

Abstract
The aim of this review is to present the properties of the slow component of the delayed rectifier potassium current (IKs) in the human ventricle. The review gives a detailed description of the physiology, molecular biology and pharmacology of the IKs current, including kinetic properties, genetic structures, agonists and antagonists. The authors also present the role of the IKs current in the human cardiac repolarization focusing on several pathophysiological situations, such as the LQT syndrome and the Torsade de Pointes arrhythmia.
Keywords: K+ current, IKs, cardiac action potential, repolarization reserve, arrhythmias, long‐QT syndrome
INTRODUCTION
Action potential of the cardiac ventricular muscle (Fig. 1), which normally shows a more than a hundred millisecond plateau phase, provides the opportunity of the heart for rhythmic contraction to pump blood through the vessels to supply the different organs. This unique electrophysiological property of the ventricle, unlike other excitable tissues like nerve or skeletal muscle lacking plateau phase, is due to the simultaneously opening and closing transmembrane ion channels providing the possibility of positively and negatively charged ions to pass, and thereby carrying inward and outward currents through the cell membrane. Inward depolarizing currents in the heart are mainly carried by sodium and calcium, whose ions move from the extracellular space to the cytosol. Outward repolarizing currents mainly but not exclusively are carried by potassium ions that flow from the inside of the myocytes to the extracellular space. In the cardiac muscle, there are several potassium channels, which distinctly differ from each other in respect of activation range, channel gating kinetic properties, and pharmacological response to various drugs. Changes or modulation of the potassium channels may lead to both antiarrhythmic and proarrhythmic consequences, therefore, better understanding of the function of these channels has great importance; it is a necessity in the therapy or prevention of life‐threatening cardiac arrhythmias.
Figure 1.
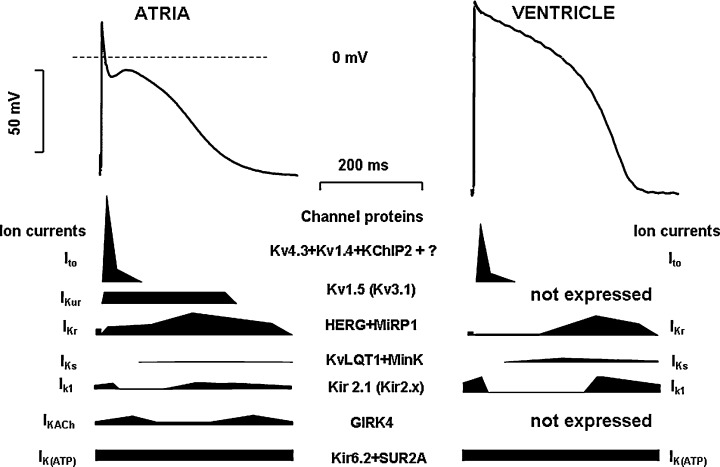
Action potential waveforms and underlying ionic currents in human atrial (left) and ventricular (right) myocytes. Ito= transient outward current; IKur= ultrarapid delayed rectifierpotassium current; IKr= rapid delayed rectifierpotassium current; IKs= slow delayed rectifierpotassium current; IK1= inward rectifierpotassium current; IK,ACh= acetylcholine‐sensitive potassium current; IK(ATP)= ATP‐dependent potassium current. Structural channel protein components are given for all currents. Cartoons show schematic current profileof the corresponding potassium current.
ELECTROPHYSIOLOGICAL PROPERTIES OF THE IKs CURRENT
The delayed rectifier potassium current (IK) is a major outward current responsible for ventricular muscle action potential repolarization. 1 , 2 This current was first described by Noble and Tsien using the two‐microelectrode voltage‐clamp technique in sheep cardiac Purkinje fibre strands. 3 Since its discovery, it has been examined in single isolated myocytes obtained from various regions of the heart in several mammalian species. 4 , 5 , 6 , 7 In most species, IK can be separated into rapid (IKr) and slow (IKs) components that differ from one another in terms of their sensitivity to drugs, rectification characteristics, and kinetic properties. 2 , 4 , 6 , 8 , 9 , 10
In dog cardiac ventricular cells, it was shown that IKs activated slowly (τ≈800 ms) at voltage more positive than 0 mV, and deactivated rapidly and monoexponentially (τ≈150 ms). 6 In the guinea pig, where it was first described, IKs activated very slowly, not saturating even after 5–7 s at +50 mV, and deactivated slowly, within 500–1000 ms. 11 First, it was not observed in the rabbit, 2 but later studies revealed a large and consistent IKs in rabbit ventricle as well. 7 , 12
Because of the species differences regarding the existence and properties of IKs, it would only be proper to ask as to how these findings can be extrapolated to humans. In view of the known difficulties in obtaining human tissue in general and undiseased human ventricular tissue for research in particular, few studies have so far characterized IKs in human ventricle. Some of the available data on IKs have been obtained in ventricular myocytes dissociated from diseased human hearts. Beuckelmann et al. 13 reported that IKs was absent or hardly detectable in human left ventricular myocytes. Li et al. 14 described both IKr and IKs in right ventricular myocytes obtained from explanted diseased human hearts. In this study, IKs was examined using CdCl2 to eliminate the inward Ca current (ICa), and BaCl2 to block the inward rectifier potassium current (Ik1). 14 Since Cd2+ substantially changes the kinetic properties and amplitude of IKs, 5 and Ba2+, due to the voltage dependent block and unblock of IK1 channels, 15 , 16 elicited a tail current similar to IKs tail current, 16 the results of the above study by Li et al. regarding the properties of IKs 14 must be interpreted with caution.
In cardiac ventricular myocytes isolated from undiseased human donor hearts, IKs exhibited slow and voltage independent activation at more positive than 0 mV (τ≈ 900 ms, Fig. 2A,B) and had fast and monoexponential deactivation kinetics (Fig. 2D). 16 This deactivation was voltage‐dependent, being faster at more negative (at ‐50 mV, τ≈ 90 ms), and slower at more positive voltages (at 0 mV, τ≈ 350 ms) (Fig. 2D). 16
Figure 2.
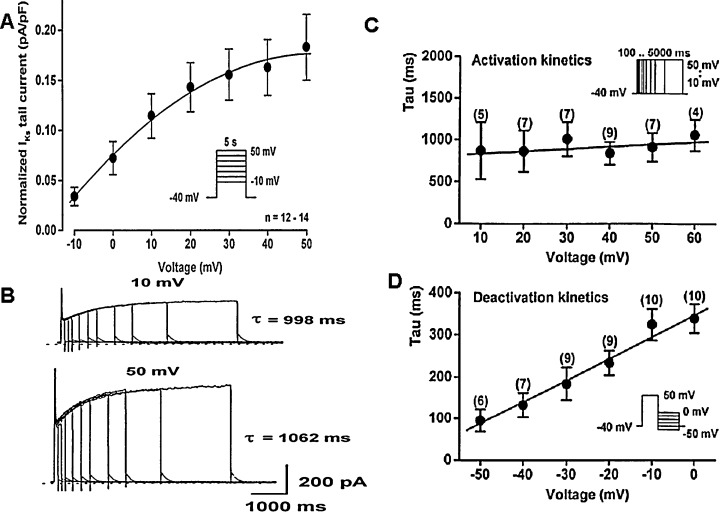
The properties of slow delayed rectifierpotassium current IKs in undiseased human ventricular myocytes. Panel A. The cell sized normalized current–voltage relation of IKs tail currents. Vertical axis represents peak tail current normalized to cell size measured in pA/pF, while horizontal axis gives the membrane potential measured in millivolts. Inset shows the applied voltage protocols, n indicates the number of experiments. Panels B–D. The voltage dependence of activation and deactivation kinetics of human IKs. Panel B shows original IKs tail current activation kinetic traces measured at depolarizing voltages to +30 mV and after returning to −40 mV. Panels C and D show the voltage dependence of the activation (C) and deactivation (D) time constants. Vertical axis: The activation and deactivation time constants (τ) in milliseconds; Horizontal axis: The membrane potential measured in millivolts. Number in parentheses represents the number of experiments at each voltages (from Ref. 16, used with permission).
