

Abstract
In neuronopathic Hunter syndrome, neurobehavioral symptoms are known to be serious but have been incompletely described. While families face significant stress stemming from this complex and far-reaching array of symptoms, neither caregiver burden nor the neurobehavioral symptoms have been measured comprehensively. We delineated these neurobehavioral characteristics and their impact on the caregiver using multiple approaches. Methods: As part of the initial phase of developing a Hunter-specific behavioral assessment tool, we used multiple methods to obtain data on patient behaviors and caregiver burden, with the intention of drafting item sets for the tool. We utilized 1) caregiver descriptions from focus groups and individual interviews, 2) observations from video-recorded play of affected children, 3) descriptions from historic chart review, 4) consultation with patient advocacy groups and international experts, 5) reports from a caregiver advisory board, and 6) literature review. Results: Neurobehavioral symptoms were diverse and categorized as focus/attention, impulsivity/heightened activity, sensation seeking, emotional/behavioral function, social interaction, and sleep. A significant reported challenge was susceptibility to misinterpretation of some behaviors as defiant or aggressive, particularly if physical. Caregiver burden involved social isolation, exhaustion, stress, and financial and vocational strain. These new descriptions will aid in developing quantitative measures of change in neurobehavioral symptoms and family burden. These descriptions will be the foundation of a neurobehavioral rating scale, which is very much needed to aid in patient management and assess interventions for individuals with neuronopathic Hunter syndrome.
Keywords: Mucopolysaccharidosis II, Hunter syndrome, Neurobehavioral symptoms, Behavioral decline, Neuronopathic phenotype
1. Introduction
We investigated the neurobehavioral symptoms specific to the neuronopathic phenotype of Hunter syndrome (mucopolysaccharidosis type II, MPS II, OMIM 309900), as well as how these symptoms affect the caregiver and family. MPS II is a rare X-linked lysosomal disorder that progressively impacts nearly every organ system, resulting in serious disability and shortened lifespan [[1], [2], [3]]. The deficiency of the lysosomal enzyme iduronate-2-sulfatase leads to accumulation of glycosaminoglycans, which results in a progressive multi-system disorder affecting both somatic and the central nervous system (CNS) [1,4,5]. Affected children often appear normal at birth and age that clinical signs emerge is variable [[6], [7], [8], [9]]. Individuals with MPS II can have a spectrum of clinical involvement from neuronopathic (severe) to nonneuronopathic (attenuated) [3,8,[10], [11], [12], [13], [14], [15]]. The CNS signs of neuronopathic individuals can typically be detected by 4 years of age, but can be as young as 1 year of age.
The neuronopathic form of MPS II involves neurocognitive decline, intense neurobehavioral symptoms, and early death often in the second decade of life [[1], [2], [3],7,8,10,12,[14], [15], [16], [17], [18]]. Clinical heterogeneity of MPS II is evident in individuals who show significant neurocognitive impairment without decline, requiring significant educational supports [10]. Neurobehavioral symptoms are generally evident by age 2–4 and may worsen in intensity or frequency with age, but subside as cognitive regression occurs [3,8,19]. Nonneuronopathic individuals typically have normal intelligence but can have deficits in attention, with variable problems with executive function or visual-motor skills [13,20]. Regardless of neurologic involvement, somatic disease is present and can be severe [3,10,13,21].
The neurobehavioral symptoms of neuronopathic MPS II are disruptive and distressing to families, have a significant impact on the quality of life for the child and their family [8,[22], [23], [24]], and may limit the ability to engage consistently in supportive therapies such as occupational therapy or speech/language therapy. These symptoms have been summarized in the literature as hyperactivity, behavior difficulties (e.g., destructiveness, aggressiveness, defiance), poor emotional regulation (temper tantrums, excitable, anxious), sleep disturbance, and perseverative chewing behavior [8,12,18,19,25,26]. While informative, these descriptions are nonspecific, as our collective clinical and collaborative experiences indicate the symptoms are more complex, diverse, and nuanced than these broad terms reflect. Further detailing how the symptoms present, and how they affect caregivers, is an important area for focus, as many families express that neurobehavioral abnormalities from neuronopathic MPS disorders are the most challenging aspect to manage among the complicated array of disease manifestations [8,27]. A major obstacle to understanding these symptoms is the inadequacy of current broad-based pediatric behavioral assessment tools, whose items do not account for the unique constellation of symptoms, nor deficits, that can be associated with MPS II [25]. However, a Hunter-specific tool would be crucial for clarifying the timing and sequence of behavioral decline, and ultimately for measuring response to therapies. The present study reports data obtained from the early phase of item development for a Hunter-specific behavioral rating scale. As part of this early phase we used a multi-faceted methodology to characterize the body of neurobehavioral symptoms that may occur in neuronopathic MPS II and to describe the impact of these symptoms on the caregiver.
2. Materials and methods
2.1. Design
A multi method approach was utilized to investigate the neurobehavioral symptoms and caregiver impact (Table 1). Data were obtained via caregiver focus groups, video recordings of affected boys in an unstructured play setting, individual caregiver interviews either in person or by phone, medical record review, a meeting of representatives from patient advocacy groups and international experts, individual expert consultation, and literature review [28]. The focus group approach was selected for obtaining caregiver data because it has been used to understand the subjective experience of individuals and families in the MPS community, most recently for understanding quality of life, caregiver burden, and patient-reported outcomes [[29], [30], [31]]. Literature review of PubMed and Google Scholar involved the search terms mucopolysaccharidosis, “Hunter syndrome,” behavior, and “cognitive decline.” Experts were selected for consultation based on publications and/or international presentations on Hunter syndrome, or a history of advisory board work on Hunter syndrome. Data were also informed with our collective clinical experience. In addition to these methods, data were shared and incorporated from a caregiver advisory board meeting that focused on the CNS aspects of MPS II.
