

Abstract
Background: Previously identified atherosclerotic genetic factors have been studied mostly in case‐control studies and in nonuniform ethnic populations, whereas data on the cumulative contribution of genetic factors to an earlier onset of a first myocardial infarction (MI) are limited. We hypothesized that several genetic atherosclerotic single nucleotide polymorphisms (SNPs) may exert an additive effect on the earlier occurrence of coronary atherothrombotic disease after adjustment for clinical factors.
Methods: Eighteen atherosclerotic high‐risk SNPs were selected based upon meta‐analyses of 614 published reports, and were incorporated into a carriership model. Multivariate regression analysis was used to identify the independent contribution of selected genotypes to the age at onset of a first MI in a cohort of 814 white (n = 622) and nonwhite (n = 192) patients enrolled in the Thrombogenic Factors and Coronary Events Study.
Results: The analysis demonstrated that selected genotypes were significantly associated with an earlier occurrence of a first MI among white patients (an average of 0.6 year reduction per carried genotype; P = 0.027), whereas the contribution of genotypes to MI onset among nonwhite patients was not significant (an average of 0.7 year increase per carried genotype; P = 0.16), with a significant ethnic × genotype interaction effect (P = 0.02).
Conclusions: Our findings suggest that currently identified atherosclerotic genetic factors confer an independent additive contribution to the earlier onset of coronary atherothrombotic disease among white patients. The lack of a significant association between these genotypes and outcome in other ethnic groups suggests that cardiovascular genetic risk should be studied directly in these populations.
Keywords: genetic polymorphisms, myocardial infarction, age
Genetic factors that contribute to coronary artery disease (CAD) are operative throughout the lifetime of an individual, and therefore may be independently associated with an earlier age at onset of coronary atherothrombotic disease. However, previous studies that have identified genetics factors as predictors of CAD and myocardial infarction (MI) used mostly a case‐control design, in which the relationship between genetic risk and the age at onset of disease could not be readily quantified. We hypothesized that several independent genetic single nucleotide polymorphism (SNP) variants may exert a small, yet additive, effect on the age at onset of a first MI after adjustment for clinical factors. To study this possibility, we used a carriership approach, in which the cumulative burden of previously identified atherosclerotic genetic risk factors was related to the age of a first MI in patients enrolled in the Thrombogenic Factors and Coronary Events Study (THROMBO). 1
METHODS
Population
Subjects were drawn from a population of 1045 postinfarction patients enrolled in the THROMBO study. The details of this study have been previously reported. 1 Briefly, between October 1, 1994 and June 30, 1997, 1161 patients were enrolled following an index MI and followed for a mean period of 28 months. Blood was drawn for genotyping in 1012 patients at the time of the index infarction. Patients with a previous MI prior to the index event (n = 192) and with absent data regarding baseline clinical characteristics (n = 6) were excluded from the analysis, resulting in a population of 814 patients with a first MI. Study patients comprised mainly three self‐reported ethnic populations: white non‐Latinos (n = 622) African‐Americans (n = 115), and Latinos (n = 64) living in the United States; other populations including Asians or Pacific Islanders, American Indians, native Alaskans and Indians comprised a relatively small proportion of study patients (n = 13). In the main analysis of the study we evaluated genetic risk among the white and nonwhite groups. In a secondary analysis, the consistency of the results in the nonwhite group was evaluated separately among African‐Americans and Latinos.
Selection of Genotypes and Prioritization of Candidate Genotypes
The procedure for selecting the genotypes explored in this study has been previously described. 2 Selection was based on an investigation of 40 genetic variants in 33 genes that code for proteins involved in coagulation, oxidative stress and inflammation, vascular reactivity, insulin resistance, apolipoproteins, and metabolism of triglyceride‐rich lipoproteins. The identification of potential candidate genotypes was based on a meta‐analysis of 614 published reports on the potential polymorphisms. Ultimately, 18 SNPs were selected. As appropriate genes were being selected, the genotypes in each SNP were stratified into high‐ and low‐risk categories based on the same meta‐analysis (Table 1).