IKs is known to be enhanced by elevation of the intracellular cAMP level, therefore the current is augmented during increased sympathetic tone. 17 , 18
Considering the kinetic properties, the human cardiac slow delayed rectifier potassium current 16 best resembles those measured in the dog 6 , 8 and rabbit 7 , 12 ventricle, but significantly differs from those found in the guinea pig heart, 4 , 9 , 10 where the density of IKs is very large. The kinetic properties of IKs also differ from each other in the rabbit, dog, or human ventricle (Fig. 3, own unpublished results). Nevertheless, in this respect, the dog and rabbit seem to be more suitable species for preclinical cardiac electrophysiological evaluation of new drugs than the guinea pig.
Figure 3.
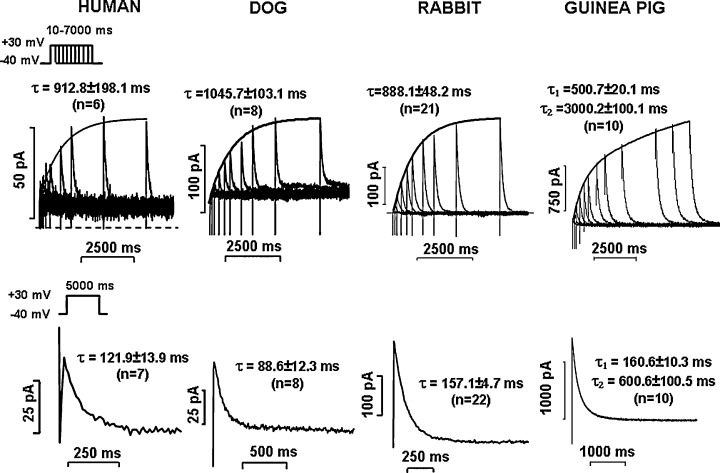
The activation (upper panels) and deactivation (bottom panels) kinetics of IKs in human, dog, rabbit, and guinea pig ventricular myocytes. Currents were measured with patch‐clamp technique as described in 12, 16, 49, and 59. Activation kinetics was studied by applying the envelope of tails protocols (see inset) from the holding potential of −40 mV. Insets show the value of activation or deactivation time constants (τ) determined by fitting the activation and deactivation curves with exponential functions. n = number of experiments. (Unpublished results from the present authors.)
MOLECULAR BIOLOGY OF THE IKs CURRENT
The structural organization of the ion channel mediating IKs was subject of extensive studies during the last decade, and at the very beginning it was controversial. Initially, it was suggested that this current was created by MinK. 19 , 20 The name itself means that minimum sequence required for a K‐current. 20 This gene (KCNE1) encodes a protein (MinK or IsK) that contains only 130 amino acids and has only one single membrane‐spanning domain with an extracellular N‐terminus. 19 MinK mRNA has been found in the mouse 19 and neonatal rat 21 heart, and the protein has been detected immunohistochemically in guinea pig ventricular myocytes. 22 This hypothesis was supported when expression of MinK protein in oocytes resulted in a current that resembled the slow delayed rectifier IKs. 23 Moreover, altered ionic selectivity and regulation of the expressed minK current after mutagenesis of the transmembrane segment and the PKC consensus site seemed to support the idea that minK encoded the IKs channel. 24 , 25 Later, positional cloning identified a novel gene KCNQ1 responsible for the chromosome 11‐associated form of the congenital LQT1 syndrome. 26 , 27 However, the expressed KvLQT1 protein forms a channel conducting a current unlike any current previously identified in cardiac preparations. This was surprising, since mutations in KvLQT1 cause the LQT1 syndrome known to be related to defects in the IKs channels. 26 , 27 Parallel two groups led by Sanguinetti 27 and Lazdunski 28 found and published in Nature that coexpression of KvLQT1 and MinK yielded a current that corresponds to the native slow IKs component. These observations, together with the biochemical data demonstrating that heterologously expressed KvLQT1 and MinK proteins can associate, have been interpreted as suggesting that MinK coassembles with KvLQT1 to form functional cardiac IKs channels (Fig. 4). In addition, the finding that mutations in the transmembrane domain of MinK alter the properties of the KCNQ1‐encoded Kv channels was interpreted as suggesting that the transmembrane segment of MinK contributes to the channel pore. 24 , 29 , 30 Thus, MinK controversy was ended by the demonstration that it acts only as β‐subunit that alters the intrinsic gating of KvLQT1; the IKs like MinK current in oocytes was due to its association with an endogenous Xenopus XKvLQT1 subunit. 27
Figure 4.
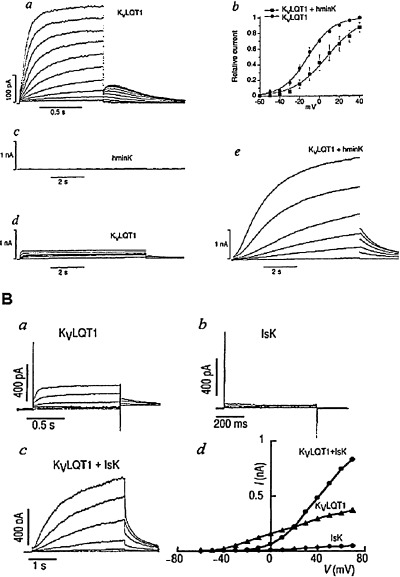
Transient expression of KvLQT1, IsK/MinK, and KvLQT1 + IsK/MinK in transfected chinese hamster ovary (CHO, Panel a) or african green monkey kidney (COS, Panel b) cells induce a current nearly identical to cardiac IKs. KvLQT1is the pore forming, while MinK/IsK is the auxiliary subunit of the IKs current. [from Ref. 27 (Panel a) and Ref. 28 (Panel b), used with permission].
KvLQT1 protein (KCNQ1 gene) is the pore‐forming subunit of the IKs channel complex with cytoplasmic N‐ and C‐terminal domains (Fig. 5). 31 , 32 The subunit consists of six transmembrane domains with helical structure. The pore region is the intervening loop inserted between segments 5 and 6, which is the responsible for the ionic selectivity and the conduction. 33 The amino acid sequence of this region (TxGYG) is highly conservative among Kv channels. The S domain contains numerous positively charged amino acids and plays a role as a voltage sensor for voltage dependent activation kinetics. 33 Two splice variants of the KvLQT1 subunit have been identified, the isoform‐1 and isoform‐2. 34 Isoform‐2 is an N‐terminally truncated version of the isoform‐1, having suppressive effects on the current generated by the full length KvLQT1 protein. Five different tetramers can be constructed from the two isoforms: two homotetramers and three heterotetramers. The maximum conductance as well the time constants for activation were shown to be different for these channels suggesting that the level of isoforms‐2 expression can be important for the IKs density. 35 For instance, the transmural heterogeneity of in repolarization was attributed to heterogeneous IKs channel density. KvLQT1 subunit alone forms voltage gated potassium channel with rapid activation kinetics, but when the MinK subunit binds to the KvLQT1 unit, the activation slows dramatically and the voltage dependence of activation shifs toward positive potentials. 36
Figure 5.