Table 1.
Assessment methods.
| Assessment method | Events (N) | Participants per event |
|---|---|---|
| Neurobehavioral symptoms | ||
| Caregiver focus groups | 3 | Event 1 N = 3 |
| Event 2 N = 2 | ||
| Event 3 N = 4 | ||
| Videos of affected children | 2 | Event 1 N = 3 |
| Event 2 N = 2* | ||
| Individual caregiver interviews | 3 | 1 |
| Patient advocacy group meeting | 1 | 3 patient advocacy groups |
| Group 1 N = 3 | ||
| Group 2 N = 1 | ||
| Group 3 N = 1 | ||
| Expert consultation | 5 | 1 |
| Medical chart review | N/A | 12 |
| Parent advisory board | 1 | N = 7 |
| Family Impact | ||
| Caregiver focus groups | 3 | Event 1 N = 3 |
| Event 2 N = 2 | ||
| Event 3 N = 4 | ||
| Individual caregiver interviews | 3 | 1 |
| Patient advocacy group meeting | 1 | 3 patient advocacy groups |
| Group 1 N = 3 | ||
| Group 2 N = 1 | ||
| Group 3 N = 1 | ||
| Expert consultation | 5 | 1 |
| Parent advisory board | 1 | N = 7 |
2.2. Participants
None of the children in any of the focus groups, advisory board, or chart review had a history of hematopoietic stem cell transplantation. All families were from the United States.
Participants for the caregiver focus groups and video recorded play were recruited through University of Minnesota IRB-approved study announcements that were distributed by the National MPS Society and Hunter syndrome patient advocacy groups to affected families, who then had the option to contact the investigators to participate. Inclusion criteria for participants in the caregiver focus groups and video recorded play included 1) confirmed diagnosis of neuronopathic MPS II by caregiver report; 2) age 4 to 9 years; and 3) ability to ambulate. This age range was selected to increase the likelihood that neurobehavioral symptoms would be actively expressed. In these groups of unrelated males, 2 of the 4 children were receiving intravenous (IV) enzyme replacement therapy (ERT) and experimental intrathecal (IT) ERT; the other two were receiving only IV ERT. Five caregivers participated.
Chart review included all available paper records on all children with untreated MPS II who underwent neuropsychological assessment as part of an evaluation for eligibility for hematopoietic stem cell transplantation (i.e., prior to the approval of enzyme replacement therapy). The behavior observations within the neuropsychological evaluation reports were used. The neurocognitive data on these 12 boys, ages 1 year 10 months to 4 years 10 months, have been previously reported [32].
The caregiver advisory board involved seven parents (two were married to each other) of seven children (two were brothers); the children were all males and ranged in age from 4 to 12 years. Within that group, parents reported that 4 were enrolled in the IT ERT clinical trial and all were receiving weekly IV ERT.
3. Results
Neurobehavioral symptoms were categorized into 6 domains, while caregiver and family impact could be categorized into 5 domains based on the data collected (Table 2).
Table 2.
Domain for neurobehavioral symptoms and impact.
| Neurobehavioral symptoms | Caregiver/family impact |
|---|---|
| Focus/attention | Family social isolation |
| Impulsivity/heightened activity | Psychological stress |
| Sensory seeking | Exhaustion |
| Emotional/behavioral function | Concern/focus on development |
| Social function | Financial and vocational strain |
| Sleep |
3.1. Neurobehavioral symptoms
Symptom expression and intensity were found to vary day-to-day, or even hour to hour. In the focus groups, caregivers reported “good days” and more challenging days. Sources of variation were not always obvious, although poor sleep, medical complications, major schedule changes, or novel circumstances were considered potential factors. A significant challenge was susceptibility to misinterpretation of some behaviors as aggressive or defiant, when these attributions applied for only a proportion of circumstances.
Literature search catalogued neurobehavioral symptoms including hyperactivity/overactivity, restlessness, impulsivity, aggression, destructiveness, temper tantrums, frustration, behavior issues/problems, obstinacy, playful exuberance, sleeplessness, seizure-like behavior and perseverative chewing [8,12,14,15,18,19]. These existing descriptions all matched findings from this study; no reports in the literature were in clear conflict with the present behavioral data obtained. However, findings from caregiver focus groups and interviews, as well as consultation with experts, yielded a larger assortment of symptoms, of which a subset were observed in videos and described in medical chart review.
3.1.1. Attention/focus
Short attention spans and high distractibility are pervasive and can limit engagement in activities. Shift in focus can be swift and lead to abrupt changes between tasks or objects. At times, poor attention can interfere with the children's ability even to participate in things they are interested in. Some caregivers described prolonged staring even when seizures have been ruled-out. Conversely, caregivers reported concerns about excessive focus on certain concepts, ideas, or visual electronic media. The children may become “stuck” on particular topics, items or phrases. Difficulty “pulling away” from excessive focus may appear as noncompliance with requests or instruction.
