

Abstract
Background
The cardiac sodium channel Nav1.5, encoded by the gene SCN5A, is associated with a wide spectrum of hereditary arrhythmias. The gain‐of‐function mutation p.I141V in SCN5A was identified in a large multigenerational family with exercise‐induced polymorphic ventricular arrhythmias. The purpose of this study was to evaluate the molecular and clinical effects of flecainide administration on patients with this syndrome.
Methods
Eleven p.I141V carriers who exhibited frequent multiformic premature ventricular complexes (PVCs) during exercise were subjected to exercise stress tests, both before and after intravenous infusion of 2 mg/kg flecainide. The in vitro effects of flecainide were evaluated using the patch‐clamp technique with HEK293 cells expressing the Nav1.5 channel.
Results
The flecainide treatment significantly reduced the frequency of PVCs during and after exercise. Next, the sensitivity of the p.I141V mutant channel to flecainide was compared to that of the wild type channel. Perfusion of flecainide inhibited the peak and window currents in both groups.
Conclusion
The clinical investigations of the affected patients, as well as the molecular and pharmacological characterization of the SCN5A p.I141V mutation, provide new evidence supporting the association of this mutation with exercise‐induced polymorphic ventricular arrhythmias. These data also demonstrate that flecainide may serve as an effective treatment for the defect in Nav1.5 that leads to an increased sodium window current.
Keywords: flecainide, Nav1.5‐I141V, PVCs, cardiac arrhythmias, cardiac sodium channel
Inherited arrhythmias represent a major cause of sudden cardiac death in individuals with structurally normal hearts. Mutations in more than 30 genes encoding ion channels and channel‐interacting proteins have been associated with an increasingly wide range of inherited cardiac arrhythmias.1, 2, 3, 4, 5 Gain and loss of function mutations in the SCN5A gene, which encodes the cardiac voltage‐gated sodium channel Nav1.5, are associated with the occurrence of several inherited cardiac arrhythmias; i.e., type‐3 congenital long QT syndrome (LQTS), Brugada syndrome (BrS), cardiac conduction disease, atrial fibrillation, and multifocal ectopic Purkinje‐related premature contractions (MEPPC).3, 6, 7 In a recent study, a mutation in the SCN5A gene, which lead to a p.I141V substitution, was identified in a large multigenerational family with exercise‐induced polymorphic ventricular arrhythmias.8 This mutation modified the biophysical properties of the Nav1.5 channel, causing an increase in cardiac cell excitability and predisposing the individual to both atrial and ventricular arrhythmias.8
Flecainide, a common class IC antiarrhythmic drug with sodium channel‐blocking properties, has been reported to reduce exercise‐induced ventricular arrhythmias in patients with cardiac ryanodine receptor and calsequestrin‐associated catecholaminergic polymorphic ventricular tachycardia (CPVT), and to decrease MEPPC.3, 9, 10 The aim of this study was to assess the molecular effect of flecainide on the function of the p.I141V mutant Nav1.5 channel, as well as its clinical effect on ventricular arrhythmias and atrioventricular conduction in p.I141V carriers.
METHODS
Cell Culture
Human Embryonic Kidney 293 (HEK293) cells were cultured at 37°C in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum, 4 mM glutamine, and 20 mg/ml gentamycin in a humidified atmosphere of 5% CO2 and 95% air. All cell medium components, except glutamine (Sigma‐Aldrich, Buchs, Switzerland), were purchased from Gibco (Basel, Switzerland).