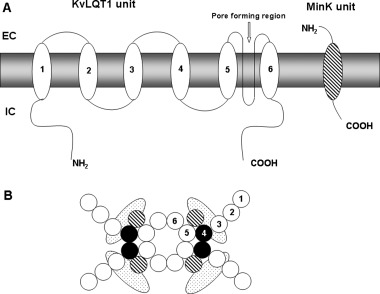
Schematic representation of the principal subunits to form IKs channel. Panel A. Relation of the two major subunits (KvLQT1 and MinK) in the cell membrane. Panel B. Formation of the aqueous pore by four subunits. In both panels, numbers indicate the six alpha helical domains traversing the cell membrane. EC and IC mean extracellular and intracellular region, respectively.
The MinK protein (KCNE1 gene) named also as IsK consists of 126–130 amino acids with a single transmembrane segment. The N‐terminal is N‐glycosylated and located on the extracellular side of the membrane. 19 Subunit MinK cannot form a pore alone, it only modifies the kinetic properties of the KvLQT1 subunit, when its transmembrane domain interacts with the pore‐forming segment of KvLQT1. 27 There are several other known members of the KCNE family (KCNE2, KCNE3 KCNE4, and KCNE5), which also interact with KCNQ1, influencing the behavior of the IKs current, and with other Kv family currents (Kv4.3 or HERG). 33 , 37 , 38 Mutations in MinK protein may also lead to development of the Long‐QT (LQT4 and LQT5) syndrome. 39 , 40 , 41
Table 1 summarizes the gene and protein structure forming the IKs channel.
Table 1.
The IKs Channel Pore Forming α and Auxiliary β‐Subunits
| Subunit | Protein | Gene | Locus in Human | Current |
|---|---|---|---|---|
| Alpha (pore) | KvLQT1 | KCNQ1 | 11p15 | IKs |
| Beta (auxiliary) | MinK, IsK | KCNE1 | 21q22 | IKs |
PHARMACOLOGY OF THE IKs CURRENT
Several activators and blockers of IKs have been developed in the last decade. The failure of the IKr blockers and the Sanguinetti hypothesis, 11 which proposed positive rate dependent prolongation after inhibition of IKs, turned the attention of the pharmaceutical industry to develop IKs blockers as new antiarrhythmics devoid of reverse rate dependent action potential duration (APD) lengthening effects. Until now, three compounds and their analogs have been described as selective blockers of the IKs channel.
The Hoechst/Aventis compound chromanol 293B has been reported as the first selective blocker of the IKs. 42 Several studies showed its efficacy on the IKs channel parallel with its purported Class III antiarrhythmic efficacy. 43 , 44 However, it was shown that chromanol 293B, at the concentration required for the full IKs block (about 30‐50 μM), affects Ito, another important repolarizing current, 43 which made the interpretation that selective IKs block lengthens repolarization substantially, and thereby elicits antiarrhythmic effect, became rather uncertain. However, in these studies, the investigators used chromanol 293B concentrations up to 100–150 μM, were the agent looses its selectivity even regarding IKr. 45 , 46 In vivo studies with chromanol 293B were also contradictory. 47 , 48 Our group reported no discernible changes in QTc interval following administration of chromanol 293B in anaesthetised dogs 49 and Langendorff perfused rabbit hearts, 12 whereas others observed T wave abnormalities and QT prolongation. 48 In the late nineties, by variation of the aromatic substituent, a more potent analog of chromanol, the HMR‐1556, was developed at Aventis. 50 This compound 51 has a higher potency and better selectivity compared to chromanol. Its EC50 value is in the submicromolar range (about 30–50 nM), 51 which made HMR‐1556 one of the most suitable tools for testing the properties of IKs current in vitro 51 , 52 , 53 and in vivo 54 , 55 as well.
The second chemical classes of IKs channel blockers are benzodiazepine derivates synthesized by Merck‐Sharpe&Dohme Pharma (MSD). Several from this family of compounds, such as L‐735,821 56 and L‐768,673, 57 proved to be very effective (EC50 about 1–20 nM) and selective IKs blockers. 58 In our own studies, L‐735,821 failed to lengthen significantly the rabbit, 12 dog, 49 and human 59 APD, while in other studies, L‐768,673 lengthened the guinea pig 58 and rabbit 60 APD. L‐768,673 was shown to prevent certain types of arrhythmias in both anesthetized and conscious dogs. When L‐768.673 was combined with the beta‐adrenoceptor blocker timolol, additional antiarrhythmic effect was reported in dogs with myocardial infarction. 61
The third class of IKs blockers was recently developed by Bristol Myers Squibb. It is represented by the benzamide derivative BMS‐208782. The compound has a high potency to inhibit IKs, (EC50 is about 9 nM), but no other meaningful pharmacological data have so far been reported regarding this compound. 62
Several other drugs, in addition to blocking different transmembrane ion channels, inhibit the IKs current as well. For example, such drugs are amiodarone, 63 azimilide, 64 bepridil, 65 cibenzoline, 66 clofilium, 67 imipramine, 68 indapamide, 69 mibefradil, 65 propafenone, 70 terfenadine, 71 tedisamil. 72
Activators of IKs were also described. DIDS 31 and mefenamic acid, 31 , 73 at relatively high concentrations (100–200 μM), are able to increase the amplitude of IKs, but these drugs at such a high concentrations affect also other transmembrane ion currents. A benzodiazepine derivate L‐364,373 (R‐L3) 73 from MSD was first reported to enhance IKs in the guinea pig when applied in the submicromolar concentration range, but the effect seems dependent either on the species/tissue used for the investigation, or on other experimental conditions, since in later studies, the same compound showed less potency in the rabbit, 74 or even lack of effectiveness in the dog ventricle. 75
The therapeutic usefulness of IKs activators is uncertain, but one might speculate that enhancing IKs would strengthen the repolarization reserve, thereby decreasing the risk of torsade de pointes arrhythmias in situations where potassium channels are inhibited by different drugs or when the channels are downregulated (for example by heart failure, diabetes) or when these channels have loss of function due to genetic causes, like in congenital long QT syndromes. 75 , 76
THE ROLE OF THE IKs CURRENT IN THE HUMAN CARDIAC REPOLARIZATION
Previously, it was generally accepted that inhibition of IKs would result in a significant lengthening of repolarization, thereby providing Class III antiarrhythmic effect to avoid reverse‐rate dependent action potential prolongation. This was suggested by Jurkiewicz and Sanguinetti, 11 who based this on their earlier observation in guinea pig ventricular myocytes, where the deactivation of IKs is slower than the normal length of the diastole. Consequently, significant accumulation of IKs could be expected during fast rate, i.e., short diastole. This accumulation of IKs was supposed to contribute to the repolarization‐shortening usually seen during short cycle length. Therefore, pharmacological block of IKs was believed to elicit more APD lengthening at fast than at normal or slow rates, and there has been an effort to develop selective IKs blockers as potential antiarrhythmic blockers agent without the risk of generating torsade de pointes arrhythmias. 11
The absence of selective IKs blockers until the last decade made it impossible to directly evaluate the physiological role of IKs in determining cardiac action potential configuration. Early studies with selective IKs blockers provided controversial results as to how inhibition of IKs would affect repolarization in the ventricle. Some studies provided evidence that selective inhibition of IKs resulted in significant APD or QT prolongation, 45 , 46 , 48 but other studies 49 , 54 did not confirm it. Repeated later investigations in the dog, 49 rabbit, 12 and human 59 ventricle in the absence of sympathetic stimulation showed that inhibition of IKs by chromanol 293B, L‐735,821 and HMR‐1556 did not cause significant lengthening of repolarization (Fig. 6) at a wide range of stimulation frequency. The explanation for these unexpected results came from the careful experimental analysis of the properties of IKs itself. Since IKs activates slowly (τ≈ 900‐1000 ms) at more positive voltages, there is only very little current that can be expected to flow during the time and voltage courses of the plateau of the normal action potential, which is usually shorter than 200 ms, and seldom gets to more positive voltages than 20–30 mV. 12 , 16 , 49 , 59 However, in situations where the APD is prolonged beyond normal, there is a possibility for more IKs activation to allow an important means of limiting excessive APD lengthening by a negative feedback mechanism (Fig. 7) providing more safety of the repolarization process. 12 , 49 , 59 In the presence of enhanced sympathetic stimulation, however, the magnitude of IKs is increased, its activation speeds up, and shifted into more negative voltages. 54 In addition, due to the simultaneous sympathetic enhancement of the L‐type calcium current (ICa), the plateau voltage of the action potential would shift upward in more positive direction, further increasing the activation of IKs. 77 Therefore, IKs block in the presence of elevated sympathetic activity may result in a significant repolarization lengthening (Fig. 8). This latter effect may be further enhanced by the lack of the outward IKs, which does not oppose the augmented inward ICa current. 77 In general, it can be concluded that the pharmacologic block of IKs does not provide antiarrhythmic benefit, but, on the contrary, it can decrease the safety of repolarization and consequently increase the proarrhythmic risk, especially if sympathetic tone is enhanced. 54 , 59
Figure 6.
