

Abstract
Background
Brugada syndrome (BrS) is an inherited arrhythmic disease linked to SCN5A mutations. It is controversial whether SCN5A mutation carriers possess a greater risk of major arrhythmic events (MAE). We examined the association of SCN5A mutations and MAE in BrS patients.
Methods
We comprehensively searched the databases of MEDLINE and EMBASE from inception to September 2017. Included studies were published cohort and case–control studies that compared MAE in BrS patients with and without SCN5A mutations. Data from each study were combined using the random‐effects model. Generic inverse variance method of DerSimonian and Laird was employed to calculate the risk ratios (RR) and 95% confidence intervals (CI).
Results
Seven studies from March 2002 to October 2017 were included (1,049 BrS subjects). SCN5A mutations were associated with MAE in Asian populations (RR = 2.03, 95% CI: 1.37–3.00, p = 0.0004, I 2 = 0.0%), patients who were symptomatic (RR = 2.66, 95% CI: 1.62–4.36, p = 0.0001, I 2 = 23.0%), and individuals with spontaneous type‐1 Brugada pattern (RR = 1.84, 95% CI: 1.05–3.23, p = 0.03, I 2 = 0.0%).
Conclusions
SCN5A mutations in BrS increase the risk of MAE in Asian populations, symptomatic BrS patients, and individuals with spontaneous type‐1 Brugada pattern. Our study suggests that SCN5A mutation status should be an important tool for risk assessment in BrS patients.
Keywords: Brugada syndrome, genetic, major arrhythmic events, SCN5A, sudden cardiac death
1. INTRODUCTION
Brugada syndrome (BrS) is an autosomal dominant inherited arrhythmia syndrome characterized by ST‐segment elevation in the right precordial leads without obvious evidence of ischemia, electrolyte disturbances, or structural heart disease. It predisposes patients to major arrhythmic events (MAE) including sustained ventricular tachycardia, ventricular fibrillation, appropriate implantable cardioverter defibrillator (ICD) shocks, aborted cardiac arrest, and sudden cardiac death (Brugada & Brugada, 1992; Priori et al., 2013). There are geographic differences in the prevalence of BrS: it varies from 0.5 to 4 per 1,000 in Asian countries such as Thailand, the Philippines, Japan, and Singapore (Rattanawong et al., 2016; Rattanawong, Ngarmukos, et al., 2017), whereas the prevalence is less than 0.2 per 1,000 in the western hemisphere (Kamakura, 2013). It is more common in men and can be induced by fever, with the prevalence of fever‐induced BrS of approximately 2%–4% (Kamakura, 2013; Rattanawong et al., 2016).
The most common identifiable genetic defect in BrS lies in the SCN5A gene, which encodes the α‐subunit of the NaV1.5 cardiac sodium channel and accounts for 14%–26% of the cases (Chen et al., 1998; Yamagata et al., 2017). More than 300 mutations in the SCN5A gene have been linked to the syndrome (Juang & Horie, 2016). The only preventive measure for sudden cardiac death in BrS is ICD implantation; thus, risk stratification to select the patient in whom ICD is appropriate is crucial (Probst et al., 2010). The use of SCN5A mutation status to prognosticate the risk of MAE in BrS patients has been controversial (Adler et al., 2016): some studies showed positive results (Makarawate et al., 2017; Nishii et al., 2010; Yamagata et al., 2017), while others failed to correlate the mutation to subsequent MAE (Andorin et al., 2016; Gehi, Duong, Metz, Gomes, & Mehta, 2006; Priori et al., 2002; Probst et al., 2010). The goal of this systematic review and meta‐analysis was to examine the association of SCN5A mutations and MAE in BrS patients.
2. METHOD
2.1. Search strategy
Two investigators (WV and PC) independently searched for published studies indexed in MEDLINE and EMBASE databases from inception to September 2017 using a search strategy that included the terms “SCN5A,” “mutation,” and “Brugada.” Only English language publications were included. A manual search for additional pertinent studies and review articles using references from retrieved articles was also completed.
2.2. Inclusion criteria
The eligibility criteria included the following:
Cohort study (prospective or retrospective) or case–control study reporting the incidence of MAE in BrS patients with and without SCN5A mutations
Calculation of relative risk, hazard ratio, odds ratio, incidence ratio, or standardized incidence ratio with 95% confidence intervals (CI) or provision of sufficient raw data for these calculations were provided.
Use of BrS participants without SCN5A mutations as controls.
Study eligibility was independently determined by two investigators (JC and PM) and any discrepancies were resolved by mutual consensus. Newcastle‐Ottawa quality assessment scale was used to evaluate each study's quality. The scale uses a star system (0–9) to evaluate three domains: recruitment and selection of the participants, similarity and comparability between the groups, and ascertainment of the outcome of interest among cohort studies. Higher scores represent higher study quality (Stang, 2010).
2.3. Data extraction
A standardized data collection form was used to obtain the following information from each study: title of study, name of first author, year of study, year of publication, country of origin, number of participants, demographic data of participants, method used to identify cases and controls, methods used to diagnose the outcomes of interest (SCN5A mutation and MAE), methods to verify if the variants were disease‐causing, and average duration of follow‐up with confounders that were adjusted effect estimates with 95% CI and covariates that were adjusted in the multivariable analysis.
To ascertain the accuracy, all investigators independently performed this data extraction process. Any data discrepancy was resolved by referring back to the original articles.
2.4. Statistical analysis
We performed a meta‐analysis of the included cohort studies using a random‐effects model. The extracted studies were excluded from the analysis if they did not present an outcome in each intervention group or did not have enough information required for continuous data comparison. We pooled the point estimates from each study using the generic inverse‐variance method of DerSimonian and Laird (1986). The heterogeneity of effect size estimates across these studies was quantified using the I 2 statistic and Q statistic. For the Q statistic, substantial heterogeneity was defined as p < 0.10. The I 2 statistic ranges in value from 0% to 100% (I 2 < 25%, low heterogeneity; I 2 = 25%–50%, moderate heterogeneity; and I 2 > 50%, substantial heterogeneity; Higgins, Thompson, Deeks, & Altman, 2003). A sensitivity analysis was performed to assess the influence of the individual studies on the overall results by omitting one study at a time. We used a sequential omitting strategy, as described by Patsopoulos, Evangelou, and Ioannidis (2008), to examine whether overall estimates were influenced by the substantial heterogeneity observed. Publication bias was assessed using a funnel plot and Egger's regression test (p < 0.05 was considered significant; Sterne & Egger, 2001). Potential sources of heterogeneity from clinical characteristics were analyzed with subgroup analysis and were compared with meta‐regression. Pooled risk ratio (RR), sensitivity analysis, funnel plot, and forest plot were performed using Review Manager (RevMan) [Computer program]. Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014. Egger test was performed using the Stata SE 14.1 software from StataCorp LP.