3.1.2. Impulsivity and heightened activity
Impulsivity and activity level are extremely elevated as compared with age expectation. Constant moving or pacing are seen, as are climbing inappropriately, jumping off of unsafe surfaces, and grabbing or breaking things. Hyperactive/impulsive behavior can also be highly physical, manifesting as running into, hitting, throwing, or knocking down objects or other people. These behaviors are frequently mistaken by others to be intentionally aggressive (Fig. 1).
Fig. 1.

Potential for error in appraisal of neurobehavioral symptoms.
Fig. 1. A subset of neurobehavioral symptoms often reported in neuronopathic MPS II (left) may commonly be interpreted as aggressive due to the high level of physical involvement of the behaviors and the potential for injury. However, findings from this study indicate that these behaviors may be displayed in a variety of non-aggressive circumstances (right), such as when seeking sensory stimulation or attempting to engage socially. This illustration is one example of potential for error in appraisal of behaviors; this misattribution was also reported for other appraisal categories (e.g., noncompliance) across symptoms.
A symptom obtained across data sampling methods, which was often ascribed to hyperactivity/impulsivity, was a tendency to run abruptly without warning and without a clear destination, as though the children are eloping. This “fleeing behavior” can occur without heeding physical cues of the environment. That is, running may, for some, be expressed regardless of sidewalk, park boundary, street, or body of water. Another neurobehavioral symptom ascribed to hyperactivity was a fair amount of repetitive motion, such as opening and closing doors or hinged objects, or moving their bodies or extremities in repetitive ways.
3.1.3. Sensory seeking
Significant sensory input is sought in many forms. It was often reported to be mistaken for aggression or hyperactivity. For physical sensory input, the children may run-crash into objects or people. Of challenge in some of the children is what is appraised by caregivers as a high pain tolerance, which does not help deter physical sensory seeking behavior. Oral fixations and chewing were seen across sampling methods. Biting was reported to be a serious concern, with reports of non-aggressive biting that is oral sensory seeking or communication attempts. Auditory and visual sensory seeking was displayed as preference for loud noises, busy spaces and bright lights. Banging on objects loudly, yelling, slamming, and flicking lights were described.
3.1.4. Emotional/behavioral function
Emotional and behavioral responses tend to suggest abnormality of reactivity. Caregivers reported that their children are frequently labeled as “autistic” due to their unusual and often unmanageable emotional and behavioral expressions. Sometimes emotional reactions are stronger than the circumstances call for, but other times emotional response appears far too mild, or absent. In addition, there are times when emotions are incongruent with the situation (e.g., inappropriate laughter when someone gets hurt).
There may be sudden outbursts of emotion, without clear trigger. These outbursts may reflect frustration, anger, happiness, sadness, or others. Tantrums may occur without identifiable cause; other times, the source is clear. In general, tantrums, high frustration, and anger outbursts were described as significantly problematic and difficult to manage or soothe.
Other reports consistent with abnormality of emotional reactivity included poor cooperation, defiance, and oppositional behavior. Also consistent with this reactivity, aggressive behaviors were described, in the forms of hitting, pinching, biting, pushing, or throwing.
3.1.5. Social
Social skills may be lacking, as is awareness of social cues. Eye contact is difficult to establish and even harder to maintain. Still the children are social and interested in others, but cannot consistently connect in ways that meet general social expectations. They invade others' space and may purposefully collide with others as a means to initiate interaction, again leading to mistaken appraisals as aggressive. Biting may be a communication attempt. Insufficient interest in interactively playing with others was reported; instead parallel play was observed and described. Some caregivers indicated that inattention or excessive focus (i.e., inability to stop an engrossing activity) interferes with cooperative play.
3.1.6. Sleep
Poor sleep in the form of shorter periods of sleep, difficulty settling, and night-waking, was reported by all caregivers. Some caregivers felt sleep apnea was a major contributor to sleep problems. Caregivers expressed concern that pain interferes with sleep but their children cannot communicate it.
3.2. Impact on caregivers and families
Caregivers described symptom management as their way of life, often reporting that questions about the severity of stress are inadequate, because “severity” is relative. They reflected that others may be shocked and overwhelmed by symptoms that affected families find to be comparatively minor. Caregiver descriptions of their own responses to neurobehavioral symptoms suggested internal beliefs about strength and resilience, given the intensity of disease manifestations that they manage. Still, significant disease impact and burden were readily acknowledged.
3.2.1. Withdrawal
Strategic social withdrawal by families is pervasive. A caregiver summarized that MPS II “shrinks our worlds.” Families resort to isolation to minimize safety risks to their affected child or others, as well as judgment or interference from uninformed onlookers. For example, they skip activities (e.g. going out for dinner, community events), or do them at off-times (e.g. go to the park at 5 am).
3.2.2. Psychological stress about symptom management and attribution
Caregivers face chronic worry about safety and an inability to relax. They reported that they live on constant alert, can never leave their child alone, and usually need more than one person watching the child due to the vast array of risks and dangers that abound. Most caregivers “safety proof” their houses (e.g. Plexiglas over the TV, no knick knacks lying around, install locks and gates). Substantial pre-planning is necessary to engage in any activities outside the home, whether mundane or pleasurable events. Often multiple adults are needed to supervise an outing.
