

Abstract
Background: Cardiac myopathy manifesting as arrhythmias is common in the neurological disease, myotonic dystrophy type 1 (DM1). The purpose of the present study was to evaluate heart rate variability (HRV) in patients with DM1.
Methods: In a multicenter study, history, ECG, and genetic testing were performed in DM1 patients.
Results: In 289 patients in whom the diagnosis of DM1 was confirmed by a prolonged cytosine‐thymine‐guanine (CTG) repeat length the most common ambulatory ECG abnormality was frequent ventricular ectopy (16.3%). The 24‐hour time domain parameters of SDNN (SD of the NN interval) and SDANN (SD of the mean NN, 5‐minute interval) declined as age and CTG repeat length increased (SDNN: −8.5 ms per decade, 95% confidence intervals [CI]−12.9, −4.2, −8.7 ms per 500 CTG repeats, CI −15.7, −1.8, r = 0.24, P < 0.001; SDANN: −8.1 ms per decade, CI −12.4, −3.8, −8.8 ms per 500 CTG repeats, CI −15.7, −1.9, r = 0.23, P < 0.001). Short‐term frequency domain parameters declined with age only (total power: −658 ms2 per decade, CI: −984, −331, r = 0.23, P < 0.001; low frequency (LF) power −287 ms2 per decade, CI: −397, −178, r = 0.30, P < 0.001; high frequency (HF) power: −267 ms2 per decade, CI: −386, −144, r = 0.25, P < 0.001). The LF/HF ratio increased as the patient aged (0.5 per decade, CI: 0.1, 0.9, r = 0.13, P = 0.03).
Conclusions: In DM1 patients a decline in HRV is observed as the patient ages and CTG repeat length increases. A.N.E. 2003; 8(3):227‐232
Keywords: myotonic dystrophy, heart rate variability, genetics, arrhythmias
Myotonic dystrophy type 1 (dystrophia myotonica type 1, [DM1]) is a progressive, systemic, autosomal dominant neuromuscular disease. 1 The heart is commonly involved in DM1 primarily with myocardial fibrosis and degeneration often preferentially affecting the conduction system. 2 , 3 Patients with DM1 typically manifest cardiac involvement with arrhythmias including atrioventricular block, atrial and ventricular tachyarrhythmias, and sudden death. 4 , 5 , 6 , 7 , 8 Whether cardiac autonomic nervous system (ANS) abnormalities influence or accompany the myocardial degenerative changes in patients with DM1 is not clear. 9 , 10 , 11 , 12 , 13 , 14
The genetic basis of DM1 is an expanded trinucleotide cytosine‐thiamine‐guanine (CTG) repeat found on chromosome 19 in the 3′‐untranslated region of a serine‐threonine protein kinase, known as DMPK. 15 , 16 , 17 Individuals without DM1 have between 5 and 35 copies of the repeat, whereas in patients with DM1 between 50 and 2000 repeats have been observed. 1 The severity of skeletal muscle weakness in DM1 patients increases with increasing CTG repeat length. 18 , 19 The severity of cardiac conduction involvement and arrhythmias in DM1 patients increases with both increasing age and CTG repeat length. 20
The purpose of the present report is to evaluate the relationship between ANS function measured using heart rate variability (HRV) indices and clinical and genetic factors in patients with DM1.
METHODS
Patients
The Arrhythmias in DM1 Study is a multicenter registry designed to identify and evaluate individuals with DM1 and prospectively follow them to determine the arrhythmic outcome. The study was initiated in March 1997 with patients recruited at 23 neuromuscular specialty clinics associated with the Muscular Dystrophy Association (see Appendix). All adult patients (age ≥ 18 years) who carry a diagnosis of DM1 based on one or more characteristics of clinical findings, family history, electromyography, and muscle biopsy were invited to participate. The presence or absence of cardiac disease was not a consideration used to select patients for study entry. The Institutional Review Board at Indiana University and at each participating site approved the study. Informed consent was obtained from each study participant.
Clinical Evaluation, Resting ECG, and Genetic Testing
Neurological and cardiac characteristics were ascertained from the medical history and by physical examination done by the physician investigators. The severity of skeletal muscle weakness was scored using a standardized and validated 5‐point muscular disability rating scale. 21
Each patient underwent a standard 12‐lead ECG using local equipment. The ECGs were analyzed by a single investigator (W.J.G.) blinded to other study results.
