointing to an EMT-related functional link in breast cancer cells. Indeed, SPCA2 knockdown was demonstrated to cause a 50% post-translational reduction in total and cell surface expression of E-cadherin.
E-cadherin mediates key signaling pathways including the Hippo-YAP pathway, which is involved in regulation of organ size by regulating cell proliferation, apoptosis, and stem cell self-renewal. E-cadherin maintains Hippo signaling by phosphorylation, inactivation, and nuclear exclusion of YAP (Yes-activated protein) and its paralog, TAZ (transcriptional activator with PDZ binding motif) [58]. Dang et al. showed that restoring SPCA2 expression in TNBC cell lines MDA-MB-231 and HS578T significantly increased E-cadherin levels, elevated resting Ca2+ levels, and SICE [21, 53]. Concomitantly, YAP phosphorylation increased, and transcription of mesenchymal markers (N-cadherin, SNAI1, SNAI2/SLUG, Vimentin, and ZEB1) was suppressed. Consistent with the shift away from mesenchymal phenotypes, restoring SPCA2 in TNBC cell lines also decreased wound-closure and cell migration in vitro. In vivo, ectopic expression of SPCA2 in a bioluminescent model MDA-MB-231 Luc injected in the mammary fat pad of NSG mice, decreased lung metastasis [21, 53]. Cancer stem cells (CSCs) are rare immortal cells within a tumor that can both self-renew by dividing and giving rise to many cell types that constitute the tumor. CSCs also have been shown to be involved in fundamental processes of cell proliferation and metastatic dissemination. CSCs are generally resistant to chemotherapy and radiotherapy [59]. Ectopic expression of SPCA2 in TNBC cell lines reduced CSC markers OCT4, NANOG, and SOX2 [53].
In summary, subtype-specific expression and SPCA isoform switching has functional significance for breast cancer progression and patient outcomes. In our current model (Figure 3), constitutively high expression of SPCA2 in luminal and HER2+ breast cancers breast cancer cell lines and tumors inappropriately activates Ca2+ influx through Orai1 and potentially other Ca2+ influx channels to drive MAP kinase signaling, cell proliferation, anchorage-independent growth and tumor formation as evidenced in a murine xenograft model. It is worth noting that SPCA2 is significantly more effective in activating Orai channels compared to the SPCA1 isoform [50, 51]. As a consequence, high SPCA2 expression negatively correlates with patient survival outcomes in luminal breast cancers. Conversely, low SPCA2 levels were associated with poor prognosis in TNBC patients. Restoring SPCA2-mediated calcium signaling, elevated cytosolic Ca2+ levels, restored E-cadherin expression, reversed epithelial-mesenchymal transition, reduced cancer stem cell markers, decreased migration in vitro, and metastasis in vivo.
Figure 3:

Isoform switching of SPCA pumps accompanies reversible epithelial to mesenchymal transitions in breast cancer cells. In the epithelial state, cells are attached to one another and to the basement membrane. SPCA2 elevates cytoplasmic Ca2+ levels (red) and is required for E-cadherin biogenesis, which maintains cell adhesion. Loss of SPCA2 results in corresponding loss of surface E-cadherin expression, promoting tumor cell detachment, migration and invasion. Disruption of Hippo-YAP signaling downstream from E-cadherin results in expression of mesenchymal genes such as Zeb1 and vimentin. SPCA2 is replaced by SPCA1 in mesenchymal state, which maintains lower cytoplasmic resting Ca2+ levels (blue).