Significant psychological stress stems from difficulty managing or soothing emotional and behavioral challenges. Caregivers feel at a loss to help their children via standard techniques. Neurocognitive limitations interfere with the effectiveness of “time out” or other cause-effect concepts. Many caregivers feel extreme stress when clear triggers cannot be identified for emotional or behavioral outbursts or tantrums; often there is worry about pain as the source.
Psychological stress also arises from others' misinterpretation of their child's behaviors, and these opportunities are plentiful. For example, others may assume the affected child is aggressive, cruel, or inconsiderate due to behaviors that are in actuality sensory seeking, impulsive, or social interaction attempts. Others may critique insufficient parenting as the cause of the behaviors. Low attention and distractibility may be misconstrued as poor understanding, inability, or noncompliance. Caregivers often feel in the position of defending, explaining, and advocating in this regard; however, it occurs so often that some caregivers reported giving up and ignoring others who do not understand, as it feels both overwhelming and unjust.
3.2.3. Exhaustion
The disrupted sleep is exhausting to caregivers who reported that they have no choice but to “push through” the day. Reduced sleep has the expected negative effects on their children's functioning (i.e., poorer attention, more emotional and behavioral challenges, further difficulty learning), which translates to more stress and complications in supporting their children.
Caregivers exert significant work attending to their children's basic needs, which becomes more challenging as neurobehavioral symptoms increase and the children grow physically stronger. This combination of worsening neurobehavioral symptoms and physical maturation contributes to difficulty physically coaxing them through activities. Whether the children's basic needs have been adequately addressed is a source of stress and worry.
3.2.4. Concerned focus on development and fearful vigilance of neurobehavioral change
Caregivers feel an urgency to help their children attain some skills. It is defeating and frustrating when caregivers believe their child would be able to master a task if attention were adequate. They particularly reported significant anxiety that the combination of poor attention and accumulating boredom with repeated cognitive testing for clinical trials may be interfering with accuracy of results.
Caregivers reported tension over the desire for a reduction in symptom severity but a knowledge that this reduction often indicates disease progression. They feel great stress watching for signals of symptom change. If symptoms are worse on a day following a poor night's sleep, caregivers described “overthinking” that this worsening is due to disease progression rather than fatigue.
3.2.5. Financial and vocational strain
Financial strain is substantial, due to accumulating costs of treatments, supportive interventions (e.g., speech/language, occupational, physical, applied-behavior analysis), additional therapies (e.g., music or equine therapies), chronic medical complications requiring hospital care, specialized child care or child care for siblings, and physical accommodations in daily life (e.g., specialized strollers, feeding chairs, safety proofing). It was acknowledged that financial strain may involve different factors depending on country of origin and/or healthcare funding. Vocational stress is a serious concern and adds to financial hardship for many families. Some caregivers leave their jobs due to the high time demands of multiple treatments, as well as the unpredictable nature of the need for urgent medical intervention and extended care for somatic complications. This job loss leads to not only reduction in income but also change in professional identity or goals. Other caregivers reported continuing to work in a vocational setting where stress arises from either frequent absences or worry about their child's wellness while they are away from home.
4. Discussion
Using a multi-faceted methodology, we have assembled a comprehensive description of the body of neurobehavioral symptoms that may appear in the neuronopathic phenotype of MPS II, along with the specific day-to-day impact of these symptoms on the caregivers and family. Our findings indicate that the clinical heterogeneity of MPS II is also seen in the behavioral realm, as the diversity and intensity of neurobehavioral symptom expression is quite varied. This study builds upon important existing work by detailing the unique and complex assortment of behaviors that have contributed to historical descriptions such as hyperactivity, aggression, and obstinacy. Our findings indicate that some neuronopathic MPS II-specific symptoms are possibly obscured by current broad-based descriptors such as hyperactivity or aggression.
Uncovering the diversity of symptoms associated with existing descriptors potentially has important implications for understanding how aspects of the CNS may be differentially affected by disease or therapies. For example, if a proportion of aggressive behaviors are not truly aggressive (e.g., physical sensation seeking, Fig. 2), or a proportion of hyperactive behaviors are not truly hyperactive (e.g., repetitive movements, banging loudly), then different neuroanatomical correlates or pathophysiological processes may be implicated. Understanding direct and indirect causes of behavioral decline; the timing, sequence, and evolution of symptoms; and neurobehavioral response to therapies is critical in light of associated profound caregiver and family burden, as well as caregiver preferences that greater attention be given to neurobehavioral disease when assessing therapeutic outcomes.
Fig. 2.
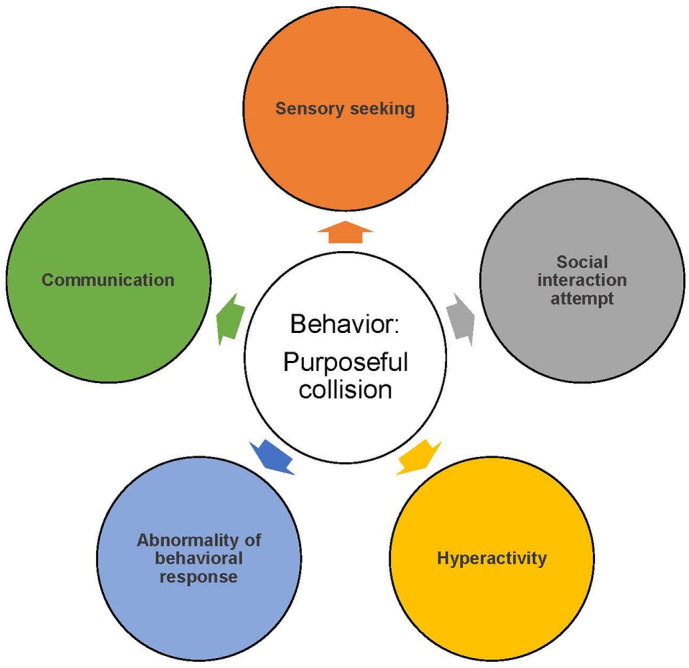
Potential attributions for a neurobehavioral symptom.