Cellular Electrophysiology
HEK293 cells were transfected with DNA complexed to JetPEI (Polyplus Transfection, Basel, Switzerland), according to the manufacturer's instructions. DNA concentrations were 1 μg of pCDNA3.1‐Nav1.5 wild type (WT) or p.I141V, and 1 μg of pIRES‐hβ1‐CD8. The transfection and cell splitting procedures have been previously described.8 Twenty‐four hours after transfection, the resulting sodium current was recorded at room temperature (23–25°C), and under whole‐cell voltage clamp conditions with an Axopatch 200B amplifier (Axon Instruments, Inc., Union City, CA, USA) interfaced to a personal computer and driven by the PClamp 10 software (Molecular Devices Corp., Sunnyvale, CA, USA). For whole cell recordings, a P/4 protocol was used to cancel the residual capacitive currents. The cells were bathed with an extracellular solution containing (in mmol/L): NaCl 50, NMDG‐Cl 80, CsCl 5, MgCl2 1.2, CaCl2 2, HEPES 10, glucose 5. The pH was adjusted to 7.4 with CsOH. Glass pipettes (tip resistance: 1.5 to 2.5 MΩ) were filled with an intracellular medium containing (in mmol/L): CsCl 60, aspartic acid 50, CaCl2 1, MgCl2 1, HEPES 10, EGTA 11, Na2ATP 5. The pH was adjusted to 7.2 with CsOH. The studied cell was locally superfused with extracellular medium with or without 10 μM flecainide, and, if indicated, 30 μM tetrodotoxin (TTX). For the window current measurements, the presented recordings were normalized to the peak current obtained at −20 mV and were TTX‐subtracted (30 μM). All products were purchased from Sigma, except TTX, which was obtained from Alomone (Jerusalem, Israel).
Patients
The study group consisted of 13 family members who were identified to be heterozygous carriers of the gain‐of‐function SCN5A mutation p.I141V.8 The p.I141V mutation is associated with the manifestation of frequent multiformic premature ventricular complexes (PVCs) during periods of physical exercise. The mutation and the implicated family members were described in detail in a previous study.8 The patients were otherwise healthy and did not have any hypertension, diabetes, or valvular or coronary artery disease.
Study Scheme
A total of 9 of the 13 patients were using beta‐adrenergic‐blocking agents during the study. The medications used included 2.5 to 15 mg of bisoprolol (n = 5), 47.5 to 190 mg of metoprolol (n = 3) and 40 mg of propranolol (n = 1). One of the patients was also using amiodarone. The patients were subjected to an exercise stress two times in the same day. A baseline exercise stress test was carried out in the morning. The test was repeated in the afternoon 15 minutes after a 15‐minute intravenous infusion of flecainide (Tambocor, 3M Health Care Ltd., Leicester, United Kingdom), at a dosage of 2 mg/kg of body weight. An electrocardiogram (ECG) was continuously monitored during the infusion.
Exercise Stress Test
Exercise tests were performed with a bicycle ergometer. The initial load was 30 W, followed by increments of 15 W each minute. The workload targets for both exercise stress tests (pre‐ and postflecainide infusion) were identical. A continuous 12‐lead ECG was printed at a paper speed of 25 mm/s and an amplification of 0.1 mV/mm throughout the exercise test. Both the maximum workload achieved and the heart rate at which the PVCs first appeared were recorded whenever applicable. The number of PVCs that occurred 5 minutes prior to exercise, during exercise, and 8 minutes postexercise, as well as the maximum number of consecutive PVCs in ventricular salvos were recorded. Blood cell count, plasma creatinine, potassium, and sodium levels were measured before the first exercise test. To assess the effects of the single dose of flecainide on heart rhythm, a 24‐hour ambulatory ECG was recorded on all of the patients on an outpatient basis starting at the discharge from the hospital after the second exercise stress test. Rhythm, mean heart rate, number of PVCs, and number and maximum length of ventricular tachycardias (≥3 consecutive ventricular complexes) were recorded. A second 24‐hour ambulatory ECG was performed after a washout period of at least 1 week. The study was approved by the Ethics Review Committee and was in accordance with the institutional guidelines and the Declaration of Helsinki. An informed consent was obtained from all patients.
Data Analysis and Statistical Methods
Currents were analyzed with Clampfit software (Axon Instruments, Inc.). Data were analyzed using a combination of pClamp10, Excel (Microsoft) and Prism (Graphpad). Comparisons between study phases were performed with paired‐samples and impaired two‐tailed Student's t‐tests or ANOVA test. Data are expressed as mean ± SD. For the molecular characterization of flecainide effect, data are expressed as mean ± SEM. A P value <0.05 was considered significant.
