

Abstract
Background
Andersen–Tawil Syndrome (ATS) is a channelopathy caused by mutations in KCNJ2 gene. It is characterized by symptoms of ventricular arrhythmias, periodic paralysis or muscle weakness, and dysmorphic features. ATS can present with the triad of symptoms, any combination or none of them. Risk factors for dangerous arrhythmias are unknown. The study assessed the impact of K897T polymorphism in hERG1 gene and H558R polymorphism in SCN5A gene coexisting with R218Q mutation in KCNJ2 in one family on clinical manifestation.
Methods
Family members underwent clinical assessment, ECG and genotyping. Holter monitoring was performed in mutation carriers and additionally in one family member with no mutation, but with K897T polymorphism.
Results
Proband with ATS mutation, K897T and H558R polymorphisms and proband's sister with ATS mutation and K897T polymorphism presented following symptoms: loss of consciousness, bidirectional and polymorphic ventricular tachycardia and about 5000 ventricular extrasystoles. Symptoms presented by the member with only the ATS mutation and by member with ATS mutation and H558R polymorphism were not as severe. U wave appeared in all examined family members regardless of the mutation presence. Studied individuals with ATS mutation had the T‐peak–U‐peak interval longer than 200 ms. In all ATS mutation carriers it was longer than in family members with no mutation. T‐peak–T‐end interval was the longest (>120 ms) in members with coexisting mutation and K897T polymorphism.
Conclusion
ATS severity possibly depends on other genes’ polymorphisms. In the presented family, it could depend on the presence of K897T polymorphism in hERG1.
Keywords: electrophsiology—long QT syndrome, clinical, molecular biology/genetics basic
Long QT Syndrome 7 (LQT7 syndrome), known as an Andersen–Tawil Syndrome (ATS), is a potassium channelopathy caused by an autosomal dominant mutation in the KCNJ2 gene. It is characterized by the triad of symptoms: ventricular arrhythmias, periodic paralysis or muscle weakness, and dysmorphic features of face and fingers.1 Penetration and expression of ATS mutations are variable. Some patients present with the whole triad of symptoms, the combination or none of them.2 We describe a family, with the R218Q mutation in the KCNJ2 gene (herein refered as the “mutation”) coexisting with the K897T polymorphism in the human ether‐a‐go‐go related (hERG1) gene and the H558R polymorphism in the SCN5A gene. To the best of our knowledge, to date, there are no published reports on such families.
The K897T polymorphism in the hERG1 gene has been reported as an important modifier of the IKr current, probably leading to QT interval prolongation—as it is observed in the LQT2 syndrome.3 The H558R polymorphism in the SCN5A gene has been associated with the LQT3 syndrome and possibly can influence repolarization and depolarization of cardiac muscle cells.4 In the case when ATS mutation coexists with the K897T polymorphism in the hERG1 and the H558R polymorphism in the SCN5A the risk of ventricular arrhythmias and sudden cardiac death (SCD) remains unknown.
METHODS
Family members of the patient with the ATS mutation, the K897T polymorphism in the hERG1 gene and the H558R polymorphism in the SCN5A gene underwent clinical assessment, 12 leads ECG and genotyping. Holter ECG monitoring was performed in all studied carriers of the ATS mutation and additionally one family member without the ATS, but with the K897T polymorphism in the hERG1 gene. The family tree has been presented in Figure 1.
Figure 1.