Conclusions
Treatment of breast cancer has improved in recent decades, yet many patients succumb to this disease. There is an urgent need to enhance patient survival by identifying new biomarker-based drug targets and elucidating mechanisms of resistance against current chemotherapeutic agents. Although breast cancer subtype-specific dysregulation of key components of the Ca2+ toolkit has been extensively reported (Table 2), therapeutic targeting is not straightforward. Since both elevation and depletion of Ca2+ levels can drive malignant phenotypes, the specific molecular mechanisms driving these changes need to be clearly understood. This is exemplified by the opposite survival prognosis of alterations in SPCA2 levels depending on receptor status. Thus, targeting of SPCA2 in breast cancer needs to subtype specific. Inhibitors that decrease SPCA2-driven Ca2+ signaling in luminal and HER2+ breast cancers and enhancers that reactivate deficient SPCA2-mediated calcium signaling in basal/TNBC could be effective therapeutic tools. Subtype-specific targeting of calcium signaling could be another arrow in the armory of precision medicine approaches in the treatment of cancer.
Highlights:
Prolonged elevation or depletion of intracellular calcium is oncogenic
Dysregulated expression of the Ca2+ toolkit varies with breast cancer subtype
Isoform switching of SPCA pumps occurs during epithelial mesenchymal transition
SPCA2 confers opposite survival prognosis depending on hormone receptor status
Therapeutic targeting of Ca2+ pumps and channels must be tailored to cancer subtype
Funding Source:
The authors are supported, in part, by a grant from the NIH (R01 DK108304)
Abbreviations:
- ER
estrogen receptor
- PR
progesterone receptor
- HER2
Human epidermal growth factor receptor 2
- TNBC
Triple Negative Breast Cancer
- TRP
Transient receptor potential
- SOCE
Store Operated Calcium Entry
- SICE
Store Independent Calcium Entry
- SERCA
Sarco/endoplasmic reticulum Ca2 +ATPase
- MCU
Mitochondrial uniporter
- SPCA
Secretory pathway Calcium ATPase
- EMT
Epithelial-Mesenchymal Transition
- MET
Mesenchymal-Epithelial Transition
- YAP
Yes-Associated protein
- TAZ
Transcriptional co-activator with PDZ-binding motif
- CSC
Cancer stem cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures of Potential Conflicts of Interest: none
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019, CA: a cancer journal for clinicians, 69 (2019) 7–34. [DOI] [PubMed] [Google Scholar]
- [2].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA: a cancer journal for clinicians, 68 (2018) 394–424. [DOI] [PubMed] [Google Scholar]
- [3].Bodai BI, Tuso P, Breast cancer survivorship: a comprehensive review of long-term medical issues and lifestyle recommendations, The Permanente Journal, 19 (2015) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sharma GN, Dave R, Sanadya J, Sharma P, Sharma K, Various types and management of breast cancer: an overview, Journal of advanced pharmaceutical technology & research, 1 (2010) 109. [PMC free article] [PubMed] [Google Scholar]
- [5].Alkabban FM, Ferguson T, Cancer, Breast, StatPearls [Internet], StatPearls Publishing, Place Published, 2018. [Google Scholar]
- [6].Mariotto AB, Etzioni R, Hurlbert M, Penberthy L, Mayer M, Estimation of the number of women living with metastatic breast cancer in the United States, Cancer Epidemiology and Prevention Biomarkers, 26 (2017) 809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lim E, Metzger-Filho O, Winer EP, The natural history of hormone receptor–positive breast cancer, Oncology, 26 (2012). [PubMed] [Google Scholar]