Fig. 2. This illustration depicts the multiple potential explanations, represented in colored circles, for an example neurobehavioral symptom at the figure's center, i.e., purposeful collision. While purposeful collision may broadly be appraised as aggressive, in MPS II it can be a behavioral sign of sensory seeking, attempting to engage socially, communicating, or others.
Evaluating whether interventions have an impact on disease requires first a clear measurement of the natural history, from which therapy-induced differences in course can be assessed [13,[33], [34], [35]]. Important work has been done to estimate age ranges that some categories of behaviors emerge [8,18], yet a current gap exists for systematic, prospective measurement and analysis of the full array of disease-specific symptoms [17,23,25,26,28]. A key obstacle is that existing generic assessment tools for pediatric behavioral functioning are inadequate for capturing the unique array of neurobehavioral symptoms of neuronopathic MPS II [23,25,26]. Extant tools may measure some of these symptoms, but unique symptoms may be missing, such that there is no single assessment tool that represents all of them [23,25]. Our study reports findings from our first step in developing a method for quantification of these symptoms, i.e., first clearly defining the disease-specific neurobehavioral complications that require measurement.
Neuronopathic MPS II is distinguished in the phenotypic spectrum not only by neurobehavioral symptoms but also neurocognitive complications, including regression, and shortened lifespan. It is currently unknown as to whether neurobehavioral worsening precedes, follows, or coincides with neurocognitive abnormalities [8,23,25]. Hyperactivity and difficult to manage behaviors have been proposed among a group of early markers that predict the neuronopathic phenotype [18]. Many affected children have developmental delays and speech/language delays from infancy and toddlerhood [1,3,14], which can make the earliest emergence of neurobehavioral symptoms harder to detect, as they may be attributed to the delays. In this study, caregivers reported that neurocognitive status has a major impact on neurobehavioral symptoms, and vice versa (e.g., low intellectual skills complicate attempts to curb or extinguish undesirable behaviors). The current findings aligned with the sparse reports in the literature that communication skills decline in a differential pattern [14], such that skills for verbal expression/speaking/vocalizing decline before skills in understanding what is said. Caregivers in this study indicated communication deficits are a major source of frustration and consequent outbursts when children cannot communicate their needs or desires, and caregivers face stress when they cannot understand their children's needs or how to help.
Across the phenotypic spectrum of MPS II, neurocognitive and neurobehavioral function may be affected by not just neurodegeneration but also structural CNS effects and complications from somatic disease [3,8,19]. For example, abnormalities in white matter volumes have been reported even in the nonneuronopathic phenotypes, and have been associated with poorer attention [13]. Caregivers reported their own frequent worry that behavioral worsening and/or head-banging may be evidence of pain from increased intracranial pressure, however the impact of communicating hydrocephalus is unclear on the MPS II neurobehavioral abnormalities [3,10]. Hearing loss may affect learning, communication and behavioral regulation [3,23,25]. Skeletal/joint disease-related mobility limitations [3,19] combine with disease-related loose bowel movements [14,15] and learning challenges to make toileting a particularly difficult milestone, such that inability to achieve bowel and/or bladder training has been proposed as one of seven early predictors of the neuronopathic phenotype [18]. Clarifying the relationships of the diverse somatic complications with neurobehavioral effects can only be possible with accurate measurement of the behavioral signs.
There is universal acknowledgment that these neurobehavioral symptoms are a significant source of stress for caregivers [3,8,14,26,36], and our multi-method approach expands understanding of the unrelenting burden they face. Alongside acting as their children's behavior therapists, counselors, in-home nurses, special educators, protectors and advocates, the caregivers also face social isolation, outsider judgment, and stress associated with remaining alert for safety and vigilant for symptom change, often with the backdrop of massively deprived sleep, and financial and vocational strain. Further investigation into comprehensive supportive measures to reduce caregiver burden, as has been done with other neuronopathic MPS types [30], is urgently needed. The present findings add further justification that measuring neurobehavioral response to therapy is an important observer-reported outcome to consider in selecting endpoints for CNS-modifying clinical trials.
This study is limited by small participant numbers. Caregiver feedback was critical for understanding impediments to recruitment. Most recruitment challenges were attributable to the profound effects of disease on family capacity to engage, including cancellations due to medical events; discomfort traveling due to the child's high degree of behavioral and medical needs; or being committed to other medically related travel (e.g., intrathecal trial; surgeries by experts), such that it was not feasible to add an additional research-related travel visit to a destination where medical care would not be a part. While we attempted to address this possibility with the option for phone interviews, even this solution may not have been sufficient. As a result, selection bias is a factor, as enrollment involved caregivers who have the community or family supports to travel and participate in research. This aspect has the potential to skew the assortment of symptoms that are reported. Another limitation of this study is a lack of measurement of neurodevelopment to characterize participants; however obtaining these data were both beyond the scope of this study and in conflict with other ongoing interventional studies where interval assessments are regular. Last, neither the timeline of the appearance and cessation of the behavioral symptoms, nor their frequencies, could be reported. However, reporting this level of detail with confidence would require a tool to quantify the behaviors longitudinally, which adds further rationale to our assertion that a Hunter-specific behavioral assessment tool is needed.