3. RESULTS
3.1. Description of included studies
Our search strategy yielded 382 potentially relevant articles (227 articles from EMBASE and 155 articles from MEDLINE). After exclusion of duplicates, 253 articles underwent title and abstract review. Two hundred and eleven articles were excluded at this stage as they were not cohort studies and they were not conducted in patients with BrS, leaving 42 articles for full‐length article review. Thirty‐five articles were excluded because they did not report the outcome of interest or they did not have a control group. Therefore, seven prospective cohort studies of BrS patients were included in this meta‐analysis. Figure 1 outlines the search and literature review process. The clinical characteristics and summary of included studies are provided in Table 1. The Newcastle‐Ottawa scales of the included studies are described in the Table 1.
Figure 1.
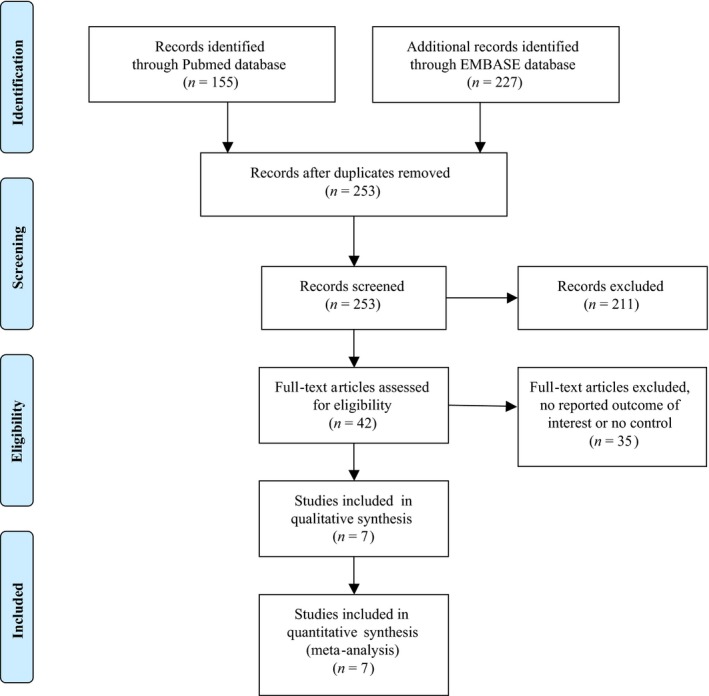
Search methodology and selection process
Table 1.
The clinical characteristics and summary of included studies
| Priori et al. | Eckardt et al. | Nishii et al. | Conte al. | Andorin et al. | Makarawate et al. | Yamagata et al. | |
|---|---|---|---|---|---|---|---|
| Country | Italy | Netherlands, Germany, and France | Japan | Belgium | European countries | Thailand | Japan |
| Study design | Prospective cohort | Prospective cohort | Prospective cohort | Retrospective cohort | Prospective cohort | Prospective cohort | Prospective cohort |
| Year of Publication | 2002 | 2005 | 2010 | 2015 | 2016 | 2017 | 2017 |
| Study subjects | BrS patient (130 probands, 70 affected family members) | BrS with type‐1 BrP patients from 4 university hospitals | BrS with type‐1 BrP patients who were admitted to 5 hospitals in Japan during January 1997 to December 2009 | BrS with spontaneous or drug‐induced Brugada type‐1 BrP who underwent ICD implantation and follow‐up at a single study | BrS with type‐1 BrP patients who are younger than 19 years from 16 European tertiary centers | Symptomatic BrS patients with type‐1 BrP with ICD implantations in KhonKaen, Thailand, between 2008 and 2011 | BrS with Type‐1 BrP patients who underwent genetic testing for SCN5A mutation and were followed up between 1988 and 2013 |
| Exclusion criteria | Right ventricular cardiomyopathy by echocardiography | Structural heart diseases, acute ischemia and metabolic or electrolyte disturbances | Abnormalities found by echocardiography and chest X‐ray | None | Structural cardiac abnormalities, electrolyte or metabolic disturbances at the time of ECG recording | Structural heart diseases | Structural heart disease |
| Number of subjects (% male, mean age) | 200 patients (76% male, mean age 41 ± 18 years) | 212 patients (71.7% male, mean age 45 ± 6 years) | 108 patients (97.2% male, mean age 46.8 ± 11.6 years) | 176 patients (67.0% male, mean age 43 years±16.8 years) | 106 patients (54.7% male, mean age 11.1 ± 5.7 years) | 40 patients (97.5% male, median age 43 years [range 22–66]) | 415 patients (97% male, mean age 46 ± 14 years) |
| Methods of mutation detection | DHPLC and/or SSCA | DNA sequencing | DNA sequencing | DNA sequencing | DHPLC and/or DNA sequencing | DNA sequencing | DHPLC, SSCA, and DNA sequencing |
| Verification if the variants were disease‐causing | A panel of 400 healthy white individuals (800 alleles) was used as control | Not described | Not described | Not described | Not described | Only H558R and R1193Q variants were reported; both of which were known to relate to Brugada syndrome | The frequencies and statuses of the mutations (using an in silico phenotype prediction algorithm) were evaluated |
| Presence of SCN5A mutations |
84 Positive (42%) 116 Negative |
57 Positive (26.8%) 126 Negative 29 No genetic testing |
17 Positive (15.7%) 91 Negative |
23 Positive (21.9%) 82 Negative |
58 Positive (54.7%) 17 Negative 31 No genetic testing |
13 Positive (32.5%) 27 Negative |
60 Positive (14.5%) 355 Negative |
| Number of symptomatic patients at diagnosis (%) | 56 (28) | 89 (42) | 65 (60) | 130 (73.8) | 21 (20) | 40 (100) | 187 (45) |
| Endpoints | Documented VF or sudden death | SCD or documented VF | Appropriate ICD shock therapy | Appropriate ICD shock therapy | SD, documented VT or VF, appropriate ICD shock | Appropriate ICD shock therapy | Appropriate ICD shock, aborted cardiac arrest, or SCD |
| Average follow‐up | Mean 34 ± 44 months | Mean 40 ± 50 months | Mean 71.9 ± 41.3 months | Mean 83.8 ± 57.3 | Median 54 months (1st–3rd quartile 15–99) | Median 24 months (range 13–52 months) | Mean 72 months (range 1–249 months) |
| Conclusion by authors | SCN5A mutation was not associated with a higher risk of events and showed 32% sensitivity and 57% specificity to identify patients with cardiac arrest | Previous histories of aborted SCD and syncope were predictors for adverse outcome | SCN5A mutation is not associated with initial episodes of VF in BS, but is associated with early and frequent recurrence of VF in symptomatic patients | Risk stratification by means of electrophysiology study might identify asymptomatic patients at risk for arrhythmic events | SCN5A mutation may be necessary but is insufficient on its own for the development of lethal arrhythmia | R1193Q variant may be a genetic marker for ventricular arrhythmia in symptomatic BrS patients with ICD treatment | BrS patients with SCN5A mutations exhibit more conduction abnormalities on ECG and have higher risk for cardiac events |
| NOS | 7 | 9 | 7 | 8 | 8 | 8 | 6 |
BrP: Brugada pattern; BrS: Brugada syndrome; DHPLC: denaturing high‐performance liquid chromatography; ECG: Electrocardiogram; ICD: Implantable cardioverter defibrillator; NOS: Newcastle‐Ottawa Scale; SCD: sudden cardiac death; SD: Sudden death; SSCA: single‐strand conformational polymorphism analysis; VF: Ventricular fibrillation; VT: ventricular tachycardia.