Ten milliliters of venous blood was collected from each patient. Analysis of the DM1 CTG repeat sequence was performed on peripheral blood lymphocytes (Athena Diagnostics Laboratory, Worcester, MA). Polymerase chain reaction (PCR) based assays were used to determine small allele lengths of 5–100 repeats. 22 Southern‐blot‐based assays were used to estimate repeat lengths greater than 100. 17 Accuracy of allele length determination is estimated at two CTG repeats for repeats of 5 to 100 and 10% for repeats greater than 100.
24‐Hour ECG Ambulatory ECG Recording
Each site was provided with a digital monitor (Space Labs Burdick, Redmond, WA) to obtain the 24‐hour ambulatory ECG. During the initial 20 minutes of recording the patients were left quiet in an examining room in a resting and supine position. Normal activity was allowed during the remainder of the recording. The digital sampling rate was 1000 Hz during the initial 20 minutes and 200 Hz during the remainder of the 24‐hour recording.
Ambulatory ECG analysis and annotation were done using commercial software (Vision Premier, Space Labs Burdick, Redmond, WA) with review and editing by a single investigator (W.J.G.). Patients with known coronary artery disease or diabetes mellitus, with less than 18 hours of useable ECG data, with atrial fibrillation or flutter, or with a pacemaker were excluded from the current report.
HRV Analysis
Long‐term time‐domain analysis was measured from the entire useable ECG recording with HRV indices derived from the normal‐to‐normal RR (NN) intervals. 23 Three HRV indices were measured including standard deviation (SD) of the NN intervals (SDNN) as an estimate of overall HRV, the SD of the mean NN intervals measured over each 5 minutes (SDANN) as an estimate of long‐term components of HRV, and the square root of the mean of the sum of the squares of the NN interval difference (RMSSD) as an estimate of short‐term components of HRV.
A 5‐minute duration, preferentially minute 6 to 10, during the initial 20 minutes of recording was used for short‐term frequency domain analysis. Fast Fourier transform was used to determine power for a low frequency component (LF, 0.04–0.15 Hz), a high frequency component (HF, 0.15–0.4 Hz), and a total frequency (TF, 0–0.4 Hz). The ratio of LF to HF (LF/HF) was used as an index of sympathovagal balance. 23
Statistical Analysis
Variables are given as mean ± SD. Step‐wise multivariate linear and logistic regression analyses were used to determine the relationship between the factors. All tests were conducted with a two‐sided alpha risk level of 0.05. We used SPSS 10.0 for Windows (SPSS, Inc., Chicago, IL) for all analyses.
RESULTS
Of the 391 patients with a clinical diagnosis of DM1 enrolled in the registry, 11 had indeterminate and 32 had normal CTG repeat length and were excluded because the diagnosis of DM1 was questionable. In the remaining 348 patients, 11 carried a diagnosis of coronary artery disease and/or diabetes mellitus and were excluded because of a possible alteration in HRV related to these conditions. In the remaining 337 patients, HRV analysis was unavailable in 48 because 10 had a permanent pacemaker, 13 had atrial fibrillation or flutter, and 25 did not have an ambulatory ECG of sufficient duration or quality. This report evaluates the remaining 289 patients. Clinical, genetic, and resting ECG characteristics (Table 1) are similar to other published series of DM1 patients and to a previous report from this registry. 20 , 24 , 25 Similar to a previous report from this registry the muscular disability rating, ECG PR interval, and ECG QRS duration were related to age and CTG repeat length using multivariate linear regression. 20 As the DM1 patient aged and as CTG repeat length increased the muscular disability and cardiac conduction disease reflected in the PR interval and QRS duration worsened.
Table 1.