Table 1.
Genotypes
| Gene | Polymorphic Alleles* (Reference SN ID) | Frequency of High‐Risk Allele in Whites/African‐ American/Asians/Latinos† | High‐Risk Genotype(s)† | Biologic Effect |
|---|---|---|---|---|
| Apolipoprotein | 112R > C | 14/25/9/NA | e2e4; e3e4; | Impaired |
| E (APOE) | 158 C > R | e4e4§ | clearance of | |
| (rs429358) | VLDL/ | |||
| (rs7412) | chylomicrons | |||
| Apolipoprotein | ins, del | 34/20/NA/NA | Del/del | Increased serum |
| B (APOB) | LDL | |||
| Interleukin‐6 | −174G > C | 43/9/NA/NA | GC; CC | Increased |
| (IL6) | (rs1800795) | systemic inflammation | ||
| Platelet beta 3 | 11T > C | 15/16/3/NA | TC; CCA | Increased |
| integrin (ITGB3) | [HPA 1a, 1b] | (PL‐A1A2; | platelet | |
| (rs5918) | 2A2) | aggregation | ||
| Angiotensin‐converting | ins, del | 46/38/NA/NA | del/del | Increased ACE levels |
| enzyme (ACE) | ||||
| Angiotensin II | 1166A > C | 29/5/NA/NA | AC; CC | Increased blood |
| receptor 1 | (rs5186) | pressure | ||
| (AGTR1) | ||||
| Cholesterol ester | 277A > G | 60/72/NA/NA | GG; GA | Higher CETP |
| Transfer protein | G = TaqIB +/B1 | activity; lower | ||
| (CETP) | (rs708272) | HDL levels | ||
| Endothelial | 894G > T | 34/16/9/NA | TT | Coronary spasm; |
| nitric oxide | (298E > D) | hypertension | ||
| synthase (NOS3) | (rs1799983) | |||
| Apolipoprotein | −75G > A | 20/10/32/NA | GA; AA | Lower HDL |
| A1 (APOA1) | (Mspl) | levels | ||
| (rs670) | ||||
| Apolipoprotein | 3238C > G | 8/NA/20/NA | CG; GG | Increased TG |
| C‐III (APOC3) | [Sstl] (rs5128) | levels | ||
| Prothrombin | 20210G > A | 2/1/0.5/2 | GA; AA | Increased |
| (F2) | (rs1799963) | prothrombin | ||
| P‐selectin | 76666 C > A | 89/99/NA/NA | AA | Increased |
| (SELP) | (715P > T) | platelet‐endo‐ | ||
| (rs6136)55 | thelium reactivity | |||
| Paraoxonase | 584 A > G | 24/NA/NA/66 | AG; GG | Increased lipid |
| (PON1) | (192Q > R) | QR; RR | oxidation | |
| 59(rs662) | ||||
| Vascular | 242T > C | 40/42/NA/8 | TC; CC | Impaired |
| NADPH oxidase | (rs4673) | endothelial | ||
| p22phox | function | |||
| (CYBA) | ||||
| Factor XIII | 163T > G | 74/74/NA/NA | GG | Predisposition to |
| (F13A1) | (34L > V) | venous and | ||
| (rs3024472) | arterial thrombosis | |||
| Angiotensinogen | 235M > T | 42/82/NA/NA | TT | Increased |
| (AGT) | (rs699) | angiotensinogen; essential hypertension | ||
| Lipoprotein | −93T > G | 2/28/NA/9 | TT (Hind III +/+) | Increased |
| lipase (LPL) | [HindIII] | LDL,TG, lower | ||
| (rs320) | HDL | |||
| Plasminogen | [−675] 4G/5G | 54/38/NA/NA | 4G/4G | Increased |
| activator | plasma PAI‐1 | |||
| inhibitor‐1 | activity | |||
| (PAI1) |
*Sequences in parentheses denote the corresponding amino acid substitutions.