- [8].Sareyeldin RM, Gupta I, Al-Hashimi I, Al-Thawadi HA, Al Farsi HF, Vranic S, Moustafa A, Gene Expression and miRNAs Profiling: Function and Regulation in Human Epidermal Growth Factor Receptor 2 (HER2)-Positive Breast Cancer, Cancers, 11 (2019) 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yao H, He G, Yan S, Chen C, Song L, Rosol TJ, Deng X, Triple-negative breast cancer: is there a treatment on the horizon?, Oncotarget, 8 (2017) 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dai X, Li T, Bai Z, Yang Y, Liu X, Zhan J, Shi B, Breast cancer intrinsic subtype classification, clinical use and future trends, American journal of cancer research, 5 (2015) 2929. [PMC free article] [PubMed] [Google Scholar]
- [11].Waks AG, Winer EP, Breast cancer treatment: a review, Jama, 321 (2019) 288–300. [DOI] [PubMed] [Google Scholar]
- [12].Del Valle HB, Yaktine AL, Taylor CL, Ross AC, Dietary reference intakes for calcium and vitamin D, National Academies Press, Place Published, 2011. [PubMed] [Google Scholar]
- [13].Cross BM, Breitwieser GE, Reinhardt TA, Rao R, Cellular calcium dynamics in lactation and breast cancer: from physiology to pathology, Am J Physiol Cell Physiol, 306 (2014) C515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Azimi I, Roberts-Thomson S, Monteith G, Calcium influx pathways in breast cancer: opportunities for pharmacological intervention, British journal of pharmacology, 171 (2014) 945–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Giorgi C, Marchi S, Pinton P, The machineries, regulation and cellular functions of mitochondrial calcium, Nature reviews Molecular cell biology, 19 (2018) 713–730. [DOI] [PubMed] [Google Scholar]
- [16].Clapham DE, Calcium signaling, Cell, 131 (2007) 1047–1058. [DOI] [PubMed] [Google Scholar]
- [17].Roderick HL, Cook SJ, Ca 2+ signalling checkpoints in cancer: remodelling Ca 2+ for cancer cell proliferation and survival, Nature Reviews Cancer, 8 (2008) 361. [DOI] [PubMed] [Google Scholar]
- [18].Marchi S, Pinton P, Alterations of calcium homeostasis in cancer cells, Current opinion in pharmacology, 29 (2016) 1–6. [DOI] [PubMed] [Google Scholar]
- [19].Grice DM, Vetter I, Faddy HM, Kenny PA, Roberts-Thomson SJ, Monteith GR, Golgi calcium pump secretory pathway calcium ATPase 1 (SPCA1) is a key regulator of insulin-like growth factor receptor (IGF1R) processing in the basal-like breast cancer cell line MDA-MB-231, Journal of Biological Chemistry, 285 (2010) 37458–37466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dang D, Prasad H, Rao R, Secretory pathway Ca2+-ATPases promote in vitro microcalcifications in breast cancer cells, Molecular carcinogenesis, 56 (2017) 2474–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Makena MR, Dang DK, Ko M, Bandral M, Rao R, Secretory pathway calcium ATPase-2 (SPCA2) regulates metastasis by suppressing mesenchymal markers in triple negative breast cancer cell lines, AACR, 2019. [Google Scholar]
- [22].Stewart TA, Yapa KT, Monteith GR, Altered calcium signaling in cancer cells, Biochimica et Biophysica Acta (BBA)-Biomembranes, 1848 (2015) 2502–2511. [DOI] [PubMed] [Google Scholar]
- [23].Cui C, Merritt R, Fu L, Pan Z, Targeting calcium signaling in cancer therapy, Acta pharmaceutica sinica B, 7 (2017) 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Maklad A, Sharma A, Azimi I, Calcium signaling in brain cancers: roles and therapeutic targeting, Cancers, 11 (2019) 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bates E, Ion channels in development and cancer, Annual review of cell and developmental biology, 31 (2015) 231–247. [DOI] [PubMed] [Google Scholar]
- [26].Monteith GR, Davis FM, Roberts-Thomson SJ, Calcium channels and pumps in cancer: changes and consequences, Journal of Biological Chemistry, 287 (2012) 31666–31673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, Trebak M, Orai3 is an estrogen receptor α-regulated Ca2+ channel that promotes tumorigenesis, The FASEB Journal, 27 (2013) 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Azimi I, Milevskiy MJ, Chalmers SB, Yapa KT, Robitaille M, Henry C, Baillie GJ, Thompson EW, Roberts-Thomson SJ, Monteith GR, ORAI1 and ORAI3 in breast cancer molecular subtypes and the identification of ORAI3 as a hypoxia sensitive gene and a regulator of hypoxia responses, Cancers, 11 (2019) 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yang S, Zhang JJ, Huang XY, Orai1 and STIM1 are critical for breast tumor cell migration and metastasis, Cancer Cell, 15 (2009) 124–134. [DOI] [PubMed] [Google Scholar]