3.2. Meta‐analysis results
Seven studies from March 2002 to October 2017 were included in this meta‐analysis involving 1,049 subjects with BrS (302 patients with SCN5A mutations and 747 patients without SCN5A mutations). Five studies revealed an increased MAE among BrS patients with SCN5A mutations (Andorin et al., 2016; Conte et al., 2015; Makarawate et al., 2017; Nishii et al., 2010; Yamagata et al., 2017) with one of the five studies (Makarawate et al., 2017) achieving statistical significance. The pooled analysis demonstrated a nonsignificant increased risk of MAE in BrS patients with SCN5A mutations compared to those without the mutation, with a pooled RR of 1.50 (95% CI: 0.93–2.41, p = 0.10, I 2 = 38.0%). A forest plot of this meta‐analysis is shown in Figure 2.
Figure 2.
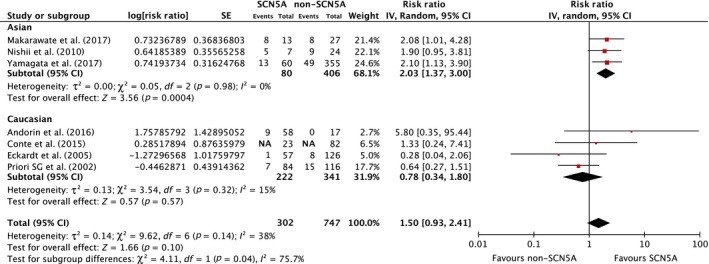
Forest plot of the included studies assessing the association between SCN5A mutation and major arrhythmic events and fatal arrhythmia among Asian, Caucasian, and overall analysis
In subgroup analysis among ethnicities, three studies (two studies from Japan and one study from Thailand) were included in Asian populations (Makarawate et al., 2017; Nishii et al., 2010; Yamagata et al., 2017) involving 486 subjects with BrS (80 patients with SCN5A mutations and 406 patients without SCN5A mutations). All three studies revealed increased MAE among BrS patients with SCN5A mutations (Makarawate et al., 2017; Nishii et al., 2010; Yamagata et al., 2017) with one study (Makarawate et al., 2017) achieving statistical significance. The pooled analysis demonstrated a significant increased risk of MAE in Asian BrS patients with SCN5A mutations compared to those without the mutation (RR = 1.78, 95% CI: 1.23–2.58, p = 0.002, I 2 = 0%). A forest plot of this meta‐analysis is shown in Figure 2.
In Caucasian cohorts, four studies were included in the analysis (Andorin et al., 2016; Conte et al., 2015; Eckardt et al., 2005; Priori et al., 2002) involving 563 subjects with BrS (222 patients with SCN5A mutations and 341 patients without SCN5A mutations). One study revealed a nonsignificant increase in MAE among BrS patients with SCN5A mutations (Andorin et al., 2016). The pooled analysis did not demonstrate an increased risk of MAE in Caucasian BrS patients with SCN5A mutations compared to those without the mutation (RR = 0.78, 95% CI: 0.34–1.80, p = 0.57, I 2 = 15%). A forest plot of this meta‐analysis is shown in Figure 2.
In symptomatic BrS patients, there were four studies (Andorin et al., 2016; Makarawate et al., 2017; Nishii et al., 2010; Yamagata et al., 2017) involving 271 subjects with BrS (60 patients with SCN5A mutations and 211 patients without SCN5A mutations). Every study revealed an increased MAE among symptomatic BrS patients with SCN5A mutations with two studies (Makarawate et al., 2017) achieving statistical significance. The pooled analysis demonstrated a significant increased risk of MAE in Asian BrS patients with SCN5A mutations compared to those without the mutation (RR = 1.78, 95% CI: 1.23–2.58, p = 0.0001, I 2 = 23%). A forest plot of this meta‐analysis is shown in Figure 3a. The pooled analysis of asymptomatic BrS patient showed an increased but non‐significant risk of MAE (RR = 1.85, 95% CI: 0.60–5.68, p = 0.28, I 2 = 0%). However, only two studies involving 290 subjects with asymptomatic BrS (78 patients with SCN5A mutations and 212 patients without SCN5A mutations) reported data suitable for meta‐analysis (Figure 3b).
Figure 3.
