

Abstract
Summary
We introduce Tibanna, an open-source software tool for automated execution of bioinformatics pipelines on Amazon Web Services (AWS). Tibanna accepts reproducible and portable pipeline standards including Common Workflow Language (CWL), Workflow Description Language (WDL) and Docker. It adopts a strategy of isolation and optimization of individual executions, combined with a serverless scheduling approach. Pipelines are executed and monitored using local commands or the Python Application Programming Interface (API) and cloud configuration is automatically handled. Tibanna is well suited for projects with a range of computational requirements, including those with large and widely fluctuating loads. Notably, it has been used to process terabytes of data for the 4D Nucleome (4DN) Network.
Availability and implementation
Source code is available on GitHub at https://github.com/4dn-dcic/tibanna.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Efficient execution of data processing and analysis pipelines is essential in many areas of research that involve large datasets. However, integration of a pipeline with local computing infrastructure is often a painstaking process. The need for such integration leads to redundant creation of seemingly identical pipelines, decreasing reproducibility and efficiency.
Cloud platforms present a number of advantages for large-scale data analysis, including scalability and efficient data sharing. However, the frequent lack of separation between workflow specifications and platform-specific configuration impedes transition to the cloud. Having to handle detailed aspects of the cloud platform renders the advantages of cloud computing impractical for many pipeline developers.
For effective separation of pipelines from platforms, pipelines should be made portable and standardized, and all platform-specific tasks must be delegated to a separate pipeline management tool. To enable this framework, there have been efforts to create a standard for portable pipelines. Common Workflow Language (CWL) (https://www.commonwl.org/) and Workflow Description Language (WDL) (https://software.broadinstitute.org/wdl/) describe the structure of a pipeline (e.g. steps, inputs and outputs), whereas Docker and Singularity (Kurtzner et al., 2017) enable generation of portable images of executable components and dependencies. Support for standard pipeline languages is becoming more widely adopted. In addition, Galaxy (Giardine et al., 2005), a GUI-based bioinformatics analysis platform, is planned to support CWL in the near future. Domain Specific Languages (DSLs) such as Nextflow (Tommaso et al., 2017) or Snakemake (Köster and Rahmann, 2012) provide better expressive power than CWL, but are bound to a single pipeline manager.
At the Data Integration and Coordination Center (DCIC) for the NIH-sponsored 4D Nucleome (4DN) Network (Dekker et al., 2017), our aim is to efficiently process a large number of datasets from multiple experimental types submitted by member laboratories. We therefore require a workflow management system that supports standardized and portable pipelines (CWL/WDL) on a public platform. We chose Amazon Web Services (AWS), the most widely used commercial cloud service. Additionally, the Network needs an open-source implementation so the scientific community can easily run the same standardized 4DN pipelines on its own data.
2 Results
We have developed and used Tibanna as an open-source pipeline manager and automated cloud resource allocator for running Docker-based pipelines on AWS. Tibanna integrates easily with other systems through its Python API.
2.1 Overview
Tibanna creates an instance of an individually pre-configured virtual machine (EC2) for each job (Fig. 1). The instance autonomously fetches data and the pipeline, runs the pipeline, stores logs and output files on the cloud and finally, terminates itself. Instead of depending on a master server or queue, each job gets its own serverless scheduler comprised of AWS Lambdas coordinated by the AWS Step Function. The 4DN DCIC uses additional Lambdas to communicate with our Data Portal.
Fig 1.
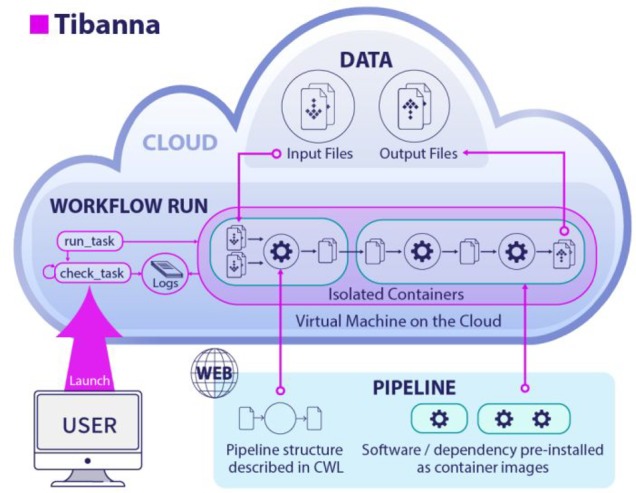
Tibanna. For each pipeline execution, Tibanna creates a workflow run by launching a customized virtual machine (EC2), runs the pipeline and destroys itself when finished. Scheduling is performed individually by serverless components
2.2 Execution unit
Tibanna runs a single execution unit, and dependencies can be managed through its dependency feature. This allows one to decide and manage execution units based on resource requirements and logical grouping, and determine what intermediate files to keep for future re-processing and curation. An execution unit may be a whole pipeline, a sub-pipeline or a single step, and these units are not defined by the pipeline structure itself. We found that tools that attempt to auto-determine execution units tend to make non-optimal choices and are difficult to integrate with desired execution designs.
2.3 How to use Tibanna
Tibanna auto-configures cloud components and permissions with a single command. Its only requirements are to provide input data on the cloud and to prepare a publicly available pipeline in CWL or WDL, along with a public Docker image. Each job is described as a JSON file or a Python dictionary object, specifying the pipeline, input files and parameters, and where the output should be collected. Submission and monitoring of jobs is performed locally from the command line or using the Python API. Logs can also be retrieved similarly or through the AWS Web Console. A user with a private key file can securely connect to running instances for more detailed monitoring. An easy-to-follow documentation comes with the software for more details.