†Frequency of high‐risk allele based on data derived from control groups in this meta‐analysis.
†Autosomal dominant or recessive effects for each of the 18 SNPs were prespecified according to the results of the meta‐analysis.
§E2 = 112R > C and 158R > C; E3 = 112R > C and 158C > R; E4 = 112C > R and 158C > R.
ACE = angiotensin‐converting enzyme; HDL = high‐density lipoprotein; LDL = low‐density lipoprotein; NA = not available; PAI‐1 = plasminogen activator inhibitor‐1; TG = triglycerides.
Carriership Analysis
The carriership analysis approach 2 is based on the assumption that each of a group of several genetic risk factors makes a small, yet additive, contribution to the development of a coronary atherothrombotic event. Each subject in the study cohort was assigned a score based on the number of prespecified priority genotypes carried by the subject. This score was related to the age at onset of a first MI using multivariate regression analysis. The identified genotype risk was expressed as an average change in the age at onset of a first MI per carried genotype (carriership score). This analytic approach permitted adjustment for genetic, clinical, and environmental covariates.
End Point
The primary end point of this study was the age at onset of a first MI associated with a risk factor compared to patients without the risk factor. Age was analyzed as a continuous end point in the multivariate regression model.
Statistical Analysis
Baseline clinical characteristics by ethnic group were compared using the two‐sided t‐test for continuous variables, and the chi‐square test for categorical variables. Multivariate regression analysis, with the end point of age as a continuous variable, was carried out to explore the independent contribution of clinical and genetic factors to the age at onset of a first MI among study patients. A stepwise forward selection procedure was used to identify important clinical, laboratory, and environmental risk factors associated with an early age of first MI, with P < 0.10 for entering covariates. The final model included 4 identified risk factors (treated hypertension, male gender, current smoking, and diabetes), the carriership score, and ethnicity. Interaction‐term analysis was used to identify the effect of clinical and genetic factors in each ethnic group. Analyses were performed with the use of SAS software (version 9.13; SAS, Cary, NC, USA). A 2‐sided P value <0.05 was used for declaring statistical significance.
RESULTS
The age distribution of white and nonwhite patients at enrollment with a first MI is shown in Figures 1A and B, respectively. Compared to white patients, nonwhite subjects experienced a first MI at a significantly younger age, had a significantly higher proportion of diabetes mellitus and treated hypertension, and a lower proportion of male subjects (Table 2A). Laboratory data, 2 months after the index MI also demonstrated significant metabolic differences between the two groups (Table 2B).
Figure 1.
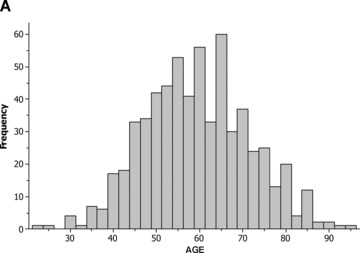
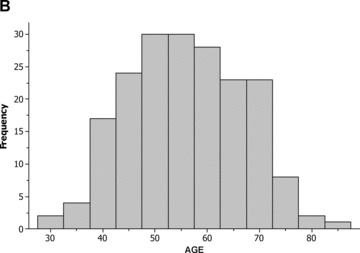
Distribution of age among (A) white patients; and (B) nonwhite patients.
Table 2.