Clinical and Resting ECG Characteristics in Patients with Myotonic Dystrophy Type 1
| Characteristics | Finding (n = 289, ±SD) |
|---|---|
| Age (range), years | 41.4 ± 12.1 (18.0–77.8) |
| Gender | 153 female (52.9%) |
| Muscular disability rating scale (1–5) | 3.1 ± 1.0 |
| 1. No clinical muscular impairment | 12 (4.2%) |
| 2. Minimal signs without distal muscular weakness | 71 (24.6%) |
| 3. Distal muscular weakness | 83 (28.7%) |
| 4. Mild to moderate proximal muscular weakness | 110 (38.1%) |
| 5. Severe proximal muscular weakness | 13 (4.5%) |
| Heart failure (related to myotonic dystrophy) | 5 (1.7%) |
| CTG repeat length (range) | 630 ± 376 (54–1965) |
| Resting 12‐lead ECG | |
| Normal ECG | 108 (37.4%) |
| PR interval, ms | 195 ± 35 |
| First‐degree AV block | 129 (44.6%) |
| QRS duration, ms | 102 ± 20 |
| QRS duration ≥ 120 ms | 46 (15.9%) |
SD = standard deviation.
24‐Hour Ambulatory ECG and HRV
Ambulatory ECG results including HRV findings are reported in Table 2. The most common ECG abnormality observed was ventricular ectopy. Pauses of more than 3 seconds were observed in seven patients, related to paroxysmal AV block in five patients and sinus node slowing in two patients. The mean time domain analysis results are consistent with values at or slightly below accepted normal values. 23 The short‐term frequency domain analysis results showed wide variability with mean values consistent with overall diminished power and sympathetic predominance compared to accepted normal values.
Table 2.
24‐hour Ambulatory ECG Characteristics of Patients with Myotonic Dystrophy Type 1
| Characteristics | Finding (n = 289, ±SD) |
|---|---|
| Duration of recording, hours | 23.5 ± 1.0 |
| Average heart rate, beats per minute | 75 ± 9 |
| Ventricular pause > 3 seconds | 7 (2.4%) |
| Ventricular ectopy | |
| VPDs > 10 per hour | 47 (16.3%) |
| Ventricular couplets | 39 (13.5%) |
| Nonsustained ventricular tachycardia | 12 (4.2%) |
| Time domain analysis (24‐hour recordings) | |
| SDNN (ms) | 131 ± 44 |
| SDANN (ms) | 117 ± 43 |
| RMSSD (ms) | 39 ± 28 |
| Frequency domain analysis (5‐minute supine, resting) | |
| Low frequency power (ms2) | 888 ± 1179 |
| High frequency power (ms2) | 520 ± 1299 |
| Total power (ms2) | 2482 ± 3454 |
| Low frequency/high frequency ratio | 4.0 ± 4.4 |
SD = standard deviation; VPDs = ventricular premature depolarizations; SDNN = SD of the NN intervals; SDANN = SD of the mean 5‐minute NN intervals; RMSSD = square root of the mean of the sum of the squares of the NN interval.
Whether a relationship existed between HRV and clinical and genetic characteristics in the DM1 population was analyzed using multivariate linear regression. SDNN, as an estimate of overall HRV, was used as the dependent variable. Only two factors, age and CTG repeat length, were independently related to SDNN (r = 0.24, P < 0.001). The linear relationship was an inverse one with SDNN decreasing as age (SDNN: −8.5 ms per decade, 95% confidence intervals [CI]: −12.9, −4.2) and CTG repeat length (SDNN: −8.7 ms per 500 repeats, CI: −15.7, −1.8) increased. Using logistic regression both age (relative risk [RR]: 1.3 per decade, CI: 1.0, 1.6) and CTG repeat length (RR: 1.4 per 500 repeats, CI: 1.0, 2.0), P = 0.04 were predictive of an increased likelihood of a depressed SDNN of less than 100 ms.
No significant relationship was observed between the severity of skeletal muscle weakness and SDNN (r = 0.10, P = NS).
Measurement of the ECG PR interval provides a quantitative assessment of the degree of cardiac conduction degeneration present in the DM1 patient. 3 , 25 A weak but significant inverse relationship was present between SDNN and the PR interval (SDNN: −3.1 ms per 20 ms increase in PR interval, CI: −6.0, −0.2, r = 0.12, P = 0.04).
The relationship between age and CTG repeat length and SDNN is shown in Figure 1.
Figure 1.
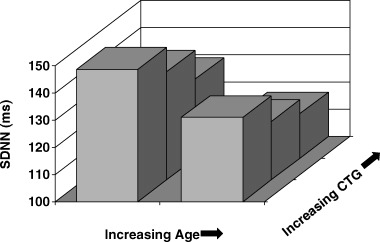
Mean SDNN values (ms) in relation to age and CTG repeat length. Shown is age divided at a median value compared with the CTG repeat length tertile. SDNN values declined as age and CTG repeat length increased (see text for linear regression analysis).