- [30].Jardin I, Lopez J, Salido G, Rosado J, Store-operated Ca2+ entry in breast Cancer cells: Remodeling and functional role, International journal of molecular sciences, 19 (2018) 4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang Y, Jiang Z, Wang B, Chang L, Liu J, Zhang L, Gu L, Expression of STIM1 is associated with tumor aggressiveness and poor prognosis in breast cancer, Pathology-Research and Practice, 213 (2017) 1043–1047. [DOI] [PubMed] [Google Scholar]
- [32].McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, Smart CE, Brown MA, Kenny PA, Roberts-Thomson SJ, ORAI1-mediated calcium influx in lactation and in breast cancer, Molecular cancer therapeutics, 10 (2011) 448–460. [DOI] [PubMed] [Google Scholar]
- [33].Motiani RK, Abdullaev IF, Trebak M, A Novel Native Store-operated Calcium Channel Encoded by Orai3 selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells, Journal of Biological Chemistry, 285 (2010) 19173–19183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Flourakis M, Lehen’kyi V, Beck B, Raphael M, Vandenberghe M, Abeele FV, Roudbaraki M, Lepage G, Mauroy B, Romanin C, Shuba Y, Skryma R, Prevarskaya N, Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells, Cell Death Dis, 1 (2010) e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pera E, Kaemmerer E, Milevskiy MJ, Yapa KT, O’Donnell JS, Brown MA, Simpson F, Peters AA, Roberts-Thomson SJ, Monteith GR, The voltage gated Ca 2+-channel Ca v 3.2 and therapeutic responses in breast cancer, Cancer cell international, 16 (2016) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Azimi I, Milevskiy MJ, Kaemmerer E, Turner D, Yapa KT, Brown MA, Thompson EW, Roberts-Thomson SJ, Monteith GR, TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells, J Cell Sci, 130 (2017) 2292–2305. [DOI] [PubMed] [Google Scholar]
- [37].Sumoza-Toledo A, Espinoza-Gabriel MI, Montiel-Condado D, Evaluation of the TRPM2 channel as a biomarker in breast cancer using public databases analysis, Boletín Médico Del Hospital Infantil de México (English Edition), 73 (2016) 397–404. [DOI] [PubMed] [Google Scholar]
- [38].Ouadid-Ahidouch H, Dhennin-Duthille I, Gautier M, Sevestre H, Ahidouch A, TRP channels: diagnostic markers and therapeutic targets for breast cancer?, Trends Mol Med, 19 (2013) 117–124. [DOI] [PubMed] [Google Scholar]
- [39].Papp B, Brouland J-P, Altered endoplasmic reticulum calcium pump expression during breast tumorigenesis, Breast cancer: basic and clinical research, 5 (2011) BCBCR. S7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Varga K, Hollósi A, Pászty K, Hegedűs L, Szakács G, Tímár J, Papp B, Enyedi Á, Padányi R, Expression of calcium pumps is differentially regulated by histone deacetylase inhibitors and estrogen receptor alpha in breast cancer cells, BMC cancer, 18 (2018) 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mahdi SH, Cheng H, Li J, Feng R, The effect of TGF-beta-induced epithelial-mesenchymal transition on the expression of intracellular calcium-handling proteins in T47D and MCF-7 human breast cancer cells, Arch Biochem Biophys, 583 (2015) 18–26. [DOI] [PubMed] [Google Scholar]
- [42].Reinhardt TA, Lippolis JD, Mammary gland involution is associated with rapid down regulation of major mammary Ca2+-ATPases, Biochem Biophys Res Commun, 378 (2009) 99–102. [DOI] [PubMed] [Google Scholar]
- [43].Lee WJ, Roberts-Thomson SJ, Monteith GR, Plasma membrane calcium-ATPase 2 and 4 in human breast cancer cell lines, Biochem Biophys Res Commun, 337 (2005) 779–783. [DOI] [PubMed] [Google Scholar]