RESULTS
Findings at Baseline
Thirteen family members (6 males, 7 females) carrying the SCN5A p.I141V mutation were studied. Their mean age was 54 ± 16 years (range, 33 to 74), and five of them had encountered a syncopal spell. Nine patients were in sinus rhythm, whereas two patients had an ectopic atrial rhythm and two were in atrial fibrillation. One of the patients in sinus rhythm showed ventricular pre‐excitation.
The resting heart rate, duration of the P wave, PQ interval and QRS duration, as well as the QT interval are presented in Table 1. The mean baseline QTc interval of the 13 patients studied was 448 ± 21 ms (Table 1). During the 5‐minute rest prior to the exercise stress test, the number of PVCs was less than 1 per minute in all but one patient (Table 2, Fig. 1A and 1B). In the baseline exercise stress test, patients exercised for 10 ± 3 minutes. During exercise, eight patients had more than 100 PVCs and exhibited salvos of three to eight consecutive PVCs. Seven of these eight patients were using beta‐blockers. The average threshold sinus rate at which the PVCs appeared and the total number of PVCs during the workload are presented in Table 2. Figure 1C illustrates the ECG during workload and demonstrates multiformic PVCs, bigeminy, and a short ventricular tachycardia. Figure 1D is the resting ECG during the recovery phase.
Table 1.
Electrocardiographic Parameters at Resting ECG
| After Flecainide Infusion | |||
|---|---|---|---|
| Baseline (n = 13) | 2 mg/kg (n = 11) | P Value | |
| Heart rate (min‐1) | 76±8 | 76±11 | NS |
| P (ms) | 107±19 | 118±13 | NS |
| PQ (ms) | 162±28 | 184±28 | <0.05 |
| QRS (ms) | 93±24 | 97±8 | NS |
| QT (ms) | 397±37 | 410±31 | <0.01 |
| QTc (ms) (range) | 448±21 (417–474) | 465±23 (416–493) | NS |
NS = not significant.
Table 2.
Exercise Stress Test Results
| After Flecainide Infusion | |||
|---|---|---|---|
| Baseline (n = 13) | 2 mg/kg (n = 11) | P Value | |
| Duration of exercise (min) | 10.0±3.3 | 10.2±3.5 | NS |
| Threshold heart rate at which PVCs first appeared (min−1) | 110±34 | 115±39 | NS |
| Maximum heart rate (min−1) | 162±26 | 163±23 | NS |
| Number of PVCs during 5‐minute rest prior to exercise (beats) | 8±25 (0–88) | 0±1 (0–2) | NS |
| Number of PVCs during exercise (beats) | 167±189 (0–536) | 4±7 (0–17) | 0.02 |
| Longest NSVT during exercise (beats) | 3±2 (0–8) | 1±1 (0–3) | 0.01 |
| Number of PVCs during 8‐minute rest after exercise (beats) | 90±96 (0–286) | 7±15 (0–48) | 0.02 |
NS = not significant; NSVT = nonsustained ventricular tachycardia; PVCs = premature ventricular complexes.
Figure 1.
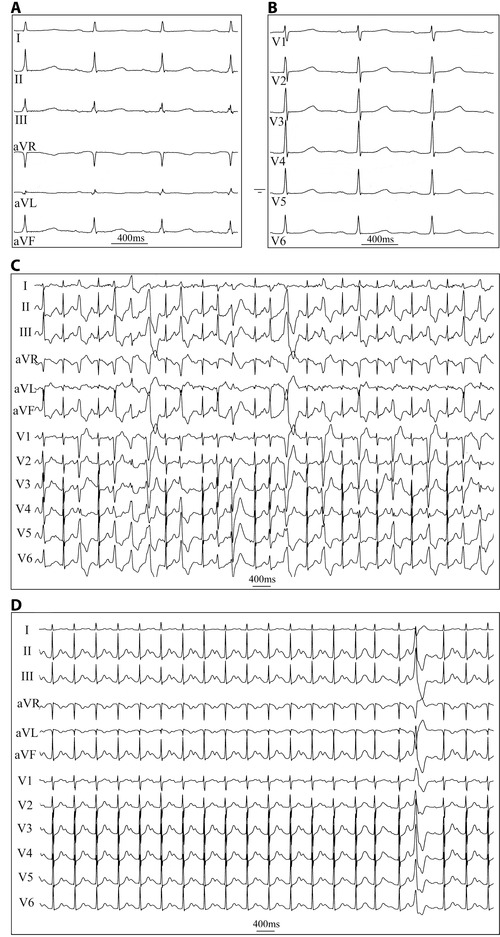
Representative ECG registrations at rest and during exercise stress test before flecainide infusion (A) ECG of a 52‐year‐old female showing sinus rhythm at rest (limb leads, ECG paper speed 50 mm/s) (B) chest leads (C) ECG during the 7th minute of exercise at workload of 120 W demonstrating multiformic PVCs, bigeminy, and a short ventricular tachycardia (25 mm/s) (D) resting ECG during the recovery phase, 1‐minute after cessation of exercise showing sinus rhythm and a single PVC (25 mm/s).