In a similar fashion to the relationship with SDNN, SDANN was inversely and independently related to age (SDANN: −8.1 ms per decade, CI: −12.4, −3.8) and CTG repeat length (SDANN: −8.8 ms per 500 repeats, CI: −15.7, −1.9), r = 0.23, P < 0.001. There was no significant relationship observed between RMSSD and age and CTG repeat length (r = 0.13, P = NS).
All short‐term frequency domain parameters were correlated to age but not CTG repeat length or other clinical variables by linear regression analysis. The relationship between age and power was an inverse one with total power (−658 ms2 per decade, CI: −984, −331, r = 0.23, P < 0.001), LF power (−287 ms2 per decade, CI: −397, −178, r = 0.30, P < 0.001), and HF power (−267 ms2 per decade, CI: −389, −144, r = 0.25, P < 0.001) decreasing as the DM1 patients aged. The relationship between LF/HF and age was weaker but remained significant with increasing sympathetic predominance (LF/HF: 0.5 per decade, CI: 0.1, 0.9, r = 0.13, P = 0.03) as the DM1 patients aged.
DISCUSSION
The present study demonstrates that in a large group of unselected DM1 patients the factors of age and the genetic abnormality of CTG length repeat correlate with HRV. The relationship is such that as the DM1 patient ages and/or as their CTG repeat length prolongs several time‐domain HRV indices decline. This study demonstrates an association between HRV and the severity of the genetic abnormality in DM1. The relationship of HRV with age and CTG repeat length is similar to the correlation seen between the severity of skeletal muscle weakness and cardiac conduction abnormalities with age and CTG repeat length. 18 , 19 , 20 These similar findings suggest that the pathophysiological process responsible for the genetic‐influenced, time‐dependent skeletal and cardiac muscle degeneration could as well depress cardiac autonomic function. Whether this depression in autonomic function is secondary to a direct cardiac effect or an indirect effect is not clear.
In this study, frequency domain HRV power declined with an overall increasing sympathetic predominance as the DM1 patient aged without an apparent association with CTG repeat length. A decline in the frequency domain HRV parameters with age has been previously shown in a normal population. 26 , 27
Previous Studies
Previous studies disagree on whether ANS abnormalities occur in patients with DM1. Mechler and Mastaglia found abnormal skeletal muscle blood flow response to pharmacological challenge in DM1 patients that they believed was secondary to the abnormalities in the vascular adrenergic response. 28 Other workers have reported abnormalities in pupillary light response related either to ANS abnormalities 9 or to smooth muscle dysfunction. 11 Several authors could not find significant abnormalities in cardiovascular autonomic reflexes in DM1 patients. 9 , 10 , 11 , 12 Inoue and colleagues reported abnormal short‐term frequency domain findings in 10 DM1 patients compared to age‐ and gender‐matched controls. 14 They found that total, LF, and HF power were all depressed in the DM1 patient. The small number of DM1 patients studied limits the interpretation of all of these previous reports.
Study Limitations
We did not include a control group to compare with the DM1 patients. The goal of the present study was to evaluate the ambulatory ECG in a large, clinically diverse population with DM1 to determine what factors associate with HRV.
The short‐term frequency domain data that were collected showed wide variations in parameter values. This wide variation could be responsible for the lack of a statistically significant association with CTG repeat length as was seen with the time‐domain HRV indices. Because the study was done at multiple centers there may have been variations in patient activity during frequency‐domain data recording despite the careful instructions given.
We cannot ascertain the etiology of the decline in HRV as the DM1 patient ages and as their CTG repeat length increases. The sympathetic predominance observed could be secondary to increasing weakness and a sedentary lifestyle. This seems less likely in that we did not observe a direct relationship between the severity of skeletal muscle weakness and HRV parameters. We did observe a relationship between the severity of cardiac conduction abnormalities reflected by the PR interval and HRV parameters supporting cardiac degenerative changes leading to a decline in HRV. Cardiac denervation determined by I‐123 metaiodobenzyl guanidine (MIBG) without perfusion abnormalities has been reported in a DM1 patient. 13
CONCLUSIONS
Sudden, presumptively arrhythmic, death is responsible for up to one third of deaths in DM1 patients. 7 , 8 The present study demonstrates that HRV declines as the DM1 patient ages and as CTG repeat length increases. This sympathetic predominance could play a role in a propensity to lethal arrhythmias in the DM1 patient.