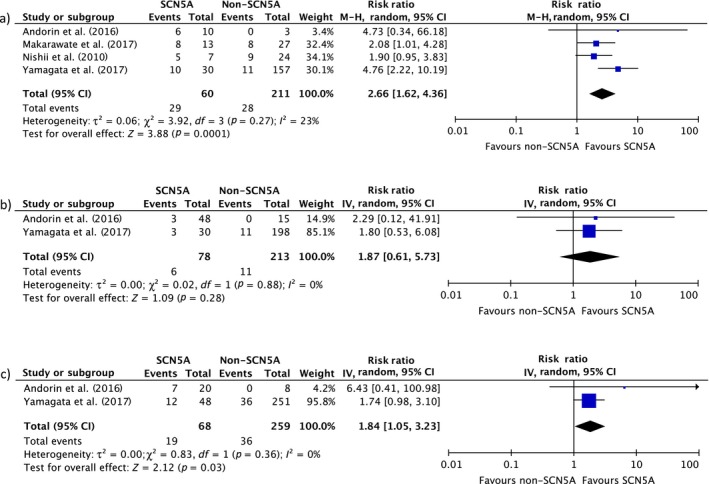
Forest plot of the included studies assessing the association between SCN5A mutation and major arrhythmic events and fatal arrhythmia in subgroup analysis of (a) symptomatic BrS, (b) asymptomatic BrS, and (c) spontaneous type‐1 BrS
Two studies reported on spontaneous type‐1 Brugada pattern (Andorin et al., 2016; Yamagata et al., 2017) involving 327 subjects with BrS and SCN5A status (68 patients with SCN5A mutations and 259 patients without SCN5A mutations). The pooled analysis demonstrated a significant increased risk of MAE in BrS patients who presented with spontaneous type‐1 Brugada pattern with SCN5A mutations compared to those without the mutation (RR = 1.84, 95% CI: 1.05–3.23, p = 0.03, I 2 = 0.0%). A forest plot of this meta‐analysis is shown in Figure 3c.
3.3. Sensitivity analysis
To assess the stability of the result, we conducted a sensitivity analysis by omitting one study at a time. We used a sequential omitting strategy, as described by Patsopoulos and colleagues, to examine whether overall estimates were influenced by the substantial heterogeneity observed (Patsopoulos et al., 2008). In the overall analysis, when omitting the study reported by Eckardt et al., the pooled analysis demonstrated a significantly increased risk of MAE in BrS patients with SCN5A mutations compared to those without the mutation, with a pooled RR of 1.65 (95% CI: 1.09–2.51, p = 0.019, I 2 = 24.3%) as well as the study reported by Priori et al. 1.89 (95% CI: 1.31–2.74, p = 0.001, I 2 = 0.0%) (Figure 5). In the subgroup analysis, none of the results was significantly altered.
Figure 5.
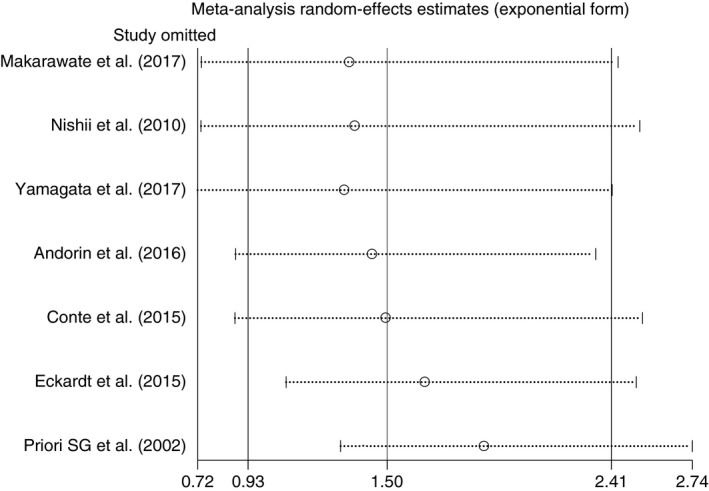
Sensitivity analysis graph to explore source of heterogeneity by omitting one study at a time
3.4. Publication bias
To investigate potential publication bias, we examined the contour‐enhanced funnel plot of the included studies in assessing change in the log odd ratio of death or composite outcome (Figure 4). The vertical axis represents study size (standard error) while the horizontal axis represents effect size (log odds ratio). The distribution of studies on both sides of the mean was symmetrical. Egger's test was nonsignificant for small‐study bias in overall analysis (p = 0.518), symptomatic BrS (p = 0.787), Caucasians (p = 0.756), and Asians (p = 0.095). Egger's test could not be performed for asymptomatic BrS, and spontaneous type‐1 pattern subgroups as there were only two studies. Meta‐regression confirmed that the studies published before 2010 were significant sources of heterogeneity (p = 0.035) but available verification status of SCN5A disease‐causing variants was not a significant source of heterogeneity (p = 0.968).
Figure 4.
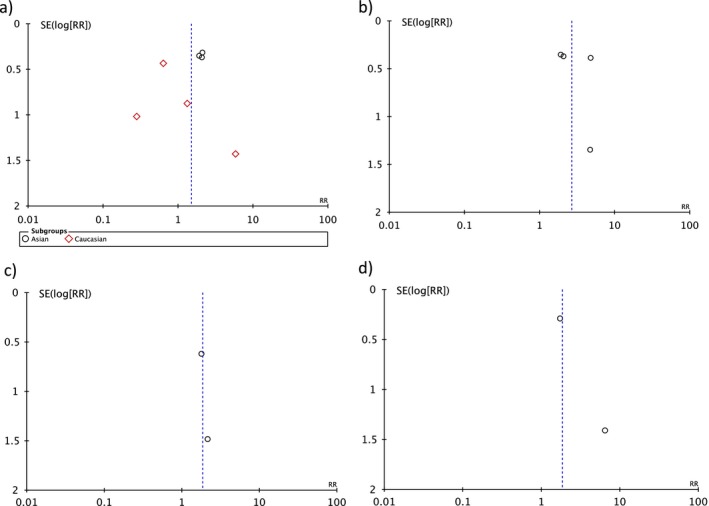
Funnel plot of SCN5A mutation and major arrhythmic events in (a) overall analysis, Asian, and Caucasian, (b) symptomatic, (c) asymptomatic, and (d) spontaneous type‐1. Circles represent observed published studies
4. DISCUSSION
We have analyzed 1,049 subjects with BrS from seven studies and showed an association between the presence of SCN5A mutations and risk for developing MAE in Asian population, patients with symptomatic BrS, and individuals with spontaneous type‐1 Brugada pattern. Increased but not statistically significant risk was found in Caucasians, all BrS individuals, and asymptomatic BrS subjects. The nonsignificant association in overall BrS may be due to interstudy and intrastudy demographic and genetic variations.