Baseline Clinical and Laboratory Characteristics
| White (n = 622) | Nonwhite (n = 192) | P Value | |
|---|---|---|---|
| A. Clinical characteristics | |||
| Age, yrs (mean ± SD) | 59 ± 12 | 56 ± 11 | 0.001 |
| Male gender (%) | 76 | 69 | 0.05 |
| Hypertension (%) | 39 | 55 | <0.001 |
| Smoking at any time (%) | 68 | 64 | 0.35 |
| Diabetes mellitus (%) | 14 | 28 | <0.001 |
| B. Laboratory values*† | |||
| Cholesterol (mg/dL) | 196 ± 44 | 204 ± 45 | 0.05 |
| HDL‐C (mg/dL) | 39 ± 12 | 42 ± 12 | 0.006 |
| LDL‐C (mg/dL) | 117 ± 37 | 127 ± 38 | 0.004 |
| Triglycerides (mg/dL) | 210 ± 121 | 175.9 ± 95 | 0.001 |
| Apo A1 (mg/dL) | 118 ± 25 | 118 ± 25 | 0.98 |
| Apo B (mg/dL) | 122 ± 27 | 129 ± 30 | 0.004 |
| Glucose (mg/dL) | 98 ± 40 | 112 ± 52 | <0.001 |
| Insulin | 18 ± 25 | 23 ± 36 | 0.04 |
| BMI (kg/m2) | 28 ± 5 | 29 ± 6 | 0.002 |
| Lp(a) (mg/dL) | 21 ± 20 | 32 ± 27 | <0.001 |
Abbreviations: ApoA1 = apolipoprotein A1; HDL‐C = high‐density lipoprotein cholesterol; LDL‐C = low density cholesterol; Lp(a) = lipoprotein (a).
*Laboratory data were obtained 2 months after the index infarction.
†Data are presented as mean ± SD.
In the nonwhite group, African‐American and Latino patients exhibited a similar mean age at onset of a first MI (56 ± 11 and 57 ± 11, respectively) and a similar proportion of patients with a history of diabetes mellitus (27% and 30%, respectively), whereas the frequency of treated hypertension was significantly higher among African‐Americans (61%) than in Latinos (40%).
High‐Risk Genotypes
Nine of the 18 prespecified SNPs exhibited a significant difference in the frequency of high‐risk genotypes between white and nonwhite study patients (Table 3). Specifically, the proportion of patient carrying high‐risk genotypes of the interleukin‐6 (IL6), platelet beta 3 integrin (ITGB3), angiotensin receptor (AGTR1), and endothelial nitric oxide synthase polymorphisms (NOS3) was higher in the former group, whereas the frequency of high‐risk genotypes of the angiotensin‐converting enzyme (ACE), apolipoprotein A‐1 (APOA1), p‐selectin (SELP), paroxonase (PON1) and angiotensinogen (AGT) polymorphisms was higher in the latter group.
Table 3.
Distribution and Age Effect of Individual High‐Risk Risk Genotypes in Study Patient
| SNP | Frequency of High‐Risk Genotype(s) (%) | Age Effect* | ||||
|---|---|---|---|---|---|---|
| White | Nonwhite | White Change (Yrs) | P Value | Nonwhite Change (Yrs) | P | |
| APOE 112R > C 158C > R | 32 | 29 | −1.8 | 0.06 | −0.8 | 0.62 |
| APOB ins, del | 9 | 9 | −0.2 | 0.93 | +4.4 | 0.07 |
| IL6‐174G > C | 61 | 22* | +0.9 | 0.35 | +2.2 | 0.20 |
| ITGB311T > C | 30 | 18* | −0.6 | 0.54 | +2.7 | 0.15 |
| ACE ins, del | 28 | 35* | +0.7 | 0.47 | +1.6 | 0.26 |
| AGTR1 1166A > C | 49 | 21* | +0.7 | 0.47 | +0.6 | 0.73 |
| CETP 277A > G | 84 | 88 | −2.7 | 0.03 | +0.1 | 0.93 |
| NOS3 894G > T | 8 | 3* | −3.4 | 0.04 | −1.6 | 0.70 |
| APOA13238C > G | 31 | 30 | −0.1 | 0.90 | −1.0 | 0.52 |
| APOC33238C > G | 20 | 31* | +0.46 | 0.69 | +2.0 | 0.20 |
| F2 20210G > A | 4 | 2 | −0.8 | 0.74 | −2.2 | 0.66 |
| SELP 76666C > A | 82 | 91* | −0.1 | 0.91 | −1.4 | 0.57 |
| PON1 584A > G | 51 | 81* | −2.2 | 0.02 | −2.3 | 0.21 |
| CYBA 242T > C | 58 | 57 | −0.8 | 0.94 | +0.9 | 0.56 |
| F13A1 163T > G | 61 | 61 | −0.2 | 0.87 | +0.5 | 0.74 |
| AGT 235M > T | 19 | 57* | −1.1 | 0.34 | +0.5 | 0.69 |
| LPL‐93T > G | 56 | 56 | −0.1 | 0.94 | +1.1 | 0.46 |
| PAI1‐675 4G/5G | 28 | 13* | −0.8 | 0.41 | −0.35 | 0.88 |
*P < 0.05.