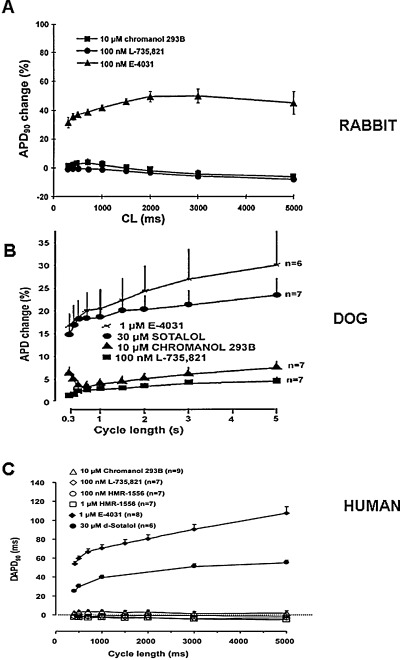
Frequency‐dependent effect of selective IKr and IKs block on action potential duration (APD) in dog (Panel A), rabbit (Panel B), and human (panel C) ventricular papillary muscles. Abscissa = pacing cycle length in milliseconds; ordinate = percentile changes in APD90[from Ref. 12 (Panel A), Ref. 49 (Panel B), and Ref. 59 (Panel C), used with permission].
Figure 7.
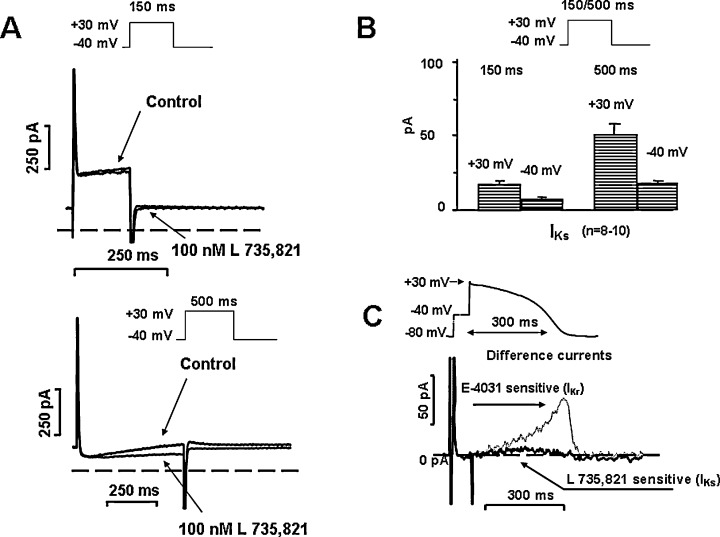
Panel A. L‐735,821 sensitive (IKs) currents in human ventricular myocytes after application of short (150 ms, upper) and long (500 ms, bottom) depolarizing test pulses. Insets show applied voltage protocols. Panel B. Average IKs currents at the end of a short (150 ms) a long (500 ms) depolarizing test pulses to +30 mV, and peak tail currents at −40 mV. Panel C. L‐735,821 sensitive (IKs) difference current recorded during an “action‐potential‐like” test pulse in human ventricular myocytes in the absence of any sympathetic agonist. The dotted line shows superposed the E‐4031 sensitive (IKr) current measured in similar condition [modified from Ref. 59, used with permission].
Figure 8.
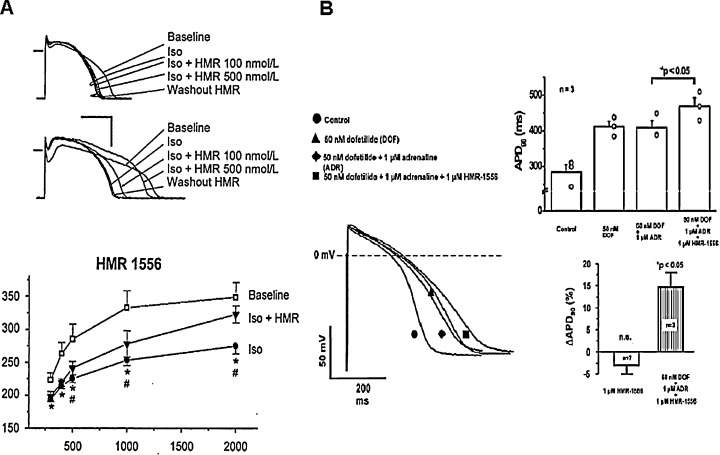
Effect of IKs block in the presence of elevated sympathetic activity. IKs blocker HMR‐1556 frequency dependently lengthens APD in dog (Panel A) and human (Panel B) ventricular myocytes, when the repolarization reserve is attenuated either by sympathetic stimulation (by isoprenaline, ISO) or combined IKr block with stimulation (by dofetilide + adrenaline, DOF + ADR) [from Refs. 12 (Panel A), Ref. 54 (Panel A) and Ref. 59 (Panel B), used with permission].
BLOCK OF THE IKs CURRENT CONTRIBUTES TO THE DEVELOPMENT OF EXPERIMENTAL TORSADE DE POINTES MODELS
The recent observations made with pharmacological block of IKs have contributed to the development of experimental LQT1 and/or torsade de pointes models. Volders et al. 78 reported that 3–4 weeks after AV ablation in dog, significant bradycardia and cardiac hypertrophy developed. In this model, several potassium currents, most notably IKs, were downregulated, and APD and QTc were prolonged. 78 In these dogs, drug treatments very often caused torsade de pointes arrhythmias, making this experimental model very useful method to test the proarrhythmic potency of drugs, which interfere with repolarization in general. 78 Similarly pharmacological block of IKs by chromanol 293B or HMR‐1556 in rabbit 79 and dogs 45 , 46 , 80 did not prolong QTc markedly, but made the heart susceptible toward eliciting torsade de pointes arrhythmias by IKr block with dofetilide. In these experiments, dofetilide alone did not or only occasionally induced torsade de pointes, but applying it after full IKs block, the drug increased the reverse‐rate dependent APD prolongation excessively, and the incidence of torsade de pointes arrhythmias were more than 70%, providing a useful model for safety pharmacology studies (Fig. 9). 79 , 80 Recently, it was also suggested the increased short‐term beat to beat APD/QT variability as a more sensitive measure of the proarrhythmic risk is related to decreased function of IKs. 81 Tachypaced rabbits showed marked downregulation of IKs, and developed torsade de pointes arrhythmias after dofetilide administration. On the contrary, bradypaced rabbits developed spontaneous torsade de pointes arrhythmia with the concomitant downregulation of both IKs and IKr channels. 82
Figure 9.