APPENDIX
The authors thank the participating investigators in the Arrhythmias in Myotonic Dystrophy Study (in alphabetical order by last name): Richard Bailly, MeritCare Medical Group, Fargo, ND; Isabelita Bella, University of Massachusetts, Worcester, MA; John Butzer, Blodgett Memorial Medical Center, Grand Rapids, MI; William Cooper, Methodist Hospital, Indianapolis, IN; Robert Cranston, Carle Clinic, Urbana, IL; L. Matthew Frank, Eastern Virginia Medical School, Norfolk, VA; David Gelber, Southern Illinois University, Springfield, IL; Laurie Gutmann, West Virginia University, Morgantown, WV; Holli Horak, Indiana University, Indianapolis, IN; Jack Kron, Oregon Health Sciences University, Portland, OR; E. Wayne Massey, Duke University, Durham, NC; Robert McMichael, Neurology Associates, Arlington, TX; Richard Moxley, University of Rochester, Rochester, NY; Michael Nigro, Michigan Institute for Neurological Disorders, Farmington Hills, MI; Shin Oh, University of Alabama, Birmingham, AL; Rahman Pourmand, Indiana University, Indianapolis, IN; Thomas Rusche, Health South Tri‐State Rehabilitation Hospital, Evansville, IN; Robert Schwendimann, Louisiana State University, Shreveport, LA; Steven Sergay, Neurology Associates, Tampa, FL; Zack Simmons, Penn State, Hershey, PA; K. Sivakumar, Barrow Neurological Institute, Phoenix, AZ; Betty Soliven, University of Chicago, Chicago, IL; Donald Tamulonis, St. Elizabeth's Health Center, Youngstown, OH; Jackie Washington, Emory University, Atlanta, GA.
Supported by grants from Medtronic, Inc. (Minneapolis, MN), the Muscular Dystrophy Association (Tucson, AZ), and the National Institutes of Health (Grant MO1‐RR00750‐27S1).
REFERENCES
- 1. The International Myotonic Dystrophy Consortium . New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1). Neurology 2000;54: 1218–1221. [DOI] [PubMed] [Google Scholar]
- 2. Kennel AJ, Titus JL, Merideth J. Pathologic findings in the atrioventricular conduction system in myotonic dystrophy. Mayo Clin Proc 1974;49: 838–842. [PubMed] [Google Scholar]
- 3. Nguyen HH, Wolfe JT, Holmes,.et al Pathology of the cardiac conduction system in myotonic dystrophy: a study of 12 cases. J Am Coll Cardiol 1988;11: 662–671. [DOI] [PubMed] [Google Scholar]
- 4. Prystowsky EN, Pritchett EL, Roses AD, et al The natural history of conduction system disease in myotonic muscular dystrophy as determined by serial electrophysiologic studies. Circulation 1979;60: 1360–1364. [DOI] [PubMed] [Google Scholar]
- 5. Hawley RJ, Milner MR, Gottdiener JS, et al Myotonic heart disease: A clinical follow‐up. Neurology 1991;41: 259–262. [DOI] [PubMed] [Google Scholar]
- 6. Phillips MF, Harper PS. Cardiac disease in myotonic dystrophy. Cardiovasc Res 1997;33: 13–22. [DOI] [PubMed] [Google Scholar]
- 7. De Die‐Smulders CE, Howeler CJ, Thijs C, et al Age and causes of death in adult‐onset myotonic dystrophy. Brain 1998;121: 1557–1563. [DOI] [PubMed] [Google Scholar]
- 8. Mathieu J, Allard P, Potvin L, et al A 10‐year study of mortality in a cohort of patients with myotonic dystrophy. Neurology 1999;52: 1658–1662. [DOI] [PubMed] [Google Scholar]
- 9. Bird TD, Reenan AM, Pfeifer M. Autonomic nervous system function in genetic neuromuscular disorders: Hereditary motor‐sensory neuropathy and myotonic dystrophy. Arch Neurol 1984;41: 43–46. [DOI] [PubMed] [Google Scholar]
- 10. Aminoff MJ, Beckley DJ, McIlroy MB. Autonomic function in myotonic dystrophy. Arch Neurol 1985;42: 16. [DOI] [PubMed] [Google Scholar]