After performing subgroup analyses, the association between SCN5A status and MAE in some groups appeared more significant with decreased heterogeneities. Overall BrS individuals analysis showed moderate heterogeneity of 38.0%, whereas subgroups analysis showed low heterogeneity (0% in Asian, 23% in symptomatic, 0% in spontaneous type‐1, 15% in Caucasian, and 0% in asymptomatic). These results reflected the effect of individual basic characteristics to the outcome of MAE, which is more homogenized when analyzed by each subgroup.
Additionally, to explore the possible sources of heterogeneity in our meta‐analysis, we used sensitivity analysis by omitting one study at a time. When omitting the studies published by Eckardt et al. and Priori et al. from the overall analysis, we found a significantly increased risk of MAE in BrS patients with SCN5A mutations compared to those without the mutations, with a pooled RR of 1.65 (95% CI: 1.09–2.51, p = 0.019) and 1.89 (95% CI: 1.31–2.74, p = 0.001, I 2 = 0.0%), consecutively (Figure 5). Heterogeneity decreased from moderate (38.0%) to low (24.3%) when we omitted only Eckardt et al. and from moderate (38.0%) to none (0.0%) when we omitted only Priori et al. These sensitivity analysis results can be explained by several possible reasons. First, since the early publications in 2002 and 2005, more than 300 novel SCN5A mutations have been discovered (Juang & Horie, 2016). Hence, Eckardt et al. (2005) and Priori et al. (2002) may not have studied several mutations included in our study and meta‐regression confirmed that the studies published before 2010 are significant sources of heterogeneity (p = 0.035). Second, their study populations were mostly asymptomatic Caucasian individuals (58% in Eckardt et al. and 72% in Priori et al.). The subgroup analyses from our study indicated significant associations of SCN5A mutations and MAE in symptomatic and Asian groups, but not in the Caucasian group. These aforementioned factors are thus suggestive of existing heterogeneity interfering with the results from our analyses. The cause of heterogeneity is also noted in the study performed by Makarawate et al. (2017) which correlated SCN5A mutation status with cardiac conduction disturbances and resultant appropriate ICD shocks. Their study included a geographically and genetically isolated population: most patients were of northeastern Thai origin; only symptomatic patients were included; and only two polymorphisms were identified (R1193Q and H558R). A degree of pathogenicity of these two variants were questionable and may be more, or less, malignant than those reported in other studies.
It is well recognized that a history of aborted cardiac arrest is one of the strongest predictors for future MAE in BrS patients (Eckardt et al., 2005; Priori et al., 2002; Probst et al., 2010; Yamagata et al., 2017); in fact, the current guidelines recommends that those who survive episodes of cardiac arrest should undergo ICD implantation (Priori et al., 2013) as the risk of subsequent cardiac events was highest among this patient subgroup and was estimated as 7.7% per year in one study (Probst et al., 2010). On the other hand, the risk of MAE in asymptomatic BrS individuals is low, approximately 0.5% per year (Probst et al., 2010). Other reported potential risks include male sex, presence of spontaneous ST‐segment elevation in the precordial leads, positive electrophysiological study, presence of atrial fibrillation, and certain electrocardiographic conduction abnormalities such as prolonged P‐wave, prolonged QRS duration, and fragmented QRS (Chen et al., 1998; Gehi et al., 2006; Priori et al., 2002; Rattanawong, Riangwiwat, et al., 2017; Yamagata et al., 2017). Priori et al. demonstrated that the presence of both syncope and spontaneous ST‐segment elevation has a sensitivity of 36% and a high specificity of 94% in predicting the occurrence of cardiac arrest in BrS patients (Priori et al., 2002). On the contrary, many studies have assessed family history of sudden cardiac death as a predictor for poorer outcomes, and the results were reproducibly unrevealing (Gehi et al., 2006; Makarawate et al., 2017; Nishii et al., 2010; Priori et al., 2002). Clinicians have long been intrigued by the concept of using SCN5A mutation status to predict MAE in BrS patients.
Adler et al. (2016) have recently reviewed the risk stratification strategy in patients with BrS and stated that, according to the large registries, the use of genetic data to risk stratify BrS patients are not well‐defined and challenging. Even though a risk score based on the mutations and other polymorphisms has been developed (Sommariva et al., 2013), the authors suggested that the tool needs to be validated before being adopted. In 2006, Gehi et al. analyzed 383 patients from two publications (Eckardt et al., 2005; Priori et al., 2002) and found no link between SCN5A mutations and increased risk of sudden cardiac death, syncope, or ICD shock (relative risk 0.60, CI: 0.29–1.26). We speculate that the nonsignificant result in their study was due to a lower number of recruited patients, lower number of included studies, and limited power to identify a minimal increase in risk. To our knowledge, our study is the first meta‐analysis to demonstrate the potential utilization of SCN5A mutation in the risk stratification scheme, particularly in certain subgroups, of BrS.
SCN5A mutations were reported in approximately 14%–26% of BrS patients and known as the most common BrS‐associated gene (Chen et al., 1998; Yamagata et al., 2017). Almost 300 SCN5A mutations have been identified in BrS, including missense mutations, nonsense mutations, nucleotide insertion/deletions, and splice site mutations, and the number of SCN5A mutations is still increasing (Juang & Horie, 2016). An SCN5A mutation does not necessarily indicate BrS (Probst et al., 2009). The functional loss of NaV1.5 cardiac sodium channel with subsequent reduced sodium current is typically described in BrS patients with SCN5A mutations (Juang & Horie, 2016). This is supported by the fact that BrS‐associated SCN5A mutations usually result in frameshift errors, splice‐site defects, or premature stop codons that lead to nonfunctional channels (Chen et al., 1998). BrS‐causing missense mutations were observed to be nonfunctional due to either disrupted protein trafficking to the cell membrane or impaired sodium conductance (George, 2005). Although some missense mutations are functional, they may cause defective gated properties of the channels involving activation and/or inactivation kinetics (Andorin et al., 2016; Rook et al., 1999). Meregalli et al. corroborated this speculation by studying 147 mutation‐positive BrS individuals and divided them into three groups: (a) those with prematurely truncated proteins (group 1); (b) those with missense mutations resulting in significantly (>90%) reduced peak sodium current (group 2); and (c) those with missense mutations resulting in mildly (≤90%) reduced peak sodium current (group 3) (Meregalli et al., 2009). They found that patients in group 1 and group 2, in which drastic peak sodium current were noted, developed a more severe conduction disorders. The underlying electrophysiological mechanisms of how altered sodium current causes BrS are still under investigation, and two models have been proposed (Meregalli, Wilde, & Tan, 2005). In the “repolarization disorder” model, the defective sodium channel reduces the myocardial sodium current and causes a disproportionate shortening of the right ventricular epicardial action potentials, leading to an exaggerated transmural (i.e., epicardium‐to‐myocardium) voltage gradient and the characteristics finding on electrocardiogram (George, 2005; Juang & Horie, 2016; Smits et al., 2002). The “depolarization disorder” model theorizes that conduction delay in the right ventricular outflow tract, with respect to the right ventricle, causes the electrocardiogram changes in BrS. The arrhythmogenicity of BrS is likely multifactorial and other pathophysiology may play a role: for instance, a recent study proposed epicardial surface fibrosis and reduced gap junction expression in the right ventricular outflow tract as arrhythmogenic mechanisms in BrS (Nademanee et al., 2015).