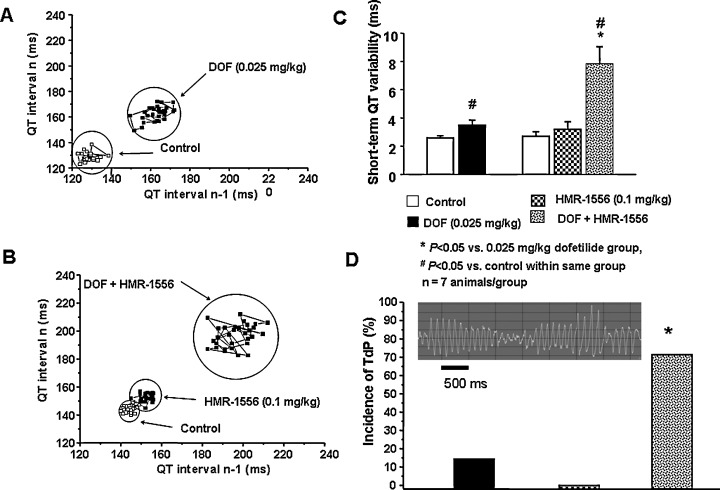
Representative examples of Poincaré plots of the QT interval from an anesthetized rabbit treated with the IKr blocker dofetilide (25 μg/kg i.v., DOF) (Panel A), and from another one treated with the IKs blocker HMR 1556 (0.1 mg/kg i.v.) followed by dofetilide (25 μg/kg i.v.) (Panel B). Short‐term QT interval variability was increased after dofetilide administration. Panel C. Combined IKr and IKs block markedly increased QT variability and provoked torsade de pointes arrhythmia (TdP) (Panel D). #P < 0.05 versus control in the same group, *P < 0.05 versus dofetilide treatment, n = 7 animals/group. (Unpublished results from the present authors and Dr. I. Baczkó; partially presented as abstract at the Heart&Rhythm Meeting‐2006; Ref. 80.)
THE REPOLARIZATION RESERVE AND THE IKs CURRENT
The concept of “repolarization reserve” was suggested by Dan Roden. 83 , 84 , 85 According to this terminology, the normal repolarization is accomplished by multiple different potassium channels providing a strong safety reserve for normal repolarization. Thus, under normal conditions, the pharmacological block or impairment of one single type of potassium channels does not necessarily lead to marked QT interval prolongation. However, in the presence of a subclinical impairment in the repolarization process, i.e., an otherwise mild potassium channel block (e.g., due to inhibition of the IKs current resulting in a decrease in repolarization reserve) 49 , 59 may precipitate marked QT prolongation, which can result in life‐threatening torsades de pointes arrhythmia.
CONGENITAL LONG QT SYNDROME AND IKS CURRENT
Congenital LQT syndrome is rather infrequent in the general population (1/5000), and is identified by QT prolongation on the surface ECG during clinical evaluation of unexplained syncope. 86 The cause of the syncope that occurs in this disorder is due to transient rapid polymorphic ventricular tachycardia known as torsades de pointes linked to delayed repolarization of the cardiac ventricular muscle. So far, seven forms of LQT (LQT1‐7) have been well characterized. They are caused by several hundred different mutations in the IKs, IKr, INa, IK1 alpha and beta subunits. 87 , 88 , 89 , 90
“Among the three most common LQTS genotyped (LQT1‐3), LQT1 probably hosts the largest percentage of genotype‐positive individuals displaying a normal/borderline resting QTc” as described in a recent review of Ackerman. 91 Similarly, simple pharmacological modulation of IKr, INa and IK1 channels also prolongs ventricular repolarization resulting in drug‐induced LQT. 92 In contrast, as mentioned before, even full pharmacological block of IKs does not lengthen repolarization in the ventricle, unless sympathetic tone is enhanced 59 , 93 or repolarization had been delayed previously by other means. 49 Based on these observations, it was concluded that IKs (KvLQT1 + MinK) channel is not a major contributing factor to “normal” repolarization but it is an important source of the repolarization reserve that opposes excessive lengthening of APD and consequently protects against torsades de pointes arrhythmia during possible impairment or change in normal function of other transmembrane ion channels. A recent study from Boulet et al. 94 describes a loss of function mutation in the KCNQ1 gene underlying the pore‐forming unit of IKs channel in a 40‐year‐old women. She had experienced torsade de pointes arrhythmia with QTc interval of 430 ms (which is well within the normal range in women), and this may also indicate that a genetic loss of IKs function does not necessarily prolong repolarization, as shown by the normal QTc of the patient. However, under some unfavourable conditions (e.g., hypokalemia, drug effects, downregulation of potassium channels), the impairment of the repolarization reserve could not provide the necessary protection, making this patient more vulnerable toward arrhythmia than those who lack defective IKs channels. In accordance with this speculation, Kääb et al. 95 reported that patients who experienced torsades de pointes arrhythmia with QT‐prolonging drugs developed more QTc lengthening after i.v. sotalol, an IKr blocking drug, than those of the control group consisting of patients without history of torsades de pointes. The interesting observation in this study was that in both groups the baseline QTc was normal and did not differ from each other (Fig. 10). 95 Although genetic testing has not been carried out in these patients, it can be assumed that the individuals who responded to sotalol might have “subclinical‐concealed‐silent” LQTS due to defective IKs channels. Based on these results, the authors suggested that the administration of provocative drug test under controlled situation might help identify selected patients at risk for developing torsades de pointes arrhythmia.
Figure 10.
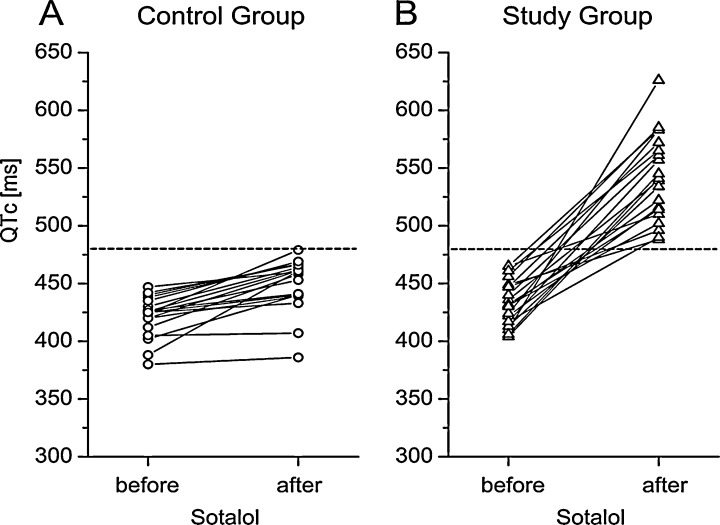
Individual QTc intervals in human control and study groups before and after sotalol. The dotted line indicates a cutoff value of 480 ms that distinguished best between the study and the control group (from Ref. 95, used with permission).
DOWN‐REGULATION OF IKS AS A POSSIBLE LINK TO DECREASED REPOLARIZATION RESERVE
IKs can be decreased not exclusively by ion channel mutation but it can be down‐regulated due to diseases such as heart failure, diabetes, and cardiac hypertrophy. Also, it is not known whether possible gene polymorphisms or simply the variation of the expression level of this channel in normal individuals play a similar role. This can be an important point, since even large variations in the IKs density cannot be expected to influence normal QTc duration significantly on the surface ECG, but they may substantially determine repolarization reserve and the stability of repolarization. It is of interest to note in this context, based on a recent study, that the coexpression of KvLQT1 cDNA with HERG cDNA increased the current‐carrying properties and trafficking of IKr channels. 96 In other words, it is possible that there is an alpha subunit interaction between IKs and IKr and such changes at the expression level of IKs can secondarily alter IKr. It would be interesting to know how mutated KvLQT1 channels could behave in this setting.