2.4 Execution example
In addition to running pipelines for 4DN, Tibanna has been used by external users to call transposon insertions from 1000 to 30× whole genome sequencing datasets. 1000 jobs were simultaneously executed, each running for 4–8 h on a spot instance with 16 cores and 32 GB memory and 250 GB disk size, costing about $1.6 per run.
2.5 Comparison to other tools
Tibanna offers several advantages compared to existing workflow management tools (Supplementary Table S1), specifically for our need to automatically execute and monitor various portable pipelines in integration with a data portal.
Local CWL executors [e.g. Cwltool, Rabix (Kaushik, 2016) and CWL-Airflow (Kotliar et al., 2018)] do not perform resource allocation. Snakemake recently started supporting local CWL executions with cloud storage but also does not handle resource management on AWS.
Tools that perform cloud resource allocation uses one of the three approaches: AWS Batch, a resizable cluster, or individual customization.
(1) AWS BatchAWS Batch (https://aws.amazon.com/batch/) is an AWS service to run batch jobs, but it is not easy to be used directly with CWL/Docker, often exhibits a mysteriously long wait time and lacks many functionalities. Examples of tools based on AWS Batch include Nextflow, Funnel (https://ohsu-comp-bio.github.io/funnel/) and Cromwell. Nextflow supports Docker, but using CWL requires conversion to the Nextflow format and the converter is not functional at this point.
Cromwell (https://software.broadinstitute.org/wdl/) was originally developed to run WDL on Google Cloud Platform (GCP), although it now supports CWL and AWS through AWS Batch. However, it leaves all the cloud configuration to the user, and due to the lack of an API, a large-scale batch run requires a cumbersome generation of many input JSON files and manual tracing of job IDs for proper output matching. Monitoring functionality that is critical for large-scale projects is not provided by either Cromwell or AWS Batch. Funnel also requires users to build a custom Amazon Machine Image (AMI).
(2) Resizable clusterStarCluster (http://star.mit.edu/cluster/docs/latest), an older tool without CWL/Docker support, and Toil (Vivian et al., 2017) work by creating a resizable cluster of EC2 instances of the same CPU and memory and an additional master instance. They require extra work for configuring and managing clusters. Nextflow also uses an auto-scalable cluster as an alternative. Arvados (https://arvados.org/) supports CWL and Docker and involves a cluster of dynamically allocated cloud instances where jobs are dispatched by SLURM. However, the initial setup process is quite complicated. Arvados also uses its own storage organization which required additional development to access and is not compatible with the file organizations used by our portal.
(3) Individual customizationTibanna instead creates a specific compute environment for an individual execution at launch, and removes it when the job is done. This approach is also adopted by Seven Bridges (https://www.sevenbridges.com/) and DNANexus (https://www.dnanexus.com/), which, as commercial portal services, also offer full automation but charge extra fees and do not easily integrate with other systems.
At 4DN DCIC, we receive datasets that vary in input file size, with vastly different memory and storage requirements. For example, the contact matrix generation sub-workflow of the 4DN Hi-C data processing pipeline involves in-memory generation of a matrix that represents non-zero interactions of all genomic locations. As the size of input datasets (i.e. reads representing contacts) varies >10-fold (up to 76 Gb), memory requirement also varies substantially, thus necessitating machines with different memory sizes for efficient computation. For this level of variability at the execution level, individual customization makes better use of the elastic compute environment of the cloud compared to cluster allocation.
3 Conclusion
Tibanna is a pipeline management system for the AWS cloud. Tibanna has been used to process 4DN data. We believe Tibanna will be useful for many other AWS projects.
Supplementary Material
Acknowledgements
We thank the members of the 4DN DCIC, the Park lab and the Avillach lab at the Department of Biomedical Informatics at Harvard Medical School for valuable feedback and the E. Alice Lee lab at Boston Children's Hospital for beta-testing Tibanna. We are especially thankful to Andy Schroeder, Peter Kerpedjiev, Martin Owens and Chuck McCallum for code review/contribution, Shannon Ehmsen for preparing the figure, and Alon Galor, Dhawal Jain, Su Wang, Geoff Nelson and Marc Rubenfield (Veritas Genetics) for comments on the manuscript.
Funding
This work was supported by the National Institutes of Health Common Fund 4D Nucleome Program U01CA200059 awarded to PJP.
Conflict of Interest: none declared.
References
- Dekker J. et al. (2017) The 4D nucleome project. Nature, 549, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik G. (2016) Rabix: an open-source workflow executor supporting recomputability and interoperability of workflow descriptions. Pac. Symp. Biocomput., 22, 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B. et al. (2005) Galaxy: a platform for interactive large-scale genome analysis. Genome Res., 15, 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster J., Rahmann S. (2012) Snakemake—a scalable bioinformatics workflow engine. Bioinformatics, 28, 2520–2522. [DOI] [PubMed] [Google Scholar]
- Kotliar M. et al. (2018) CWL-Airflow: a lightweight pipeline manager supporting Common Workflow Language. bioRxiv, 249243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzner G.M. et al. (2017) Singularity: scientific containers for mobility of compute. PLoS ONE, 12, e0177459.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommaso P.D. et al. (2017) Nextflow enables reproducible computational workflows. Nat. Biotechnol., 35, 316–319. [DOI] [PubMed] [Google Scholar]
- Vivian J. et al. (2017) Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol., 35, 314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.