- 11. Den Heijer JC, Van Dijk JG, Bollen WL, et al Assessment of autonomic function in myotonic dystrophy. J Neurol Neurosurg Psychiatry 1991;54: 531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pierangeli G, Lugaresi A, Contin M, et al Autonomic nervous system function in myotonic dystrophy. Ital J Neurol Sci 1992;13: 589–592. [DOI] [PubMed] [Google Scholar]
- 13. Machida K, Honda N, Mamiya T, et al Abnormal sympathetic innervation of the heart in a patient with myotonic dystrophy detected with I‐123 MIBG cardiac SPECT. Clin Nucl Med 1994;19: 968–972. [DOI] [PubMed] [Google Scholar]
- 14. Inoue K, Ogata H, Matsui M, et al Assessment of autonomic function in myotonic dystrophy by spectral analysis of heart‐rate variability. J Auton Nerv Syst 1995;55: 131–134. [DOI] [PubMed] [Google Scholar]
- 15. Buxton J, Shelbourne P, Davies J, et al Detection of an unstable fragment of DNA specific to individuals with myotonic dystrophy. Nature 1992;355: 547–548. [DOI] [PubMed] [Google Scholar]
- 16. Aslanidis C, Jansen G, Amemiya C, et al Cloning of the essential myotonic dystrophy region and mapping of the putative defect. Nature 1992;355: 548–551. [DOI] [PubMed] [Google Scholar]
- 17. Brook JD, McCurrach ME, Harley HG, et al Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell 1992;69: 385. [DOI] [PubMed] [Google Scholar]
- 18. Tsilfidis C, MacKenzie AE, Mettler G, et al Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nature Genetics 1992;1: 192–195. [DOI] [PubMed] [Google Scholar]
- 19. Redman JB, Fenwick RG, Jr ., Fu YH, et al Relationship between parental trinucleotide GCT repeat length and severity of myotonic dystrophy in offspring. J Am Med Assoc 1993;269: 1960–1965. [PubMed] [Google Scholar]
- 20. Groh WJ, Lowe MR, Zipes DP. Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J Cardiovasc Electrophys 2002;13: 444–448. [DOI] [PubMed] [Google Scholar]
- 21. Mathieu J, Boivin H, Meunier D, et al Assessment of a disease‐specific muscular impairment rating scale in myotonic dystrophy. Neurology 2001;56: 336–340. [DOI] [PubMed] [Google Scholar]
- 22. Fu YH, Pizzuti A, Fenwick RG, et al An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255: 1256–1258. [DOI] [PubMed] [Google Scholar]
- 23. Task force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology . Heart rate variability: Standards of measurement, physiological interpretation, and clinical use. Circulation 1996;93: 1043–1065. [PubMed] [Google Scholar]
- 24. Melacini P, Villanova C, Menegazzo E, et al Correlation between cardiac involvement and CTG trinucleotide repeat length in myotonic dystrophy. J Am Coll Cardiol 1995;25: 239–245. [DOI] [PubMed] [Google Scholar]
- 25. Lazarus A, Varin J, Ounnoughene Z, et al Relationships among electrophysiological findings and clinical status, heart function, and extent of DNA mutation in myotonic dystrophy. Circulation 1999;99: 1041–1046. [DOI] [PubMed] [Google Scholar]
- 26. Hellman JB, Stacy RW. Variation of respiratory sinus arrhythmia with age. J Appl Physiol 1976;41: 734–738. [DOI] [PubMed] [Google Scholar]
- 27. Hrushesky WJ, Fader D, Schmitt O, et al The respiratory sinus arrhythmia: A measure of cardiac age. Science 1984;224: 1001–1004. [DOI] [PubMed] [Google Scholar]
- 28. Mechler F, Mastaglia FL. Vascular adrenergic receptor responses in skeletal muscle in myotonic dystrophy. Ann Neurol 1981;9: 157–162. [DOI] [PubMed] [Google Scholar]