CONCLUSION
The slow delayed rectifier potassium current (IKs) operates in mammalian ventricular muscle including human. In normal situation, this current contributes to the repolarization process only to a minimal extent, but has a vital role in the repolarization reserve. Accordingly, when repolarization is prolonged beyond normal and/or sympathetic tone is elevated, IKs current provides an important safety mechanism preventing excessive and dangerous repolarization lengthening. Therefore, down‐regulation, genetical loss of function or pharmacological inhibition of IKs is not manifested in a marked repolarization elongation, but makes the repolarization less stable and the heart vulnerable toward repolarization abnormalities and consequently torsades de pointes arrhythmias.
Acknowledgments
Acknowledgements: The authors thank their colleague Dr. I. Baczkó for providing Figure 9 . Financial support for this work was obtained by grants from the Hungarian National Research Fund (OTKA T‐048698 and NI‐61902), Hungarian Ministry of Education (Bio‐37 KPI), National Research and Development Programmes (NKFP 1A/0011/2002 and 1/A/046/2004), the Hungarian Academy of Sciences, and the János Bolyai Research Scholarship (for N.J.).
REFERENCES
- 1. Sanguinetti MC, Keating MT. Role of delayed rectifier potassium channels in cardiac repolarisation and arrhythmias. News Physiol Sci 1997;12:152–158. [Google Scholar]
- 2. Carmeliet E. Voltage‐ and time‐dependent block of the delayed K+ current in cardiac myocytes by dofetilide. J Pharmacol Exp Ther 1992;262:809–817 [PubMed] [Google Scholar]
- 3. Noble D, Tsien RW. Outward membrane currents activated in the plateau range of potentials in cardiac Purkinje fibres. J Physiol 1969;200:205–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol 1990;96:195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Follmer CH, Lodge NJ, Cullinan CA, et al. Modulation of the delayed rectifier IK by cadmium in cat ventricular myocytes. Am J Physiol 1992;262:C75–C83. [DOI] [PubMed] [Google Scholar]
- 6. Gintant GA. Regional differences in IK density in canine left ventricle: role of IK,s in electrical heterogeneity. Am J Physiol 1995;268:H605–H613. [DOI] [PubMed] [Google Scholar]
- 7. Salata JJ, Jurkiewicz NK, Jow B, et al. IK of rabbit ventricle is composed of two currents: Evidence for IKs . Am J Physiol 1996;271:H2477–H2489. [DOI] [PubMed] [Google Scholar]
- 8. Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial and endocardial myocytes. A weaker IKs contributes to the longer action potential of the M cell. Circ Res 1995;76:351–365. [DOI] [PubMed] [Google Scholar]
- 9. Heath BM, Terrar DA. Separation of the components of the delayed rectifier potassium current using selective blockers of IKr and IKs in guinea‐pig isolated ventricular myocytes. Exp Physiol 1996;81:587–603. [DOI] [PubMed] [Google Scholar]
- 10. Heath BM, Terrar DA. The deactivation kinetics of the delayed rectifier components IKr and IKs in guinea pig isolated ventricular myocytes. Exp Physiol 1996;81:605–621. [DOI] [PubMed] [Google Scholar]
- 11. Jurkiewicz NK, Sanguinetti MC. Rate‐dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ Res 1993;71:75–83. [DOI] [PubMed] [Google Scholar]
- 12. Lengyel CS, Iost N, Virág L, et al. Pharmacological block of the slow component of the outward delayed rectifier current (IKs) fails to lengthen rabbit ventricular muscle QTc and action potential duration. Br J Pharmacol 2001;132:101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beuckelmann DJ, Nabauer M, Erdmann E. Alteration of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res 1993;73:379–385. [DOI] [PubMed] [Google Scholar]
- 14. Li GR, Feng J, Yue L, et al. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circ Res 1996;78:689–696. [DOI] [PubMed] [Google Scholar]
- 15. Hirano Y, Hiraoka M. Changes in K+ currents induced by Ba2+ in guinea pig ventricular muscles. Am J Physiol 1986;251:H24–H33. [DOI] [PubMed] [Google Scholar]
- 16. Virág L, Iost N, Opincariu M, et al. The slow component of the delayed rectifier potassium current in undiseased human ventricular myocytes. Cardiovasc Res 2001;49:790–797. [DOI] [PubMed] [Google Scholar]
- 17. Bennet PB, Begenisch TB. Catecholamines modulate the delayed rectifying potassium current (IK) in guinea pig ventricular myocytes. Pflugers Archiv‐Eur. J. Physiol 1987;410:212–219. [DOI] [PubMed] [Google Scholar]
- 18. Han W, Wang Z, Nattel S. Slow delayed rectifier current and repolarization in canine cardiac Purkinje cells. Am J Physiol 2001;280:H1075–H1080. [DOI] [PubMed] [Google Scholar]
- 19. Honore E, Attali B, Romey G, et al. Cloning, expression, pharmacology and regulation of a delayed rectifier K+ channel in mouse heart. EMBO J 1991:10:2805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hausdorff SF, Goldstein S, Rushin EE, et al. Functional characteriation of a minimal K+ channel expressed from synthetic gene. Biochemistry 1991;30:3341–3346. [DOI] [PubMed] [Google Scholar]
- 21. Takumi T, Ohkubo H, Nakanishi S. Cloning of a membrane protein that induces a slow voltage‐gated potassium current. Science 1988;242:1042–1045. [DOI] [PubMed] [Google Scholar]
- 22. Freeman LC, Kass RS. Expression of a minimal K+ channel protein in mammalian cells and immunolocalization in guinea pig heart. Circ Res 1993:73:968–73. [DOI] [PubMed] [Google Scholar]
- 23. Busch AE, Herzer T, Takumi T, et al. Blockade of human IsK channels expressed in Xenopus oocytes by the novel class III antiarrhythmic NE‐10064. Eur J Pharmacol 1994;264:33–37. [DOI] [PubMed] [Google Scholar]
- 24. Goldstein SAN, Miller C. Site‐specific mutations in a minimal voltage‐dependent K+ channel alter ion selectivity and open‐channel block. Neuron 1991;7:403–408. [DOI] [PubMed] [Google Scholar]
- 25. Varnum MD, Busch AE, Bond CT, et al. The minK channel underlies the cardiac potassium current IKs and mediates species‐specific responses to protein kinase C. Proc Natl Acad Sci USA 1993;90:11528–11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Q, Curran AE, Splawski I, et al. Positional cloning of a novel potassium channel gene: KvLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996;12:17–23 [DOI] [PubMed] [Google Scholar]
- 27. Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996;384:80–83. [DOI] [PubMed] [Google Scholar]
- 28. Barhanin J, Lesage F, Guillemare E, et al. K(V)LQT1 and IsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996;384:78–80. [DOI] [PubMed] [Google Scholar]
- 29. Tai KK, Goldstein SA. The conduction pore of a cardiac potassium channel. Nature 1998:391:605–608. [DOI] [PubMed] [Google Scholar]