Effect of Flecainide on the p.I141V Carriers
A total of 11 patients without ventricular pre‐excitation or amiodarone treatment underwent flecainide infusion and a subsequent exercise stress test. An intravenous 15‐minute infusion of 2 mg/kg of flecainide resulted in minor prolongations of the PR and QT interval, which were nonsignificant when corrected for heart rate in the resting ECG (Table 1, Fig. 2A and B). In three patients, QTc was in the range of 480–490 ms after flecainide administration. The infusion did not cause Brugada‐type ST‐segment changes in any of the patients. During the exercise stress test, flecainide abolished the PVCs completely in five patients and reduced their number to less than 20 in four patients (Table 2 and Fig. 2C). Flecainide also decreased the number of PVCs during the postexercise 8‐minute recovery period to less than 1 PVC per minute in all except one patient (Table 2 and Fig. 2D). Two patients did not have any PVCs at baseline or during the postflecainide exercise stress test (Table 2 and Fig. 2).
Figure 2.
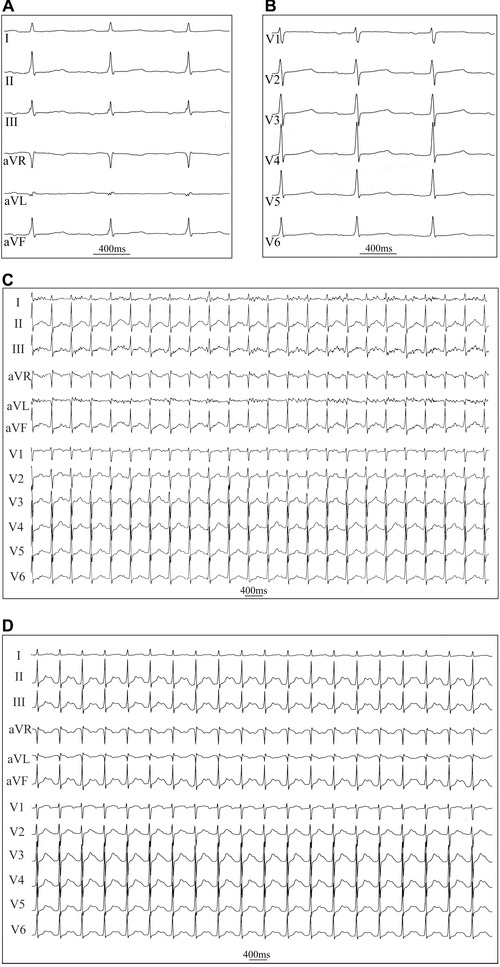
Representative ECG registrations 15 minutes after flecainide infusion (A) resting ECG of the same patient, as in Fig. 1, 15 minutes after an intravenous infusion of flecainide (2 mg/kg) showing sinus rhythm at rest (limb leads, paper speed 50 mm/s) (B) chest leads (C) ECG during the 7th minute of exercise at a workload of 120 W, demonstrating the absence of PVCs (25 mm/s) (D) resting ECG 1‐minute after cessation of exercise (25 mm/s).