- [44].Romero- Lorca A, Gaibar M, Armesilla AL, Fernandez- Santander A, Novillo A, Differential expression of PMCA2 mRNA isoforms in a cohort of Spanish patients with breast tumor types, Oncology letters, 16 (2018) 6950–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jeong J, VanHouten JN, Dann P, Kim W, Sullivan C, Yu H, Liotta L, Espina V, Stern DF, Friedman PA, PMCA2 regulates HER2 protein kinase localization and signaling and promotes HER2-mediated breast cancer, Proceedings of the National Academy of Sciences, 113 (2016) E282–E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Peters AA, Milevskiy MJ, Lee WC, Curry MC, Smart CE, Saunus JM, Reid L, Da Silva L, Marcial DL, Dray E, The calcium pump plasma membrane Ca 2+-ATPase 2 (PMCA2) regulates breast cancer cell proliferation and sensitivity to doxorubicin, Scientific reports, 6 (2016) 25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gatla HR, Muniraj N, Thevkar P, Yavvari S, Sukhavasi S, Makena MR, Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases, International journal of molecular sciences, 20 (2019) 1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Suraweera A, O’Byrne KJ, Richard DJ, Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi, Frontiers in oncology, 8 (2018) 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cross BM, Hack A, Reinhardt TA, Rao R, SPCA2 regulates Orai1 trafficking and store independent Ca2+ entry in a model of lactation, PloS one, 8 (2013) e67348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, Muend S, Kenny PA, Sukumar S, Roberts-Thomson SJ, Store-independent activation of Orai1 by SPCA2 in mammary tumors, Cell, 143 (2010) 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Smaardijk S, Chen J, Kerselaers S, Voets T, Eggermont J, Vangheluwe P, Store-independent coupling between the Secretory Pathway Ca(2+) transport ATPase SPCA1 and Orai1 in Golgi stress and Hailey-Hailey disease, Biochim Biophys Acta Mol Cell Res, 1865 (2018) 855–862. [DOI] [PubMed] [Google Scholar]
- [52].Morgan MP, Cooke MM, Christopherson PA, Westfall PR, McCarthy GM, Calcium hydroxyapatite promotes mitogenesis and matrix metalloproteinase expression in human breast cancer cell lines, Mol Carcinog, 32 (2001) 111–117. [DOI] [PubMed] [Google Scholar]
- [53].Dang DK, Makena MR, Llongueras JP, Prasad H, Ko M, Bandral M, Rao R, A Ca2+-ATPase Regulates E-cadherin Biogenesis and Epithelial–Mesenchymal Transition in Breast Cancer Cells, Molecular Cancer Research, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cook SJ, Lockyer PJ, Recent advances in Ca(2+)-dependent Ras regulation and cell proliferation, Cell Calcium, 39 (2006) 101–112. [DOI] [PubMed] [Google Scholar]
- [55].Garside VC, Kowalik AS, Johnson CL, DiRenzo D, Konieczny SF, Pin CL, MIST1 regulates the pancreatic acinar cell expression of Atp2c2, the gene encoding secretory pathway calcium ATPase 2, Exp Cell Res, 316 (2010) 2859–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fenech MA, Sullivan CM, Ferreira LT, Mehmood R, MacDonald WA, Stathopulos PB, Pin CL, Atp2c2 Is Transcribed From a Unique Transcriptional Start Site in Mouse Pancreatic Acinar Cells, J Cell Physiol, 231 (2016) 2768–2778. [DOI] [PubMed] [Google Scholar]
- [57].Brabletz T, Kalluri R, Nieto MA, Weinberg RA, EMT in cancer, Nature Reviews Cancer, 18 (2018) 128. [DOI] [PubMed] [Google Scholar]
- [58].Kim N-G, Koh E, Chen X, Gumbiner BM, E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components, Proceedings of the National Academy of Sciences, 108 (2011) 11930–11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Makena MR, Ranjan A, Thirumala V, Reddy A, Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance, Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, (2018). [DOI] [PubMed] [Google Scholar]