†Age effect was analyzed using multivariate regression modeling. Findings were adjusted for gender, a history of diabetes mellitus, a history of hypertension, and current smoking.
When the association between high‐risk genotypes of the 18 SNPs and age was analyzed (Table 3), different trends were shown for white and nonwhite patients: high‐risk genotypes of 14 SNPs (78%) were associated with a lower age at onset of a first MI among white patients, whereas among nonwhite patients most mutation (61%) exhibited a nonsignificant association with an older age at onset of a first MI. Notably, among white patients the age‐reduction effect was marginally significant or significant for four mutations (apoplipoprotein E [APOE], cholesterol ester transfer protein [CETP], NOS3, and PON1), whereas among nonwhite patients none of the 18 mutations exhibited a trend to an age‐reducing effect.
Carriership Model
The average age effect among white and nonwhite patients per carried genotype, after adjustment for clinical variables, is shown in Table 4. Overall, carriership analysis of the 18 high‐risk genotypes demonstrated a nonsignificant trend to a reduction in the age at onset of a first MI among all study patients (an average of 0.29 year reduction per carried genotype; P = 0.21). This trend, however, was attributed to a significant age‐reduction effect among white subjects (an average of 0.6 year reduction per carried genotype; P = 0.027), whereas the contribution of genotypes to the age at onset of a first MI was nonsignificant among nonwhite patients (an average of 0.7 year increase per carried genotype; P = 0.16), with a significant interaction effect between the two groups (P value for genetic × ethnic interaction = 0.02). The carriership effect among nonwhite patients was consistent for the two main ethnic populations in this group, and demonstrated a nonsignificant age increase per carried genotype for African‐Americans (an average of 0.5 year increase per carried genotype; P = 0.43) and Latinos (an average of 1.1 year increase per carried genotype; P = 0.10).
Table 4.
Multivariate Regression Analysis: Factors Associated with a Change in the Age At Onset of a First MI
| Variable | White | Nonwhite | ||
|---|---|---|---|---|
| Age Difference (Years)* | P Value | Age Difference (Years)* | P Value | |
| Male | −5.3 | <.0001 | −2.8 | 0.09 |
| Hypertension | +4.4 | <.0001 | +5.3 | <.0001 |
| Smoking at any time | −5.2 | <.0001 | −4.3 | 0.007 |
| Diabetes mellitus | −0.5 | 0.09 | +0.5 | 0.15 |
| Change per carried genotype† | −0.6† | 0.027 | +0.7† | 0.16 |
*Change (increase or decrease) in the number of years at onset of a first MI associated with factor compared to that among patients without the factor.
†Age difference reflects the average change in the number of years at onset of a first MI per carried genotype.
†P value for carriership score × ethnic interaction = 0.02.
Clinical factors significantly associated with the age at onset of a first MI were similar among the ethnic groups and included gender, current smoking, and a history of treated hypertension.