- 30. Wang KW, Tai KK, Goldstein SA. MinK residues line a potassium channel pore. Neuron 1996;16:571–577. [DOI] [PubMed] [Google Scholar]
- 31. Busch AE, Busch GL, Ford E, et al. The role of the IsK protein in the specific pharmacological properties of the IKs channel complex. Br J Pharmacol 1997;122:187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Busch AE, Suessbrich H. Role of the IsK protein in the minK channel complex. Trends Pharmacol Sci 1997;18:26–29. [DOI] [PubMed] [Google Scholar]
- 33. Melman YF, Krummerman A, McDonald TV. KCNE regulation of KvLQT1 channels: Structure–function correlates. Trends Cardiovasc Med 2002;12:182–187. [DOI] [PubMed] [Google Scholar]
- 34. Demolombe S, Baro I, Pereon Y, et al. A dominant negative isoform of the long QT syndrome 1 gene product. J Biol Chem 1998;273:6837–6843. [DOI] [PubMed] [Google Scholar]
- 35. Pereon Y, Demolombe S, Baro I, et al. Differential expression of KvLQT1 isoforms across the human ventricular wall. Am J Physiol Heart Circ Physiol 2000;278:H1908–H1915. [DOI] [PubMed] [Google Scholar]
- 36. Bryant SM, Wan X, Shipsey SJ, et al. Regional differences in the delayed rectifier current (IKr and IKs) contribute to the differences in action potential duration in basal left ventricular myocytes in guinea‐pig. Cardiovasc Res 1998;40:322–331. [DOI] [PubMed] [Google Scholar]
- 37. Melman YF, Domenech A, De La Luna S, et al. Structural determinants of KvLQT1 control by the KCNE family of proteins. J Biol Chem 2001;276:6439–6444. [DOI] [PubMed] [Google Scholar]
- 38. Lundquist AL, Manderfield LJ, Vanoye CG, et al. Expression of multiple KCNE genes in human heart may enable variable modulation of I(Ks). J Mol Cell Cardiol 2005;38:277–287. [DOI] [PubMed] [Google Scholar]
- 39. Duggal P, Vesely MR, Wattanasirichaigoon D, et al. Mutation of the gene for IsK associated with both Jervell and Lange‐Nielsen and Romano‐Ward forms of Long‐QT syndrome. Circulation 1998;97:142–146. [DOI] [PubMed] [Google Scholar]
- 40. Yong S, Tian X, Wang Q. LQT4 gene: the "missing" ankyrin. Mol Interv 2003;3:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krumerman A, Gao X, Bian JS, et al. An LQT mutant minK alters KvLQT1 trafficking. Am J Physiol Cell Physiol 2004;286:C1453–C1463. [DOI] [PubMed] [Google Scholar]
- 42. Busch AE, Suessbrich H, Waldegger S, et al. Inhibition of IKs in guinea pig cardiac myocytes and guinea pig IsK channels by the chromanol 293B. Pflügers Arch Eur J Physiol 1996;432:1094–1096. [DOI] [PubMed] [Google Scholar]
- 43. Bosch RF, Gaspo R, Busch AE, et al. Effects of the chromanol 293B, a selective blocker of the slow component of the delayed rectifier K+ current on repolarisation in human and guinea pig ventricular myocytes. Cardiovasc Res 1998;38:441–450. [DOI] [PubMed] [Google Scholar]
- 44. Schreieck J, Wang Y, Gjini V, et al. Differential effect of beta‐adrenergic stimulation on the frequency‐dependent electrophysiologic actions of the new class III dofetilide, ambasilide, and chromanol 293B. J Cardiovasc Electrophysiol 1997;8:1420–1430. [DOI] [PubMed] [Google Scholar]
- 45. Sun ZQ, Thomas GP, Antzelevitch C. Chromanol 293B inhibits slowly activating delayed rectifier and transient outward currents in canine left ventricular myocytes. J Cardiovasc Electrophysiol 2001;12:472–478. [DOI] [PubMed] [Google Scholar]
- 46. Burashnikov A, Antzelevitch C. Prominent I(Ks) in epicardium and endocardium contributes to development of transmural dispersion of repolarization but protects against development of early afterdepolarizations. J Cardiovasc Electrophysiol 2002;13:172–177. [DOI] [PubMed] [Google Scholar]
- 47. Bauer A, Becker R, Karle C, et al. Effects of the I(Kr)‐blocking agent dofetilide and of the I(Ks)‐blocking agent chromanol 293b on regional disparity of left ventricular repolarization in the intact canine heart. J Cardiovasc Pharmacol 2002;39:460–467. [DOI] [PubMed] [Google Scholar]
- 48. Emori T, Antzelevitch C. Cellular basis for complex T waves and arrhythmic activity following combined I(Kr) and I(Ks) block. J Cardiovasc Electrophysiol 2001;12:1369–1378. [DOI] [PubMed] [Google Scholar]
- 49. Varró A, Baláti B, Iost N, et al. The role of the delayed rectifier component IKs in dog ventricular muscle and Purkinje fibre repolarization. J Physiol 2000;523:67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gerlach U, Brendel J, Lang HJ, et al. Synthesis and activity of novel and selective I(Ks)‐channel blockers. J Med Chem 2001;44:3831–3837. [DOI] [PubMed] [Google Scholar]
- 51. Gogelein H, Bruggemann A, Gerlach U, et al. Inhibition of IKs channels by HMR‐1556. N-S Arch Pharmacol 2000;362:480–488. [DOI] [PubMed] [Google Scholar]
- 52. Bosch RF, Schneck AC, Csillag S, et al. Effects of the chromanol HMR‐1556 on potassium currents in atrial myocytes. N-S Arch Pharmacol 2003;367:281–288. [DOI] [PubMed] [Google Scholar]
- 53. Thomas GP, Gerlach U, Antzelevitch C. HMR 1556, a potent and selective blocker of slowly activating delayed rectifier potassium current. J Cardiovasc Pharmacol 2003;41:140–147. [DOI] [PubMed] [Google Scholar]
- 54. Volders PG, Stengl M, Van Opstal JM, et al. Probing the contribution of IKs to canine ventricular repolarization: Key role for beta‐adrenergic receptor stimulation. Circulation 2003;107:2753–2760. [DOI] [PubMed] [Google Scholar]
- 55. Nakashima H, Gerlach U, Schmidt D, et al. In vivo electrophysiological effects of a selective slow delayed‐rectifier potassium channel blocker in anesthetized dogs: potential insights into class III actions. Cardiovasc Res 2004;61:705–714. [DOI] [PubMed] [Google Scholar]
- 56. Salata JJ, Jurkiewicz NK, Sanguinetti MC, et al. The novel class III antiarrhythmic agent, L‐735,821 is a potent and selective blocker of IKs in guinea pig ventricular myocytes (abstract). Circulation 1996;94:I‐529. [Google Scholar]
- 57. Selnick HG, Liverton NJ, Baldwin JJ, et al. Class III antiarrhythmic activity in vivo by selective blockade of the slowly activating cardiac delayed rectifier potassium current IKs by (R)‐2‐(2,4‐trifluoromethyl)‐N‐[2‐oxo‐5‐phenyl‐1‐(2,2,2‐trifluoroethyl)‐2,3‐dihydro‐1H‐benzo[e][1,4]diazepin‐3‐yl]acetamide. J Med Chem 1997;40:3865–3868. [DOI] [PubMed] [Google Scholar]
- 58. Salata JJ, Selnick HG, Lynch JJ Jr. Pharmacological modulation of IKs: Potential antiarrhythmic therapy. Curr Med Chem 2004;11:29–44. [DOI] [PubMed] [Google Scholar]
- 59. Jost N, Virag L, Bitay M, et al. Restricting excessive cardiac action potential and QT prolongation: A vital role for IKs in human ventricular muscle. Circulation 2005;112:1392–1399. [DOI] [PubMed] [Google Scholar]