When compared to SCN5A‐negative BrS patients, those with SCN5A mutations tend to exhibit significantly longer conduction intervals on electrocardiogram, such as PQ or His‐to‐ventricle intervals, and more fragmented QRS, both at baseline and throughout the follow‐up (Makarawate et al., 2017; Nishii et al., 2010; Rattanawong, Riangwiwat, et al., 2017; Smits et al., 2002; Yokokawa et al., 2007). These parameters were also predictive of the presence of the mutation: for example, PQ duration of ≥210 milliseconds had a sensitivity of 48% and a specificity of 98% for detecting SCN5A mutation in BrS patients (Smits et al., 2002). The prognostic value of SCN5A status has become more apparent in recent well‐designed studies. In a study of 415 BrS patients reported by Yamagata et al. (2017), SCN5A mutation carriers tended to experience their cardiac events more frequently and at younger ages. Apart from history of aborted cardiac arrest, harboring the mutation was the only independent predictor of MAE, with a hazard ratio of 2.0 (95% CI: 1.0–3.8). They also found that mutations in the pore region of the NaV1.5 cardiac sodium channel were more associated with MAE (Yamagata et al., 2017). Hence, these studies have confirmed the genotype‐phenotype correlations in SCN5A‐positive BrS individuals, both at electrocardiographic level and clinical level.
Recently, Nademanee et al. (2011) reported right ventricular outflow tract epicardial ablation in recurrent symptomatic BrS. The indication for right ventricular outflow tract epicardial ablation in symptomatic BrS is still unclear. Right ventricular outflow tract fibrosis and conduction delay were identified in carriers of SCN5A mutations (Meregalli et al., 2009). Moreover, age‐related fibrosis has also been seen in mouse models of SCN5A mutation (Jeevaratnam et al., 2012; Royer et al., 2005). Therefore, SCN5A mutations may contribute to substrate changes which may be treatable with epicardial ablation. In our study, we found that SCN5A mutations in symptomatic BrS is twofold associated with MAE compared to symptomatic Brugada syndrome without SCN5A mutations. SCN5A mutation status may therefore enhances risk stratification of symptomatic Brugada syndrome.
4.1. Limitations
Although most recruited studies were of high quality, we recognize there are some limitations to our analysis. First, the studies are heterogeneous as discussed earlier. The potential sources of heterogeneity include age and gender of participants, definitions of MAE in each study, follow‐up duration, inclusion of mutation‐positive screened family members or of asymptomatic carriers, geographic difference, and recruiting protocol (e.g., multicenter registry vs. single center). Second, genetic heterogeneity also existed among studies. For instance, Yamagata et al. (2017) identified 55 different mutations in 60 affected individuals in their multicenter cohort, whereas Makarawate et al. (2017) found only two different mutated alleles in 13 SCN5A mutation carriers. However, since BrS is uncommon and large‐scale genetic studies have been rarely performed, the possibility of small‐study bias due to the small number of included studies and small sample size is not negligible. A larger study with a more homogeneous population is needed to confirm our results. Unfortunately, there was not enough information reported in two articles that we could use to calculate multivariate adjusted RR. Risk ratios were calculated based on number of the patients without multivariate adjustment. Third, large genomic imbalances, such as copy‐number variants (CNVs) in SCN5A, were recently shown to underlie a portion of genotype‐negative patients and should be screened (Mademont‐Soler et al., 2016; Sonoda et al., 2018). However, all of the recruited studies have used traditional methods of sequencing which could not detect CNVs; hence, the control group may include those CNV‐harboring sequencing‐negative patients and did not truly represent unaffected individuals. Fourth, three (Makarawate et al., 2017; Priori et al., 2013; Yamagata et al., 2017) of seven studies reported information on how the authors verified if the variants were disease‐causing (Table 1); however, meta‐regression confirmed that verification status of SCN5A disease‐causing variants was not a significant source of heterogeneity in overall results. Lastly, this is a meta‐analysis of observational studies with the inherent limitation of being able to confirm an association, but not a causal relationship.
5. CONCLUSION
From our study, we found that mutation status may help predict MAE and guide treatment decisions in certain subgroups of BrS, especially in Asian population, symptomatic patients, and individuals with spontaneous type‐1 Brugada pattern. The presence of SCN5A mutations may be an important tool to prognosticate risk and guide treatment in patients with BrS in the future.
CONFLICT OF INTEREST
None to declare.
ACKNOWLEDGMENT
We would like to thank Associate Professor Ammarin Thakkinstian, Ph.D. for statistical consultation.
Rattanawong P, Chenbhanich J, Mekraksakit P, et al. SCN5A mutation status increases the risk of major arrhythmic events in Asian populations with Brugada syndrome: systematic review and meta‐analysis. Ann Noninvasive Electrocardiol. 2019;24:e12589 10.1111/anec.12589
[Correction added on 05 September, 2018, after first online publication: The article title has been amended from ‘SCN5A Mutation Increases the Risk of Major Arrhythmic Events in Asian Population of Brugada Syndrome: Systematic Review and Meta‐analysis’ has been changed to ‘SCN5A mutation status increases the risk of major arrhythmic events in Asian populations with Brugada syndrome: systematic review and meta‐analysis’.]