In conclusion, the neurobehavioral manifestations of neuronopathic MPS II have a pervasive negative impact on affected children and their families and cause considerable suffering. These symptoms appear to be misunderstood with some regularity. This study's new multi-sourced descriptions are a first step in developing quantitative measures of neurobehavioral symptoms and family burden. A next step in tool development will be identifying frequencies of these behaviors in a larger sample to ensure better reliability, as has been done in other MPS-specific behavioral tools [37]. Longitudinal use of these measures will enable evaluation of change, particularly response to therapy. Together with documented changes in activities of daily living, such measures of change are critical elements for clinical trials.
Disclosures
Eisengart: Research support from Lysogene, Sangamo and Shire/Takeda; consultant to Denali Therapeutics, Regenxbio, Sangamo, Sanofi Genzyme, and Shire/Takeda; advisory boards for Amicus, bluebird bio, and Sanofi Genzyme; contract work for Shapiro Neuropsychology Consulting, LLC.
King: Research support from Alexion Pharmaceuticals, Inc., Shire/Takeda, and Sanofi Genzyme, and contract work for Shapiro Neuropsychology Consulting, LLC.
Shapiro: Partner, Shapiro Neuropsychology Consulting, LLC.
Whitley: Research support from Shire/Takeda; consultant to Shire/Takeda.
Muenzer: Consultant to BioMarin, Shire/Takeda, PTC Therapeutics, Green Cross, Sanofi Genzyme, Eloxx, Regenxbio, Denali Therapeutics, Sangamo and JCR Pharmaceuticals. Serves on advisory boards for BioMarin, Sanofi Genzyme, Green Cross, JCR Pharmaceuticals and Shire/Takeda. He is principal investigator for phase 1/2 and phase 2/3 trials that investigate intrathecal ERT for patients with neuronopathic Hunter syndrome, a phase 1/2 gene editing clinical trial for adults with Hunter syndrome and a phase 1/2 intravenous ERT clinical trial for MPS IIIA.
Funding sources
This investigator-initiated research was supported by a grant from Shire Human Genetic Therapies Inc., Lexington, MA, a member of the Takeda group of companies through # IIR-USA-001644, and by NIH U54NS065768. Historic chart review and analysis was funded by NIH U54NS065768. The Lysosomal Disease Network (U54NS065768) is a part of the Rare Diseases Clinical Research Network, an initiative of the Office of Rare Diseases Research, and the National Center for Advancing Translational Sciences. This consortium is funded through a collaboration between the National Center for Advancing Translational Sciences, the National Institute of Neurological Disorders and Stroke, and the National Institute of Diabetes and Digestive and Kidney Diseases. Data curation was supported by the National Institutes of Health's National Center for Advancing Translational Sciences, grant UL1TR002494. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health's National Center for Advancing Translational Sciences. Resources for convening the caregiver advisory board and sharing of data were provided by Denali Therapeutics (South San Francisco, CA). The advisory board was facilitated by the first author (JBE). Infrastructure support was provided by the National MPS Society, Project Alive, MPS Superhero Foundation, the University of Minnesota Department of Pediatrics, and the University of Minnesota's Center for Neurobehavioral Development. None of the funding sources had any role in study design; collection, analysis and interpretation of data; writing of the manuscript; nor the decision to submit the article for publication.
Acknowledgments
We are grateful for the support and cooperation of these courageous children and their families. We thank Cure Sanfilippo Foundation, MPS Superhero Foundation, and Project Alive for consultation and direction regarding caregiver burden. We thank Brenda Diethelm-Okita and Ashley Schneider in the Lysosomal Disease Network office at the University of Minnesota for research and administrative support. We thank Sarah Lewis (HealthPartners, Minneapolis, MN) and Katherine Spurlock (West Virginia School of Osteopathic Medicine, Lewisburg, WV) for neuropsychological assistance.