Findings in 24‐Hour Ambulatory ECG
A 24‐hour ambulatory ECG registration was performed following the single intravenous dose of flecainide and the exercise stress test. The average heart rate was 82 ± 8 bpm. A total of five patients had short episodes of ventricular tachycardia (three to six complexes). Six of the patients had more than 300 supraventricular premature complexes during the 24‐hour period. After at least a 1‐week washout period, a second 24‐hour ambulatory ECG registration was performed. Two patients did not participate in the second recording. The average heart rate was 78 ± 9 bpm. Six patients manifested short stretches of ventricular tachycardia but no one exhibited longer episodes of ventricular tachycardia than what was detected during the exercise stress tests, both before and after the flecainide infusion.
Effect of the p.I141V Mutation on Nav1.5 Channel Sensitivity to Flecainide
The sensitivity of the Nav1.5‐p.I141V channel to the effects of flecainide was evaluated. At a concentration of 10 μM, WT and mutant channels exhibited similar sensitivities to flecainide. Upon flecainide perfusion, the sodium current peak as well as the window current peaks of the p.I141V channels were similarly inhibited as compared to the WT (inhibition percentage of the sodium peak current under flecainide [%]: 64 ± 7.8 for WT, n = 8, and 67 ± 7.8 for the p.I141V, n = 8, P = NS; Fig. 3A: inhibition percentage of the sodium window current under flecainide [%]: 71 ± 5.5 for WT, n = 6, and 77 ± 2.3 for the p.I141V, n = 10, P = NS; Fig. 3B).
Figure 3.
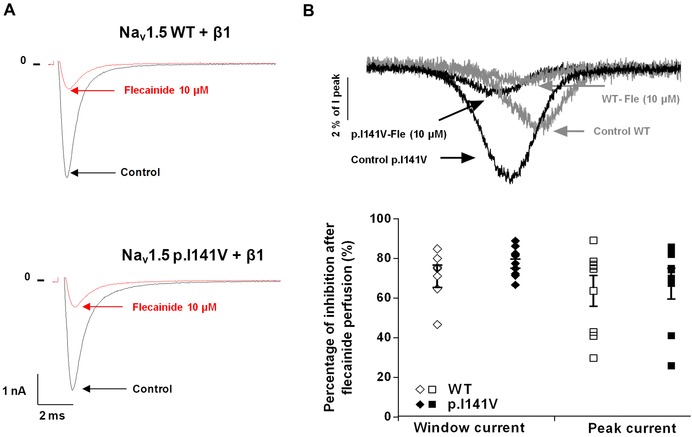
Flecainide effect on the Nav1.5‐I141V mutant (A) representative traces showing the flecainide effect on the sodium current peak of Nav1.5 WT and p.I141V channels (B) averaged normalized WT and p.I141V tetrodotoxin‐sensitive window currents, obtained with a depolarizing‐voltage ramp (from −100 and +50 mV), were normalized to the peak current at −20 mV (Ipeak) and recorded in the same cell. Effect of the perfusion of 10 μM of flecainide on the sodium window current.
DISCUSSION
Mutations in the cardiac sodium channel gene SCN5A have been linked to several arrhythmia phenotypes, including type 3 congenital LQTS, BrS, cardiac conduction defects, dilated cardiomyopathy, atrial fibrillation, and sudden infant death syndrome.7, 11, 12 Recently, several mutations of Nav1.5, such as the p.R222Q and the p.I141V mutants, were linked to cardiac hyperexcitability phenotypes associated with the alteration of the activation process of this channel.3, 6, 8, 12, 13
This study demonstrates that flecainide, when administered in an acute fashion, can effectively reduce the arrhythmic manifestations that can occur during physical exercise in carriers of this mutation.