REFERENCES
- Adler, A. , Rosso, R. , Chorin, E. , Havakuk, O. , Antzelevitch, C. , & Viskin, S. (2016). Risk stratification in Brugada syndrome: Clinical characteristics, electrocardiographic parameters, and auxiliary testing. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 13(1), 299–310. 10.1016/j.hrthm.2015.08.038 [DOI] [PubMed] [Google Scholar]
- Andorin, A. , Behr, E. R. , Denjoy, I. , Crotti, L. , Dagradi, F. , Jesel, L. , … Probst, V. (2016). Impact of clinical and genetic findings on the management of young patients with Brugada syndrome. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 13(6), 1274–1282. 10.1016/j.hrthm.2016.02.013 [DOI] [PubMed] [Google Scholar]
- Brugada, P. , & Brugada, J. (1992). Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. Journal of the American College of Cardiology, 20(6), 1391–1396. 10.1016/0735-1097(92)90253-J [DOI] [PubMed] [Google Scholar]
- Chen, Q. , Kirsch, G. E. , Zhang, D. , Brugada, R. , Brugada, J. , Brugada, P. , … Wang, Q. (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature, 392(6673), 293–296. 10.1038/32675 [DOI] [PubMed] [Google Scholar]
- Conte, G. , Sieira, J. , Ciconte, G. , de Asmundis, C. , Chierchia, G. B. , Baltogiannis, G. , … Brugada, P. (2015). Implantable cardioverter‐defibrillator therapy in Brugada syndrome: A 20‐year single‐center experience. Journal of the American College of Cardiology, 65(9), 879–888. 10.1016/j.jacc.2014.12.031 [DOI] [PubMed] [Google Scholar]
- DerSimonian, R. , & Laird, N. (1986). Meta‐analysis in clinical trials. Controlled Clinical Trials, 7(3), 177–188. 10.1016/0197-2456(86)90046-2 [DOI] [PubMed] [Google Scholar]
- Eckardt, L. , Probst, V. , Smits, J. P. , Bahr, E. S. , Wolpert, C. , Schimpf, R. , … Wilde, A. A. (2005). Long‐term prognosis of individuals with right precordial ST‐segment‐elevation Brugada syndrome. Circulation, 111(3), 257–263. 10.1161/01.CIR.0000153267.21278.8D [DOI] [PubMed] [Google Scholar]
- Gehi, A. K. , Duong, T. D. , Metz, L. D. , Gomes, J. A. , & Mehta, D. (2006). Risk stratification of individuals with the Brugada electrocardiogram: A meta‐analysis. Journal of Cardiovascular Electrophysiology, 17(6), 577–583. 10.1111/j.1540-8167.2006.00455.x [DOI] [PubMed] [Google Scholar]
- George, A. L. Jr (2005). Inherited disorders of voltage‐gated sodium channels. Journal of Clinical Investigation, 115(8), 1990–1999. 10.1172/JCI25505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins, J. P. , Thompson, S. G. , Deeks, J. J. , & Altman, D. G. (2003). Measuring inconsistency in meta‐analyses. BMJ, 327(7414), 557–560. 10.1136/bmj.327.7414.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevaratnam, K. , Rewbury, R. , Zhang, Y. , Guzadhur, L. , Grace, A. A. , Lei, M. , & Huang, C. L. (2012). Frequency distribution analysis of activation times and regional fibrosis in murine Scn5a+/‐ hearts: The effects of ageing and sex. Mechanisms of Ageing and Development, 133(9–10), 591–599. 10.1016/j.mad.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang, J. J. , & Horie, M. (2016). Genetics of Brugada syndrome. Journal of Arrhythmia, 32(5), 418–425. 10.1016/j.joa.2016.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamakura, S. (2013). Epidemiology of Brugada syndrome in Japan and rest of the world. Journal of Arrhythmia, 29(2), 52–55. 10.1016/j.joa.2013.01.004 [DOI] [Google Scholar]
- Mademont‐Soler, I. , Pinsach‐Abuin, M. L. , Riuro, H. , Mates, J. , Perez‐Serra, A. , Coll, M. , … Brugada, R. (2016). Large genomic imbalances in Brugada syndrome. PLoS One, 11(9), e0163514 10.1371/journal.pone.0163514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarawate, P. , Chaosuwannakit, N. , Vannaprasaht, S. , Sahasthas, D. , Koo, S. H. , Lee, E. J. D. , … Sawanyawisuth, K. (2017). SCN5A genetic polymorphisms associated with increased defibrillator shocks in Brugada syndrome. Journal of the American Heart Association, 6(6), 1–7. 10.1161/jaha.116.005009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meregalli, P. G. , Tan, H. L. , Probst, V. , Koopmann, T. T. , Tanck, M. W. , Bhuiyan, Z. A. , … Wilde, A. A. (2009). Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss‐of‐function sodium channelopathies. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 6(3), 341–348. 10.1016/j.hrthm.2008.11.009 [DOI] [PubMed] [Google Scholar]
- Meregalli, P. G. , Wilde, A. A. , & Tan, H. L. (2005). Pathophysiological mechanisms of Brugada syndrome: Depolarization disorder, repolarization disorder, or more? Cardiovascular Research, 67(3), 367–378. 10.1016/j.cardiores.2005.03.005 [DOI] [PubMed] [Google Scholar]
- Nademanee, K. , Raju, H. , de Noronha, S. V. , Papadakis, M. , Robinson, L. , Rothery, S. , … Behr, E. R. (2015). Fibrosis, connexin‐43, and conduction abnormalities in the Brugada syndrome. Journal of the American College of Cardiology, 66(18), 1976–1986. 10.1016/j.jacc.2015.08.862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nademanee, K. , Veerakul, G. , Chandanamattha, P. , Chaothawee, L. , Ariyachaipanich, A. , Jirasirirojanakorn, K. , … Ngarmukos, T. (2011). Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation, 123(12), 1270–1279. 10.1161/CIRCULATIONAHA.110.972612 [DOI] [PubMed] [Google Scholar]
- Nishii, N. , Ogawa, M. , Morita, H. , Nakamura, K. , Banba, K. , Miura, D. , … Ito, H. (2010). SCN5A mutation is associated with early and frequent recurrence of ventricular fibrillation in patients with Brugada syndrome. Circulation Journal, 74(12), 2572–2578. 10.1253/circj.CJ-10-0445 [DOI] [PubMed] [Google Scholar]