The significant association between genetic factors and age at onset of atherothrombotic events among white patients indicates that for each high‐risk genotype a first MI would occur, on average, a six‐tenths of year earlier than among patients with the respective wild‐type mutation, with an additive effect per carried genotype. Consistently, white patients demonstrated a gradual decline in the mean age of MI onset with increasing number of carried genotypes (Fig. 2A). The mean age at onset of a first event in this population was 63 years among subjects carrying ≤3 high‐risk mutations; 60 years among subjects carrying 6 mutations; and 57 years among subjects with ≥10 mutations. By contrast, the age at onset of atherothrombotic disease among non white subjects was not related to the number of carried mutations (Fig. 2B).
Figure 2.
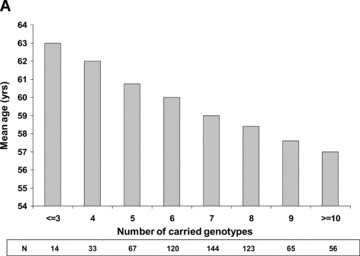
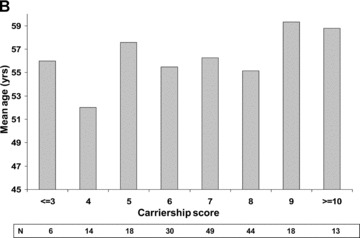
Mean age at onset of a first myocardial infarction per number of carried genotypes among (A) white patients; and (B) nonwhite patients.
DISCUSSION
In the present study we used a carriership model, based on 18 prespecified genotypes, to evaluate the cumulative contribution of atherosclerotic risk genotypes to early onset MI. The carriership score demonstrated an average of 0.6‐year reduction in the age at onset of coronary atherothrombotic disease per carried genotype among white patients, after adjustment for relevant clinical covariates. By contrast, the same prespecified genotypes did not contribute to an earlier onset of MI in other ethnic populations, including African‐Americans and Latinos.
The rationale behind the carriership approach is that the development of coronary atherothrombotic disease is only modestly affected by genetics, and is probably influenced more strongly instead by clinical and environmental factors. 3 Therefore, multiple genetic factors that are operative throughout each subject's life may have a small, yet additive, influence on the development of the disease. Our data in white subjects are consistent with this hypothesis. We have shown that individuals having a genetic factor that contributes to CAD experience a significant, albeit modest, reduction in the age at onset of a first MI. However, cumulative genetic burden conferred an additive effect in this population. For example, an environmental factor such as a history of smoking was associated with a significant 5.2‐year reduction in the age at onset of a first MI in white patients compared to an average reduction of 0.6 years, conferred by a single genetic factor. However, multiple genetic factors were associated with an additive age‐reduction effect. Thus, the contribution of ≥ 9 genetic factors in an individual to the onset of disease may be larger than the effect of smoking.