The need for an effective treatment of this SCN5A‐associated syndrome arises from the abundant ventricular arrhythmias that occur during physical effort, which markedly decrease the exercise tolerance of carriers of the p.I141V mutation. Another indication for antiarrhythmic drug therapy is to prevent paroxysmal atrial fibrillation. Flecainide has been proven to reduce exercise‐induced PVCs in CPVT and Andersen‐Tawil syndrome.9, 14, 15 For this reason, the efficacy and safety of the acute administration of flecainide to patients with the p.I141V mutation were evaluated in this study. Flecainide was highly effective in preventing the exercise‐induced ventricular arrhythmias in these patients. It reduced the number of PVCs during exercise by 98% and PVCs during the postexercise rest period by 92% (Table 2). Flecainide appeared to be a safe drug in the p.I141V carriers. No significant adverse effects on ECG were noted after administration. Despite slightly prolonging atrioventricular nodal conduction, it did not significantly prolong intraventricular conduction or repolarization time, nor did it cause Brugada‐type changes on ECG. Three patients who suffered from poor exercise tolerance insisted on having long‐term therapy with flecainide after participating in the study. A dose of 200 mg of flecainide daily was prescribed to these three patients. All three were still on this treatment and were benefitting from it after a follow‐up of 9–16 months. Quinidine (a class IA antiarrhythmic drug) has also been shown to be effective in myotonia patients carrying the p.I141V homologous mutation in the gene SCN4A, which leads to similar biophysical alterations as to those described in this study.16
On the molecular level, the beneficial effects of class I antiarrhythmic drugs on cardiac hyperexcitability is related to their cardiac sodium channel‐blocking properties.3, 6, 8, 9, 10, 15 The cardiac sodium channel Nav1.5 belongs to a group of voltage‐gated sodium channels that contain the action sites for local anesthetics and class I antiarrhythmic drugs.17 These sites are located in the α‐subunits of the Nav channels, more specifically in the sixth segment in domains I, III, and IV.17, 18, 19, 20 Mutations that neutralize critical residues in these domains alter the pharmacological properties of the channel,17, 18, 19, 20 i.e., by altering the affinity with which the channel binds a drug depending on the functional conformation of the channel.17, 18, 19, 20
Since the flecainide treatment of the affected patients was proven to reduce the occurrence of PVCs, the in vitro effects of this antiarrhythmic drug on the Nav1.5‐WT and p.I141V currents were subsequently investigated. The WT and mutant channels were both sensitive to inhibition by flecainide. Both their INa peak and the peak window current peaks were inhibited to the same extent by 10 μM flecainide. It is of interest that flecainide also markedly reduced PVC numbers in p.R222Q carriers.6 It appears that Nav1.5 channels with mutations that are located closer to the proposed binding site of flecainide.17 are more sensitive than the WT channels to flecainide block. The underlying mechanisms of inhibition, however, may vary from mutation to mutation. Several studies have suggested that flecainide may decrease the frequency of PVCs through one of the following two mechanisms: (1) directly, by decreasing cardiac cell excitability (this effect is related to the inhibition of sodium channel activity)21 or (2) indirectly, by decreasing calcium sparks through the reduction of INa. 22 In this case, the main antiarrhythmic effect of flecainide likely results from its ability to drastically reduce the increased and hyperpolarized sodium window current (Fig. 3F), which is hypothesized to cause the hyperexcitability of the cells expressing the mutant channel.8
Limitations
The long‐term effectiveness and safety of flecainide in these patients needs to be assessed.
CONCLUSION
In conclusion, both our in vitro and in vivo data suggest that flecainide is a useful antiarrhythmic drug in the arrhythmic syndrome caused by the SCN5A p.I141V mutation. These findings, coupled with those from studies involving MEPPC patients,6 suggest that flecainide is well tolerated by patients suffering from these related, yet dissimilar, arrhythmic syndromes.
Prof. Hugues Abriel, MD PhD, University of Bern, Department of Clinical Research, Murtenstrasse 35, 3010 Bern, Switzerland, Phone: 41‐31‐6320928; Fax: 41‐31‐6320946; Email: Hugues.Abriel@dkf.unibe.ch.
Funding sources: This study has been supported by a grant of the Swiss National Science Foundation to H. Abriel (310030 120707).