- Patsopoulos, N. A. , Evangelou, E. , & Ioannidis, J. P. (2008). Sensitivity of between‐study heterogeneity in meta‐analysis: Proposed metrics and empirical evaluation. International Journal of Epidemiology, 37(5), 1148–1157. 10.1093/ije/dyn065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori, S. G. , Napolitano, C. , Gasparini, M. , Pappone, C. , Della Bella, P. , Giordano, U. , … Nastoli, J. (2002). Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation, 105(11), 1342–1347. 10.1161/hc1102.105288 [DOI] [PubMed] [Google Scholar]
- Priori, S. G. , Wilde, A. A. , Horie, M. , Cho, Y. , Behr, E. R. , Berul, C. , … Tracy, C. (2013). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 10(12), 1932–1963. 10.1016/j.hrthm.2013.05.014 [DOI] [PubMed] [Google Scholar]
- Probst, V. , Veltmann, C. , Eckardt, L. , Meregalli, P. G. , Gaita, F. , Tan, H. L. , … Wilde, A. A. (2010). Long‐term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation, 121(5), 635–643. 10.1161/circulationaha.109.887026 [DOI] [PubMed] [Google Scholar]
- Probst, V. , Wilde, A. A. , Barc, J. , Sacher, F. , Babuty, D. , Mabo, P. , … Schott, J. J. (2009). SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circulation: Cardiovascular Genetics, 2(6), 552–557. 10.1161/CIRCGENETICS.109.853374 [DOI] [PubMed] [Google Scholar]
- Rattanawong, P. , Ngarmukos, T. , Chung, E. H. , Vutthikraivit, W. , Putthapiban, P. , Sukhumthammarat, W. , … Sritara, P. (2017). Prevalence of Brugada ECG pattern in Thailand from a population‐based cohort study. Journal of the American College of Cardiology, 69(10), 1355–1356. 10.1016/j.jacc.2016.12.028 [DOI] [PubMed] [Google Scholar]
- Rattanawong, P. , Riangwiwat, T. , Prasitlumkum, N. , Limpruttidham, N. , Kanjanahattakij, N. , Chongsathidkiet, P. , … Chung, E. H. (2017). Baseline fragmented QRS increases the risk of major arrhythmic events in Brugada syndrome: Systematic review and meta‐analysis. Annals of Noninvasive Electrocardiology, 23(2), e12507 10.1111/anec.12507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattanawong, P. , Vutthikraivit, W. , Charoensri, A. , Jongraksak, T. , Prombandankul, A. , Kanjanahattakij, N. , … Ngarmukos, T. (2016). Fever‐induced Brugada syndrome is more common than previously suspected: A cross‐sectional study from an endemic area. Annals of Noninvasive Electrocardiology, 21(2), 136–141. 10.1111/anec.12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook, M. B. , Bezzina Alshinawi, C. , Groenewegen, W. A. , van Gelder, I. C. , van Ginneken, A. C. , Jongsma, H. J. , … Wilde, A. A. (1999). Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovascular Research, 44(3), 507–517. 10.1016/S0008-6363(99)00350-8 [DOI] [PubMed] [Google Scholar]
- Royer, A. , van Veen, T. A. , Le Bouter, S. , Marionneau, C. , Griol‐Charhbili, V. , Leoni, A. L. , … Charpentier, F. (2005). Mouse model of SCN5A‐linked hereditary Lenegre's disease: Age‐related conduction slowing and myocardial fibrosis. Circulation, 111(14), 1738–1746. 10.1161/01.CIR.0000160853.19867.61 [DOI] [PubMed] [Google Scholar]
- Smits, J. P. , Eckardt, L. , Probst, V. , Bezzina, C. R. , Schott, J. J. , Remme, C. A. , … Wilde, A. A. (2002). Genotype‐phenotype relationship in Brugada syndrome: Electrocardiographic features differentiate SCN5A‐related patients from non‐SCN5A‐related patients. Journal of the American College of Cardiology, 40(2), 350–356. 10.1016/S0735-1097(02)01962-9 [DOI] [PubMed] [Google Scholar]
- Sommariva, E. , Pappone, C. , Martinelli Boneschi, F. , Di Resta, C. , Rosaria Carbone, M. , Salvi, E. , … Benedetti, S. (2013). Genetics can contribute to the prognosis of Brugada syndrome: A pilot model for risk stratification. European Journal of Human Genetics, 21(9), 911–917. 10.1038/ejhg.2012.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda, K. , Ohno, S. , Ozawa, J. , Hayano, M. , Hattori, T. , Kobori, A. , … Horie, M. (2018). Copy number variations of SCN5A in Brugada syndrome. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 15(8), 1179–1188. 10.1016/j.hrthm.2018.03.033 [DOI] [PubMed] [Google Scholar]
- Stang, A. (2010). Critical evaluation of the Newcastle‐Ottawa scale for the assessment of the quality of nonrandomized studies in meta‐analyses. European Journal of Epidemiology, 25(9), 603–605. 10.1007/s10654-010-9491-z [DOI] [PubMed] [Google Scholar]
- Sterne, J. A. , & Egger, M. (2001). Funnel plots for detecting bias in meta‐analysis: Guidelines on choice of axis. Journal of Clinical Epidemiology, 54(10), 1046–1055. 10.1016/S0895-4356(01)00377-8 [DOI] [PubMed] [Google Scholar]
- Yamagata, K. , Horie, M. , Aiba, T. , Ogawa, S. , Aizawa, Y. , Ohe, T. , … Shimizu, W. (2017). Genotype‐phenotype correlation of SCN5A mutation for the clinical and electrocardiographic characteristics of probands with Brugada syndrome: A Japanese Multicenter Registry. Circulation, 135(23), 2255–2270. 10.1161/circulationaha.117.027983 [DOI] [PubMed] [Google Scholar]
- Yokokawa, M. , Noda, T. , Okamura, H. , Satomi, K. , Suyama, K. , Kurita, T. , … Shimizu, W. (2007). Comparison of long‐term follow‐up of electrocardiographic features in Brugada syndrome between the SCN5A‐positive probands and the SCN5A‐negative probands. American Journal of Cardiology, 100(4), 649–655. 10.1016/j.amjcard.2007.03.078 [DOI] [PubMed] [Google Scholar]