We have recently reported an unexpected association between the presence of seven of the 18 high‐risk genotypes analyzed in this study and a reduction in the risk of recurrent coronary events, in the same population of postinfarction patients who were followed‐up over an average period of 28 months after the index MI. 2 The apparently contradictory results between the previous study and the current analysis suggest that the biological mechanisms that affect the risk of recurrent coronary events may differ from those that contribute to the onset of a first coronary event. Specifically, the incremental effect of genetic factors that operate throughout the lifetime of an individual is more likely to be detected when the age at onset of a first MI is assessed. By contrast, the relative contribution of the same genetic factors to the risk of recurrent coronary events in the post‐MI patients may be smaller when compared with other factors, such as left ventricular function and infarct size, which dominate the risk during the 2‐year postinfarction period. These clinical factors tend to be more severe in older patients. Thus, younger patients, in whom genetic factors were shown to be more prevalent in the current study, may have less clinical risk in the postinfarction period, resulting in a lower risk for recurrent events in the younger population. A similar “paradox” has been shown for post‐MI smokers. 4 Cigarette smoking is recognized as a major modifiable risk factor for CAD, 5 and as a risk factor for early onset MI. 6 However, habitual cigarette smokers have been shown to present lower mortality rates following MI. 7 , 8 , 9 Similar to post‐MI patients with genetic risk factors, cigarette smokers who present with MI tend to be younger with less diffuse CAD and fewer comorbidities compared to nonsmokers, and these differences have been invoked to explain the lower recurrent event rate in this population. 10 , 11
Previous studies that assessed genetic risk have generated inconsistent data, possibly due to the fact that genotypes were studied in mixed ethnic populations, in whom the frequency of genetic polymorphisms and their association with outcome exhibit a significant variation. 12 , 13 , 14 Our results suggest that the effects of the 18 prespecified SNPs vary among ethnic groups, with a significant ethnic × genetic interaction effect. There are several explanations why a genetic variant that influences susceptibility in one population does not have the same effect in a different population. The interactions of a genotype with other genes and/or with environmental factors might differ among groups. A genotype associated with risk might not itself be causal, but instead be in linkage disequilibrium, the nonrandom association of alleles at different polymorphic sites of a chromosome, with a causal variant. An average haplotype block size of approximately 11 kb was reported in Africans and African‐Americans, compared with 22 kb in European and Asian samples. 15 Therefore, variation in the risk associated with a genetic factor among different populations might reflect variable linkage disequilibrium, with the presence of the associated variant and the causal variant on different haplotype blocks, rather than variation in a true genetic effect. It is also possible that clinical and/or environmental risk factors assume a relatively greater importance among nonwhite patients, thereby attenuating the risk associated with genetic factors in this population. The overwhelming majority of reported gene‐disease associations has been studied mainly in white patients, mostly of European ancestry. 16 In a report by Ioannidis et al., only 48 (7%) of 667 studies that have evaluated gene‐disease associations, were performed in populations of African origin. 17 Furthermore, the genetic‐cardiovascular association studies that were included in the current meta‐analyses comprised mostly white patients. We have shown that nine of the 18 studied SNPs exhibited a significant variation in frequency among ethnic groups, and the genotypes were associated with an early development of atherothrombotic disease only in white patients. Thus, it is possible that data derived from currently published genetic association studies pertain mostly to white patients, whereas the effect of these atherosclerotic risk genotypes in other ethnic groups, including African‐Americans and Latinos, is different. This may be due to unidentified factors, such as gene‐environment interaction or linkage disequilibrium, which should be studied directly in these populations.
STUDY LIMITATIONS
The present study is unique, since we have used the age at onset of the first MI as a quantitative variable for analyzing genetic risk. However, our SNP‐screening approach has all the limitations inherent in association studies. As discussed above, the likelihood of finding an association is influenced by the frequency of the SNP variant in the population and by the ethnic heterogeneity of the population. Finding an association in one or more individual candidate SNP variants (single‐marker associations) with the age‐related end point does not necessarily indicate causality, since the identified SNP may be in linkage disequilibrium with an unidentified causal genetic variant. In addition, atherosclerotic risk genotypes were selected in the current study through an extensive literature search. It should be noted, however, that meta‐analysis studies may reflect publication bias in which positive studies are more likely to be published in highly impacted journals.
CONCLUSIONS
Our genetic carriership approach suggests that currently identified atherosclerotic risk genotypes confer an additive contribution to the early development of atherothrombotic disease in white patients, resulting in an average of 0.6‐year reduction in the age at onset of disease per carried genotype in this population. The lack of a significant association between these genotypes and outcome in other ethnic populations suggests that cardiovascular genetic risk should be studied directly among various ethnic groups, in whom the effects of atherosclerotic genotypes may be modified differently by environmental or other genetic factors.