REFERENCES
- 1. Abriel H, Zaklyazminskaya EV. Cardiac channelopathies: Genetic and molecular mechanisms. Gene 2013;517:1–11. [DOI] [PubMed] [Google Scholar]
- 2. Kaufman ES. Mechanisms and clinical management of inherited channelopathies: Long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, and short QT syndrome. Heart Rhythm 2009;6:S51–S55. [DOI] [PubMed] [Google Scholar]
- 3. Laurent G, Saal S, Amarouch MY, et al. Multifocal ectopic Purkinje‐related premature contractions: A new SCN5A‐related cardiac channelopathy. J Am Coll Cardiol 2012;60:144–156. [DOI] [PubMed] [Google Scholar]
- 4. Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin‐B mutation causes type 4 long‐QT cardiac arrhythmia and sudden cardiac death. Nature 2003;421:634–639. [DOI] [PubMed] [Google Scholar]
- 5. Watanabe H, Koopmann TT, Le Scouarnec S, et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 2008;118:2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mann SA, Castro ML, Ohanian M, et al. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J Am Coll Cardiol 2012;60:1566–1573. [DOI] [PubMed] [Google Scholar]
- 7. Liu M, Yang KC, Dudley SC Jr. Cardiac sodium channel mutations: Why so many phenotypes? Nat Rev Cardiol 2014;11:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Swan H, Amarouch MY, Leinonen J, et al. Gain‐of‐function mutation of the SCN5A gene causes exercise‐induced polymorphic ventricular arrhythmias. Circ Cardiovasc Genet 2014;7:771–781. [DOI] [PubMed] [Google Scholar]
- 9. Khoury A, Marai I, Suleiman M, et al. Flecainide therapy suppresses exercise‐induced ventricular arrhythmias in patients with CASQ2‐associated catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2013;10:1671–1675. [DOI] [PubMed] [Google Scholar]
- 10. van der Werf C, Kannankeril PJ, Sacher F, et al. Flecainide therapy reduces exercise‐induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol 2011;57:2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res 2011;108:884–897. [DOI] [PubMed] [Google Scholar]
- 12. Amarouch M‐Y, Abriel H. Cellular hyper‐excitability caused by mutations that alter the activation process of voltage‐gated sodium channels. Front Physiol 2015;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nair K, Pekhletski R, Harris L, et al. Escape capture bigeminy: Phenotypic marker of cardiac sodium channel voltage sensor mutation R222Q. Heart Rhythm 2012;9:1681–1688. [DOI] [PubMed] [Google Scholar]
- 14. Delannoy E, Sacher F, Maury P, et al. Cardiac characteristics and long‐term outcome in Andersen‐Tawil syndrome patients related to KCNJ2 mutation. Europace 2013;15:1805–1811. [DOI] [PubMed] [Google Scholar]
- 15. Watanabe H, Chopra N, Laver D, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 2009;15:380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petitprez S, Tiab L, Chen L, et al. A novel dominant mutation of the Nav1.4 alpha‐subunit domain I leading to sodium channel myotonia. Neurology 2008;71:1669–1675. [DOI] [PubMed] [Google Scholar]
- 17. Ragsdale DS, McPhee JC, Scheuer T, et al. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage‐gated Na+ channels. P Natl Acad Sci USA 1996;93:9270–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yarov‐Yarovoy V, Brown J, Sharp EM, et al. Molecular determinants of voltage‐dependent gating and binding of pore‐blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J Biol Chem 2001;276:20–27. [DOI] [PubMed] [Google Scholar]
- 19. Yarov‐Yarovoy V, McPhee JC, Idsvoog D, et al. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage‐dependent gating and drug block. J Biol Chem 2002;277:35393–35401. [DOI] [PubMed] [Google Scholar]
- 20. Liu G, Yarov‐Yarovoy V, Nobbs M, et al. Differential interactions of lamotrigine and related drugs with transmembrane segment IVS6 of voltage‐gated sodium channels. Neuropharmacology 2003;44:413–422. [DOI] [PubMed] [Google Scholar]
- 21. Liu N, Denegri M, Ruan Y, et al. Short communication: Flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Circ Res 2011;109:291–295. [DOI] [PubMed] [Google Scholar]
- 22. Sikkel MB, Collins TP, Rowlands C, et al. Flecainide reduces Ca(2+) spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovas Res 2013;98:286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]