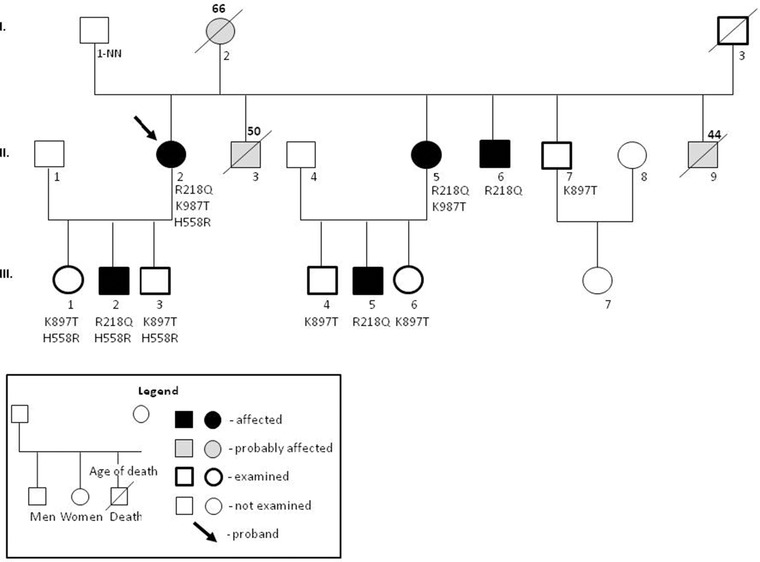
Family tree.
The Methodology of Genotyping
Ten milliliters of blood was collected in EDTA. The genomic DNA was isolated by phenol extraction. Screening for the mutations of three major genes causing LQTS: KCNQ1, KCNH2, and SCN5A was performed by polymerase chain reaction using exon‐flanking intronic primers designed by our laboratory. Amplicons were analyzed by the next generation sequencing with the GS Junior System (Roche) according to Amplicon Library Preparation Method Manual. Data was analyzed using GS Amplicon Variant Analyzer (AVA) software (Roche). We performed a direct sequencing analysis of the whole translated region of the KCNJ2 gene. DNA was amplified using site‐specific primers. Amplicons were subjected to bidirectional capillary‐based sequencing using 3130XL Genetic Analyzer, Applied Biosystems. Data analysis was done using Applied Biosystems DNA Sequencing Analysis Software version 5.3.1 (Applied Biosystems, Foster City, CA94404 USA).
RESULTS
The proband (II 2) was a 56‐year‐old woman (height: 154 cm, weight: 50 kg). In childhood, she was treated for epilepsy. She presented mandibular hypoplasia, small hands, and clinodactyly. Moreover, she was experiencing sudden, transient paralysis episodes manifested as falls associated with stress and physical activity since the age of 11. She had U wave on ECG. QTc measured 410 ms and QTUc measured 69 ms. Holter study detected premature ventricular contractions (PVC) at 4882/24 hours. Additionally, episodes of polymorphic ventricular tachycardia (PVT) and bidirectional ventricular tachycardia (BiVT) were recorded (Fig. 2).
Figure 2.
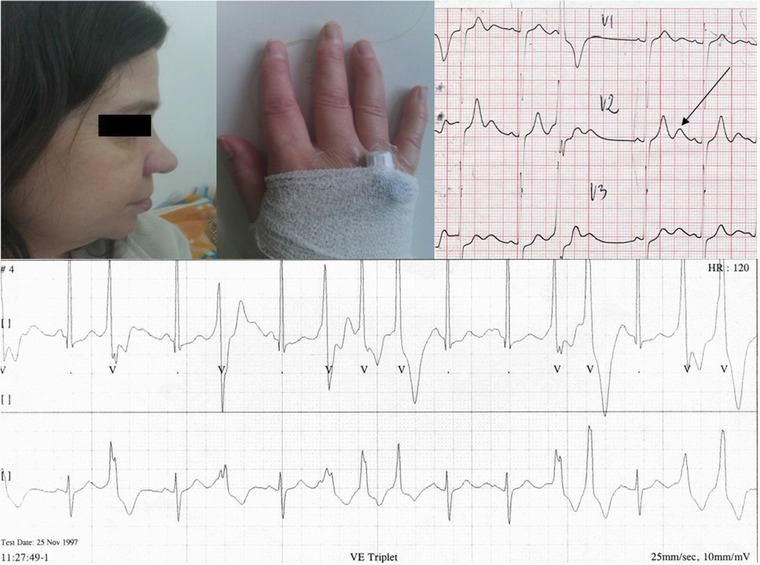
(Right upper panel) Proband's photos. Mandibular hypoplasia and clinodactyly. (Left upper panel) Proband's electrocardiogram. Right precordial leads V1–V3. Sinus rhythm, premature ventricle contractions (PVC) with RBBB morphology. Normal QT/QTc interval: 410 ms, prolonged QT + U interval: 690 ms. Visible marked U wave (tall and broad), with postextrasystolic U wave augmentation (up to 4 mm) and U‐wave alternans. The large U wave is observed in PVC. (Bottom panel) ECG Holter strip. Premature ventricle contractions (PVC), bigeminy and triplet. All PVCs begins on the top of the U wave. The U wave is separated from T wave (see after the 4th sinus beat). “U on P” sign (U wave masquerading P wave) (after the 4th sinus beat). Pseudo “Tee‐pee sign.” During a PVC, there is a prolongation of the descending limb of the T + U wave (see the 1st and 2nd PVCs).