REFERENCES
- 1. Moss AJ, Goldstein RE, Marder VJ, et al Thrombogenic factors and recurrent coronary events. Circulation 1999;99:2517–2522. [DOI] [PubMed] [Google Scholar]
- 2. Moss AJ, Ryan D, Oakes D, et al Atherosclerotic risk genotypes and recurrent coronary events after myocardial infarction. Am J Cardiol 2005;96:177–182. [DOI] [PubMed] [Google Scholar]
- 3. Sankar P, Cho MK, Condit CM, et al Genetic research and health disparities. JAMA 2004;291:2985–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barbash GI, Reiner J, White HD, et al Evaluation of paradoxical beneficial effects of smoking in patients receiving thrombolytic therapy for acute myocardial infarction: Mechanism of the Smoker's Paradox from the GUSTO‐I trial, with angiographic insights. J Am Coll Cardiol 1995;26:1222–1229. [DOI] [PubMed] [Google Scholar]
- 5. Grundy SM, Pasternak R, Greenland P, et al Assessment of cardiovascular risk by use of multiple‐risk factor assessment equations. A statement for healthcare professionals from the AHA/ACC. Circulation 1999;100:1481–1492. [DOI] [PubMed] [Google Scholar]
- 6. Gottlieb S, Fallavollita J, McDermott M, et al Cigarette smoking and the age at onset of a first non‐fatal myocardial infarction. Coron Artery Dis 1994;8:687–694. [DOI] [PubMed] [Google Scholar]
- 7. Lee KL, Woodlief LH, Topol EJ, et al Predictors of 30‐day mortality in the era of reperfusion for acute myocardial infarction. Circulation 1995;91:1659–1668. [DOI] [PubMed] [Google Scholar]
- 8. De Chillou C, Riff P, Sadoul N, et al Influence of cigarette smoking on rate of reopening of the infarct‐related coronary artery after myocardial infarction: A multivariate analysis. J Am Coll Cardiol 1996;27:1662–1668. [DOI] [PubMed] [Google Scholar]
- 9. Gottlieb S, Boyko V, Zahger D, et al Smoking and prognosis after acute myocardial infarction in the thrombolytic era. J Am Coll Cardiol 1996;28:1506–1513. [DOI] [PubMed] [Google Scholar]
- 10. Kelly TL, Gilpin E, Ahnve S, et al Smoking status at the time of acute myocardial infarction and subsequent prognosis. Am Heart J 1985;110:535–541. [DOI] [PubMed] [Google Scholar]
- 11. Grines CL, Topol EJ, O'Neill WW, et al Effect of cigarette smoking on outcome after thrombolytic therapy for myocardial infarction. Circulation 1995;91:298–303. [DOI] [PubMed] [Google Scholar]
- 12. Cohen J, Pertsemlidis A, Kotowski IK, et al Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet 2005;37:161–165. [DOI] [PubMed] [Google Scholar]
- 13. Sehgal AR. Overlap between whites and blacks in response to antihypertensive drugs. Hypertension 2004;43:566–571. [DOI] [PubMed] [Google Scholar]
- 14. Sethi AA, Nordestgaard BG, Tybjaerg‐Hansen A. Angiotensinogen gene polymorphism, plasma angiotensinogen, and risk of hypertension and ischemic heart disease: A meta‐analysis. Arterioscler Thromb Vasc Biol 2003;23:1269–1275. [DOI] [PubMed] [Google Scholar]
- 15. Gabriel SB, Schaffner SF, Nguyen H, et al The structure of haplotype blocks in the human genome. Science 2002;296:2225–2229. [DOI] [PubMed] [Google Scholar]
- 16. Bamshad M. Genetic influences on health. Does race matter? JAMA 2005;294:937–946. [DOI] [PubMed] [Google Scholar]
- 17. Ioannidis JPA, Ntzani EE, Trikalinos TA. Racial” differences in genetic effects for complex diseases. Nat Genet 2004;36:1312–1318. [DOI] [PubMed] [Google Scholar]