References
- 1.Neufeld E., Muenzer J. The Metabolic and Molecular Bases of Inherited Disease. Vol. 8. 2001. The mucopolysaccharidoses; pp. 3421–3452. [Google Scholar]
- 2.Jones S., Almassy Z., Beck M., Burt K., Clarke J., Giugliani R., Hendriksz C., Kroepfl T., Lavery L., Lin S.-P. Mortality and cause of death in mucopolysaccharidosis type II—a historical review based on data from the Hunter Outcome Survey (HOS) J. Inherit. Metab. Dis. 2009;32:534–543. doi: 10.1007/s10545-009-1119-7. [DOI] [PubMed] [Google Scholar]
- 3.Muenzer J., Beck M., Eng C., Escolar M., Giugliani R., Guffon N., Harmatz P., Kamin W., Kampmann C., Koseoglu S. Multidisciplinary management of Hunter syndrome. Pediatrics. 2009;124:e1228–e1239. doi: 10.1542/peds.2008-0999. [DOI] [PubMed] [Google Scholar]
- 4.Bigger B.W., Begley D.J., Virgintino D., Pshezhetsky A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018;125:322–331. doi: 10.1016/j.ymgme.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Clarke L.A. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology. 2011;50:v13–v18. doi: 10.1093/rheumatology/ker395. [DOI] [PubMed] [Google Scholar]
- 6.Martin H.R., Poe M.D., Reinhartsen D., Pretzel R.E., Roush J., Rosenberg A., Dusing S.C., Escolar M.L. Methods for assessing neurodevelopment in lysosomal storage diseases and related disorders: a multidisciplinary perspective. Acta Paediatr. 2008;97:69–75. doi: 10.1111/j.1651-2227.2008.00651.x. [DOI] [PubMed] [Google Scholar]
- 7.Martin R., Beck M., Eng C., Giugliani R., Harmatz P., Muñoz V., Muenzer J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome) Pediatrics. 2008;121:e377–e386. doi: 10.1542/peds.2007-1350. [DOI] [PubMed] [Google Scholar]
- 8.Roberts J., Stewart C., Kearney S. Management of the behavioural manifestations of Hunter syndrome. Br. J. Nurs. 2016;25 doi: 10.12968/bjon.2016.25.1.22. [DOI] [PubMed] [Google Scholar]
- 9.Lampe C., Atherton A., Burton B.K., Descartes M., Giugliani R., Horovitz D.D., Kyosen S.O., Magalhães T.S., Martins A.M., Mendelsohn N.J. JIMD Reports. vol. 14. Springer; 2014. Enzyme replacement therapy in mucopolysaccharidosis II patients under 1 year of age; pp. 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broomfield A., Davison J., Roberts J., Stewart C., P H., Beesley C., Tylee K., Rust S., Schwahn B., Jameson E., Vijay S., Santra S., Sreekantam S., Ramaswami U., Chakrapani A., Raiman J., Cleary M., Jones S. Ten Years of Enzyme Replacement Therapy in Paediatric Onset Mucopolysaccharidosis II in England. Molecular Genetics and Metabolism. 2019 doi: 10.1016/j.ymgme.2019.07.016. (In Press) [DOI] [PubMed] [Google Scholar]
- 11.Vollebregt A.A., Hoogeveen-Westerveld M., Kroos M.A., Oussoren E., Plug I., Ruijter G.J., van der Ploeg A.T., Pijnappel W.P. Genotype–phenotype relationship in mucopolysaccharidosis II: predictive power of IDS variants for the neuronopathic phenotype. Dev. Med. Child Neurol. 2017;59:1063–1070. doi: 10.1111/dmcn.13467. [DOI] [PubMed] [Google Scholar]
- 12.Shapiro E.G., Jones S.A., Escolar M.L. Developmental and behavioral aspects of mucopolysaccharidoses with brain manifestations—neurological signs and symptoms. Mol. Genet. Metab. 2017;122:1–7. doi: 10.1016/j.ymgme.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Yund B., Rudser K., Ahmed A., Kovac V., Nestrasil I., Raiman J., Mamak E., Harmatz P., Steiner R., Lau H., Vekaria P., Wozniak J.R., Lim K.O., Delaney K., Whitley C., Shapiro E. Cognitive, medical, and neuroimaging characteristics of attenuated mucopolysaccharidosis type II. Mol. Genet. Metab. 2015;114:170–177. doi: 10.1016/j.ymgme.2014.12.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young I., Harper P. The natural history of the severe form of Hunter's syndrome: a study based on 52 cases. Dev. Med. Child Neurol. 1983;25:481–489. doi: 10.1111/j.1469-8749.1983.tb13794.x. [DOI] [PubMed] [Google Scholar]
- 15.Wraith J.E., Beck M., Giugliani R., Clarke J., Martin R., Muenzer J. Initial report from the Hunter outcome survey. Genet. Med. 2008;10:508. doi: 10.1097/gim.0b013e31817701e6. [DOI] [PubMed] [Google Scholar]
- 16.Muenzer J., Jones S.A., Tylki-Szymańska A., Harmatz P., Mendelsohn N.J., Guffon N., Giugliani R., Burton B.K., Scarpa M., Beck M. Ten years of the Hunter Outcome Survey (HOS): insights, achievements, and lessons learned from a global patient registry. Orphanet J. Rare Dis. 2017;12 doi: 10.1186/s13023-017-0635-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barone R., Pellico A., Pittalà A., Gasperini S. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Ital. J. Pediatr. 2018;44 doi: 10.1186/s13052-018-0561-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holt J., Poe M.D., Escolar M.L. Early clinical markers of central nervous system involvement in mucopolysaccharidosis type II. J. Pediatr. 2011;159 doi: 10.1016/j.jpeds.2011.03.019. 320–326. e322. [DOI] [PubMed] [Google Scholar]