Due to arrhythmia and episodes of loss of consciousness, she was diagnosed with LQTS at the age of 21. Beta‐blockers were started. ICD has been implanted because beta‐blockers did not prevent episodes of loss of consciousness. Interestingly, she had no therapeutic ICD discharges, but she kept losing consciousness. Therefore, although epilepsy diagnosis was not confirmed, the phenytoin was added to her therapy with beta‐blockers with satisfactory results. Genetic testing revealed the K897T polymorphism in the hERG1 gene and the H558R polymorphism in the SCN5A gene. Her typical phenotype of ATS has led to genetic testing, which revealed the R218Q mutation in the KCNJ2.
The H558R polymorphism in the SCN5A gene was detected in all children of the proband. Moreover, K897T polymorphism in the hERG1 gene was inherited by the proband's daughter (III 1) and son (III 3), who both presented no symptoms through the first and the second decades of their lives. The ATS mutation was also inherited by her second son (III 2) who had mandibular hypoplasia, palpitations, and episodes of muscle weakness. His Holter study revealed 132 PVCs in 24 hours and one episode of PVT. Moreover, ADHD was diagnosed in both sons (III 2 and III 3). The interpretation of the inheritance pattern at the level of proband's parents is at least troublesome if not impossible. The proband's father (I 1) was unknown. Mother (I 2) died suddenly at the age of 66, after several syncopal episodes and pacemaker implantation. There is high probability that the proband's mother had the ATS. She had characteristic dysmorphic features and a history of syncope—as described by the proband. She also had U wave in ECG. The ATS mutation was also detected in the proband's stepsister (II 5), who shares the same mother, but has a different father, which makes this hypothesis even more likely. The K897T polymorphism in the SCN5A gene was also detected in a proband's stepsister (II 5) with the ATS mutation. She presented with the typical ATS phenotype, had a history of palpitations, U‐wave on ECG, muscle weakness, and multiple syncopal episodes. Her Holter study revealed 4284 PVCs, BiVT, and PVT (Fig. 3). Her son, who inherited the ATS‐mutation (III 5), had clinodactyly and was reported to have 74 PVCs in the Holter study. The proband's stepbrother, with the ATS mutation (II6), had syndactyly of toes and low‐set ears. He also reported a history of episodic forearms numbness. His Holter study revealed 119 PVCs in 24 hours, episodes of bigeminy and trigeminy. The stepbrother with the K897T polymorphism in the hERG1 gene (II 7) had only one episode of atrial fibrillation in the Holter study. Two proband's stepbrothers died suddenly at night (II 3, II 9) at the age of 50 and 44 years, respectively. The first had a history of alcohol abuse. His appearance had typical features of the ATS dysmorphia, in particular: small mandible, hypertelorism, and clinodactyly. We have no relevant information about the second stepbrother.
Figure 3.