- 19.Holt J.B., Poe M.D., Escolar M.L. Natural progression of neurological disease in mucopolysaccharidosis type II. Pediatrics. 2011;127:e1258–e1265. doi: 10.1542/peds.2010-1274. [DOI] [PubMed] [Google Scholar]
- 20.Crowe L., Yaplito-Lee J., Anderson V., Peters H. Cognitive and behaviour profiles of children with mucopolysaccharidosis type II. Cogni. Neuropsychol. 2017;34:347–356. doi: 10.1080/02643294.2017.1401530. [DOI] [PubMed] [Google Scholar]
- 21.Shapiro E.G., Rudser K., Ahmed A., Steiner R.D., Delaney K.A., Yund B., King K., Kunin-Batson A., Eisengart J., Whitley C.B. A longitudinal study of emotional adjustment, quality of life and adaptive function in attenuated MPS II. Mol. Genet. Metab. Rep. 2016;7:32–39. doi: 10.1016/j.ymgmr.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Needham M., Packman W., Quinn N. Health-related quality of life in patients with MPS II. J. Genet. Couns. 2015;24:635–644. doi: 10.1007/s10897-014-9791-7. [DOI] [PubMed] [Google Scholar]
- 23.Eisengart J.B. 15th International Symposium on MPS and Related Diseases, San Diego, CA. 2018, August. The impact of neurobehavioral symptoms from clinical trials through adulthood. [Google Scholar]
- 24.Guffon N., Heron B., Chabrol B., Feillet F., Montauban V., Valayannopoulos V. Diagnosis, quality of life, and treatment of patients with Hunter syndrome in the French healthcare system: a retrospective observational study. Orphanet J. Rare Dis. 2015;10 doi: 10.1186/s13023-015-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shapiro E., Eisengart J., Delaney K., Nestrasil I. Assessment of brain function and structure in MPS diseases. In: Tomatsu S., Lavery C., Giugliani R., Harmatz P., Scarpa M., Wegrzyn G., Orii T., editors. Mucopolysaccharidoses Update. Nova Science Publishers, Inc.; Hauppauge, NY: 2018. pp. 425–464. [Google Scholar]
- 26.Cross E., Hare D.J. Behavioural phenotypes of the mucopolysaccharide disorders: a systematic literature review of cognitive, motor, social, linguistic and behavioural presentation in the MPS disorders. J. Inherit. Metab. Dis. 2013;36:189–200. doi: 10.1007/s10545-012-9572-0. [DOI] [PubMed] [Google Scholar]
- 27.Malcolm C., Hain R., Gibson F., Adams S., Anderson G., Forbat L. Challenging symptoms in children with rare life-limiting conditions: findings from a prospective diary and interview study with families. Acta Paediatr. 2012;101:985–992. doi: 10.1111/j.1651-2227.2012.02680.x. [DOI] [PubMed] [Google Scholar]
- 28.King K.E., Shapiro E.G., Whitley C.B., Muenzer J., Eisengart J.B. Feasibility of quantifying behavior in early progressive MPS II. Mol. Genet. Metab. 2019;126:S85. [Google Scholar]
- 29.Oliveira M., Schwartz I., Costa L., Maia H., Ribeiro M., Guerreiro L., Acosta A., Rocha N. Quality of life in mucopolysaccharidoses: construction of a specific measure using the focus group technique. BMC Res. Notes. 2018;11 doi: 10.1186/s13104-018-3157-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shapiro E., Lourenço C.M., Mungan N.O., Muschol N., O’Neill C., Vijayaraghavan S. Analysis of the caregiver burden associated with Sanfilippo syndrome type B: panel recommendations based on qualitative and quantitative data. Orphanet J. Rare Dis. 2019;14 doi: 10.1186/s13023-019-1150-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lampe C., Jain M., Olaye A., Meesen B., Decker C., Mengel E. Relationship between patient-reported outcomes and clinical outcomes in patients with Morquio A syndrome. J. Inborn Errors Metab. Screen. 2015;3 2326409815576188. [Google Scholar]
- 32.Lewis S., Shapiro E., Whitley C., Delaney K., Eisengart J. Neurocognitive profiles of untreated Hunter syndrome. Mol. Genet. Metab. 2017;1:S83. [Google Scholar]
- 33.Delaney K.A., Rudser K.R., Yund B.D., Whitley C.B., Haslett P.A., Shapiro E.G. JIMD Reports-Case and Research Reports. vol. 13. Springer; 2013. Methods of neurodevelopmental assessment in children with neurodegenerative disease: Sanfilippo syndrome; pp. 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro E.G., Nestrasil I., Delaney K.A., Rudser K., Kovac V., Nair N., Richard C.W., Haslett P., Whitley C.B. J. Pediatr. 2016;170:278–287. doi: 10.1016/j.jpeds.2015.11.079. e274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shapiro E., Bernstein J., Adams H.R., Barbier A.J., Buracchio T., Como P., Delaney K.A., Eichler F., Goldsmith J.C., Hogan M. Neurocognitive clinical outcome assessments for inborn errors of metabolism and other rare conditions. Mol. Genet. Metab. 2016;118:65–69. doi: 10.1016/j.ymgme.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hare D.J., Mahon L., Rust S. Assessment and management of over-activity and sleep disorder in mucopolysaccharidoses. J. Child Sci. 2018;8:e124–e127. [Google Scholar]
- 37.Shapiro E.G., Nestrasil I., Ahmed A., Wey A., Rudser K.R., Delaney K.A., Rumsey R.K., Haslett P.A., Whitley C.B., Potegal M. Quantifying behaviors of children with Sanfilippo syndrome: the Sanfilippo Behavior Rating Scale. Mol. Genet. Metab. 2015;114:594–598. doi: 10.1016/j.ymgme.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]