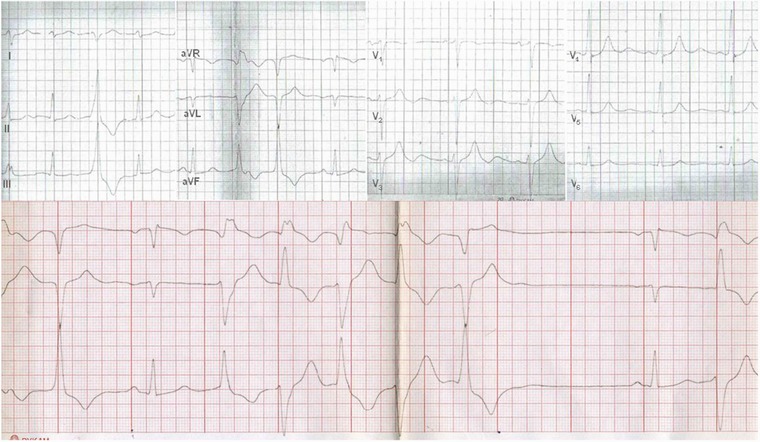
Proband's sister electrocardiogram. (Upper panel) Sinus rhythm, 75 bpm. Premature ventricle contractions. QTc interval: 425 ms, prolonged descending portion of T wave is best seen in leads V2–V3. Marked U wave is seen in leads V2–V6. The U wave is tall (with the amplitude of 4 mm), and long lasting: 250 ms. (Bottom panel) Sinus rhythm with slow bidirectional nonsustained ventricular tachycardia (150 bpm).
The summary of genotype, phenotype, clinical data, ECG and Holter studies’ findings of the family members who, at the time of this study were still alive, are presented in the Table 1.
Table 1.
Clinical and Genetic Data on Family Members
| II 2 | II 5 | II 6 | II 7 | III 1 | III 2 | III 3 | III 4 | III 5 | III 6 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age | 52 | 39 | 45 | 41 | 29 | 20 | 18 | 11 | 8 | 5 |
| R218Q | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| K897T | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 0 | 1 |
| H558R | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 |
| QT | 440 | 400 | 340 | 360 | 360 | 396 | 378 | 300 | 330 | 300 |
| QTc | 431 | 459 | 366 | 413 | 402 | 397 | 385 | 381 | 441 | 416 |
| QTU | 640 | 600 | 600 | 560 | 520 | 660 | 560 | 480 | 530 | 320 |
| QTUc | 628 | 688 | 666 | 642 | 581 | 667 | 560 | 566 | 684 | 585 |
| U wave | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Leads | V1–V4 | V2–V5 | V2–V3 | V2–V3 | V2 | V1–V5 | V2–V3 | V3 | V2–V4 | V2 |
| U wave inferior | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| U wave V2–V4 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 |
| U wave V5 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| U wave time | 180 | 200 | 180 | 160 | 160 | 240 | 140 | 100 | 160 | 80 |
| T‐peak–T‐end | 130 | 150 | 100 | 100 | 100 | 120 | 110 | 80 | 120 | 90 |
| T‐peak–U‐peak | 230 | 250 | 220 | 200 | 200 | 230 | 200 | 130 | 220 | 80 |
| VEx/24 h >1000 | Yes | Yes | No | No | – | No | – | – | No | – |
| VT bidirectional | Yes | Yes | No | No | – | No | – | – | No | – |
| VT polymorphic | Yes | Yes | No | No | – | Yes | – | – | No | – |
| AF | No | No | No | Yes | – | No | – | – | No | – |
| Dysmorphic features | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| Neurological symptoms | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Loss of consciousness | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
DISCUSSION
LQTS belong to the group of channelopathies. In these diseases, mutations disrupt the assembly and function of the proteins of the ion channels.5 The mutations causing ATS are located in the KCNJ2 gene. KCNJ2 encodes the α‐subunit of the potassium channel Kir2.1, which is a component of the inward rectifier IK1 current.6, 7 Multiple controversies about the ATS involve the risk assessment (i.e., predictors of the occurrence of dangerous arrhythmias), treatment and ECG interpretation.8 Studying the risks of cardiac arrest and sudden cardiac death (SCD) is difficult due to the lack of reliable databases and low prevalence of the ATS itself. Establishing the methods of risk assessment is crucial, so proper treatment and possibly primary prevention (i.e., ICD implantation) can be implemented. However, the risk of SCD in ATS has been estimated as extremely low.9, 10 ATS patients with a history of BiVT or PVT are at a higher risk of developing dangerous arrhythmias. On the other hand, there are carriers of the ATS mutation with no symptoms at all. Consequently, we postulate, that there are modifying factors, such as genetic polymorphisms, which allow ATS specific mutations to produce symptoms.
The mutations in the hERG1 gene, which encodes the α‐subunit of the voltage‐gated potassium ion channel (named Kv11.1) are responsible for the LQT2 syndrome. As a result, the delayed rectifier IKr current is reduced during the plateau phase, and ventricular repolarization becomes delayed. This reduction of the IKr curent leads to QT interval prolongation.11 Mutations in the SCN5A gene, which is encoding major cardiac voltage‐gated sodium channel (named NaV1.5), are responsible for the LQT3 syndrome. “Gain of function” mutations cause an increase in the “persistent sodium current,” which results in the changes of the cardiac action potential plateau phase length and causes delayed repolarization.12 In both syndromes, LQT2 and LQT3 life‐threatening ventricular arrhythmias occur, particularly “torsades de pointes,” which can lead to SCD.13
Only a number of all known single nucleotide polymorphisms (SNPs) has been associated with specific diseases.14, 15 Furthermore, the knowledge of SNPs associated with LQTS and their role is limited. As previously described, SNPs in the hERG1 gene can reduce IKr current and become responsible for LQT2 syndrome in humans.16 On the other hand, the K897T polymorphism in the hERG1 gene, is one of the most common polymorphisms in the human Kv11.1 channel protein. It affects the channel function and QT interval on ECG,17 although its net effect on that channel function is controversial. Some studies have reported prolongation of the QT intervals in women affected by the K897T polymorphism.3 Other studies have found no effect or only a small shortening of the QT interval in the carriers.15 Researchers suggested, that changed allele, when it coexists with the LQT2 mutation, acts as a genetic modifier and further exaggerate IKr current reduction.18 Thus, it can promote clinical expression of the LQT2 mutation. Separately, it has been reported that the H558R polymorphism in SCN5A gene could modify expression of an arrhythmia causing mutation, although data on this subject remains insufficient.4 Moreover, the H558R polymorphism was reported as associated with the shorter PR and QR intervals possibly with atrial fibrillation.19, 20
In some of the members of the family, the ATS mutation coexisted with the polymorphisms in the hERG1 (K897T) and the SCN5A (H558R) genes. The question remains if these SNPs influenced the clinical presentation of the ATS mutation? Looking again at our data (Table 1) we note that dysmorphic features appeared in all affected by the ATS mutation individuals. Both, the proband with coexisting ATS mutation, K897T and H558R polymorphisms and the proband's sister, with the ATS mutation and K897T polymorphism, but without the H558R polymorphism, had similar symptoms—episodes of loss of consciousness, BiVT, PVT and about 5000 PVC in Holter studies. On the other hand, the history of the symptoms in the family member with the ATS mutation only (no K897T or H558R polymorphisms) and in the family member with the ATS mutation and additional H558R polymorphism were not so severe. Moreover, the presence of the U‐wave on ECG was not characteristic of the ATS mutation. It did show up in all examined family members regardless whether they carried the ATS mutation. All the members with the ATS mutation had T‐peak–U‐peak interval longer than 200 ms and in all ATS mutation carriers it was longer than in family members who had no ATS mutation. T‐peak–T‐end interval was the longest (>120 ms) in members with the coexisting ATS mutation and hERG1 (K897T) polymorphism. In two of all studied family members, it was longer than 120 ms while it was shorter than that in others.
Conclusion
We found that more severe symptoms of the ATS can be caused by the polymorphisms (SNPs) in other genes. In the presented family the ATS severity may depend on the presence of K897T polymorphism in the hERG1 gene.
REFERENCES
- 1. Donaldson MR, Yoon G, Fu YH, et al. Andersen‐Tawil syndrome: A model of clinical variability, pleiotropy, and genetic heterogeneity. Ann Med 2004;36(Suppl 1):92–97. [DOI] [PubMed] [Google Scholar]
- 2. Nguyen HL, Pieper GH, Wilders R. Andersen‐Tawil syndrome: Clinical and molecular aspects. Int J Cardiol 2013;170:1–16. [DOI] [PubMed] [Google Scholar]
- 3. Pietilä E, Fodstad H, Niskasaari E, et al. Association between HERG K897T polymorphism and QT interval in middle‐aged Finnish women. J Am Coll Cardiol 2002;40(3):511–514. [DOI] [PubMed] [Google Scholar]
- 4. Ye B, Valdivia CR, Ackerman MJ, et al. A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol Genomics 2003;12:187–193. [DOI] [PubMed] [Google Scholar]
- 5. Ashcroft F. From molecule to malady. Nature 2006;440:440–447. [DOI] [PubMed] [Google Scholar]
- 6. Plaster NM, Tawil R, Tristani‐Firouzi M, et al. Mutations in Kir.2.1 cause the developmental ad episodic electrical phenotypes of Andresen's syndrome. Cell 2001;105:511–519. [DOI] [PubMed] [Google Scholar]
- 7. Andelfinger G, Tapper AR, Welch RC, et al. KCNJ2 mutation result in Andersen syndrome with sex‐specific cardiac and skeletal muscle phenotypes. Am J Hum Genet 2002:71:663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kukla P, Biernacka EK, Baranchuk A, et al. Electrocardiogram in Andersen‐Tawil syndrome. New electrocardiographic criteria for diagnosis of type‐1 Andersen‐Tawil syndrome. Curr Cardiol Rev 2014:10(3);222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Venance SL, Cannon SC, Fialho D, et al. The primary periodic paralyses: Diagnosis, pathogenesis and treatment. Brain 2006;129:8–17. [DOI] [PubMed] [Google Scholar]
- 10. Donaldson MR, Jensen JL, Tristani‐Firouzi M, et al. PIP(2) binding residues of Kir2.1 are common targets of mutations causing Andersen syndrome. Neurology 2003;60:181–186. [DOI] [PubMed] [Google Scholar]
- 11. Thomas D, Kiehn J, Katus HA, et al. Defective protein trafficking in hERG‐associated hereditary long QT syndrome (LQT2): Molecular mechanisms and restoration of intracellular protein processing. Cardiovasc Res 2003;60:235–241. [DOI] [PubMed] [Google Scholar]
- 12. Wang Q, Shen J, Li Z, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet 1995;4:1603–1607. [DOI] [PubMed] [Google Scholar]
- 13. Crotti L, Celano G, Dagradi F, et al. Congenital long QT syndrome. Orphanet J Rare Dis 2008;3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li G, Pan T, Guo D, Li L‐C. Regulatory variants and disease: The E‐cadherin‐160C/A SNP as an example. Mol Biol Int 2014; 2014:967565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kujovich JL. Factor V Leiden thrombophilia. Genet Med 2011;13(1):1–16. [DOI] [PubMed] [Google Scholar]
- 16. Newton‐Cheh C, Guo CY, Larson MG, et al. Common genetic variation in KCNH2 is associated with QT interval duration: The Framingham Heart Study. Circulation 2007;116:1128–1136. [DOI] [PubMed] [Google Scholar]
- 17. Laitinen P, Fodstad H, Pippo K, et al. Survey of the coding region of the HERG gene in long QT syndrome reveals six novel mutations and an amino acid polymorphism with possible phenotypic effects. Hum Mutat 2000;15:580–581. [DOI] [PubMed] [Google Scholar]
- 18. Crotti L, Lundquist AL, Insolia R, et al. KCNH2‐K897T is a genetic modifier of latent congenital long‐QT syndrome. Circulation 2005;112(9):1251–1258. [DOI] [PubMed] [Google Scholar]
- 19. Magnani JW, Brody JA, Prins BP, et al. Sequencing of SCN5A identifies rare and common variants associated with cardiac conduction: Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium. Circ‐Cardiovasc Gen 2014;7(3):365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen L, Zhang W, Fang C, et al. Polymorphism H558R in the human cardiac sodium channel SCN5A gene is associated with atrial fibrillation. J Int Med Res 2011;39(5):1908–1916. [DOI] [PubMed] [Google Scholar]