- 60. Xu X, Rials SJ, Wu Y, et al. Left ventricular hypertrophy decreases slowly but not rapidly activating delayed rectifier potassium currents of epicardial and endocardial myocytes in rabbits. Circulation 2001;103:1585–1590. [DOI] [PubMed] [Google Scholar]
- 61. Lynch JJ Jr, Salata JJ, Wallace AA, et al. Antiarrhythmic efficacy of combined I(Ks) and beta‐adrenergic receptor blockade. J Pharmacol Exp Ther 2002;302:283–289. [DOI] [PubMed] [Google Scholar]
- 62. Lloyd J, Schmidt JB, Rovnyak G, et al. Design and synthesis of 4‐substituted benzamides as potent, selective, and orally bioavailable I(Ks) blockers. J Med Chem 2001;44:3764–3767. [DOI] [PubMed] [Google Scholar]
- 63. Kodama I, Kamiya K, Toyama J. Cellular electropharmacology of amiodarone. Cardiovasc Res 1997;35:13–29. [DOI] [PubMed] [Google Scholar]
- 64. Takacs J, Iost N, Lengyel C, et al. Multiple cellular electrophysiological effects of azimilide in canine cardiac preparations. Eur J Pharmacol 2003;470:163–170. [DOI] [PubMed] [Google Scholar]
- 65. Chouabe C, Drici MD, Romey G, et al. Effects of calcium channel blockers on cloned cardiac K+ channels IKr and IKs . Therapie 2000;55:195–202. [PubMed] [Google Scholar]
- 66. Satoh H. Comparative actions of cibenzoline and disopyramide on IKr and IKs currents in rat sino‐atrial nodal cells. Eur J Pharmacol 2000;407:123–129. [DOI] [PubMed] [Google Scholar]
- 67. Li RA, Miake J, Hoppe UC, et al. Functional consequences of the arrhythmogenic G306R KvLQT1 K+ channel mutant probed by viral gene transfer in cardiomyocytes. J Physiol 2001;533:127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Valenzuela C, Sanchez‐Chapula J, Delpon E, et al. Imipramine blocks rapidly activating and delays slowly activating K+ current activation in guinea pig ventricular myocytes. Circ Res 1994;74:687–799. [DOI] [PubMed] [Google Scholar]
- 69. Turgeon J, Daleau P, Bennett PB, et al. Block of IKs, the slow component of the delayed rectifier K+ current, by the diuretic agent indapamide in guinea pig myocytes. Circ Res 1994;75:879–886. [DOI] [PubMed] [Google Scholar]
- 70. Delpon E, Valenzuela C, Perez O, et al. Propafenone preferentially blocks the rapidly activating component of delayed rectifier K+ current in guinea pig ventricular myocytes. Voltage‐independent and time‐dependent block of the slowly activating component. Circ Res 1995;76:223–235. [DOI] [PubMed] [Google Scholar]
- 71. Salata JJ, Jurkiewicz NK, Wallace AA, et al. Cardiac electrophysiological actions of the histamine H1‐receptor antagonists astemizole and terfenadine compared with chlorpheniramine and pyrilamine. Circ Res 1995;76:110–119. [DOI] [PubMed] [Google Scholar]
- 72. Jost N, Virag L, Hala O, et al. Effect of the antifibrillatory compound tedisamil (KC‐8857) on transmembrane currents in mammalian ventricular myocytes. Curr Med Chem 2004;11:3219–3228. [DOI] [PubMed] [Google Scholar]
- 73. Salata JJ, Jurkiewicz NK, Wang JX, et al. A novel benzodiazepine that activates cardiac slow delayed rectifier K+ currents. Mol Pharmacol 1998;54:220–230. [DOI] [PubMed] [Google Scholar]
- 74. Xu X, Salata JJ, Wang J, et al. Increasing I(Ks) corrects abnormal repolarization in rabbit models of acquired LQT2 and ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2002;283:H664‐H670. [DOI] [PubMed] [Google Scholar]
- 75. Magyar J, Horvath B, Banyasz T, et al. L‐364,373 fails to activate the slow delayed rectifier K(+) current in canine ventricular cardiomyocytes. N-S Arch Pharmacol 2006;373:85–90. [DOI] [PubMed] [Google Scholar]
- 76. Seebohm G, Pusch M, Chen J, et al. Pharmacological activation of normal and arrhythmia‐associated mutant KCNQ1 potassium channels. Circ Res 2003;93:941–947. [DOI] [PubMed] [Google Scholar]
- 77. Li GR, Yang B, Feng J, et al. Transmembrane ICa contributes to rate‐dependent changes of action potentials in human ventricular myocytes. Am J Physiol 1999;276:H98–H106. [DOI] [PubMed] [Google Scholar]
- 78. Volders PG, Sipido KR, Vos MA, et al. Downregulation of delayed rectifier K+ currents in dogs with chronic complete atrioventricular block and acquired torsades de pointes. Circulation 1999;100:2455–2461. [DOI] [PubMed] [Google Scholar]
- 79. So PP, Hu XD, Backx PH, et al. Blockade of IKs by HMR 1556 increases the reverse rate‐dependence of refractoriness prolongation by dofetilide in isolated rabbit ventricles. Br J Pharmacol 2006;148255–148563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lengyel C, Udvary E, Varro A. Estimation of the proarrhythmic risk by short term variability of QT‐interval and/or action potential duration in dog (abstract). Heart&Rhythm 2006;S306. [Google Scholar]
- 81. Thomsen MB, Verduyn SC, Stengl M, et al. Increased short‐term variability of repolarization predicts d‐sotalol‐induced torsades de pointes in dogs. Circulation 2004;110:2453–2459. [DOI] [PubMed] [Google Scholar]
- 82. Tsuji Y, Zicha S, Qi XY, et al. Potassium channel subunit remodelling in rabbits exposed to long‐term bradycardia or tachycardia: Discrete arrhythmogenic consequences related to differential delayed‐rectifier changes. Circulation 2006, 113:345–355. [DOI] [PubMed] [Google Scholar]
- 83. Roden DM. Long QT syndrome: Reduced repolarization reserve and the genetic link. J Intern Med 2006;259:59–69. [DOI] [PubMed] [Google Scholar]
- 84. Roden DM. Taking the idio out of idiosyncratic – predicting torsade de pointes. Pacing Clin Electrophysiol 1998;21:1029–1034. [DOI] [PubMed] [Google Scholar]
- 85. Roden DM, Yang T. Protecting the heart against arrhythmias: Potassium current physiology and repolarization. Circulation 2005;112:1376–1378. [DOI] [PubMed] [Google Scholar]
- 86. Kass RS, Moss AJ. Long QT syndrome: novel insights into the mechanisms of cardiac arrhythmias. J Clin Invest 2003;112:810–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long‐QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1 and KCNE2. Circulation 2000;102:1178–1185 [DOI] [PubMed] [Google Scholar]
- 88. Chouabe C, Neyround N, Richard P, et al. Novel mutations in KvLQT1 that affect IKs activation through interactions with IsK . Cardiovas Res 2000;45:971–980. [DOI] [PubMed] [Google Scholar]
- 89. Gouas L, Bellocq C, Berthet M, et al. New KCNQ1 mutations leading to haploinsufficiency in a general population. Defective trafficking of a KvLQT1 mutant. Cardiovasc Res 2004;63:60–68. [DOI] [PubMed] [Google Scholar]
- 90. Huang L, Bitner‐Glindzicz M, Tranebjaerg L, et al. A spectrum of functional effects for disease causing mutations in the Jervell and Lange‐Nielsen syndrome. Cardiovasc Res 2001;51:670–680. [DOI] [PubMed] [Google Scholar]
- 91. Ackerman MJ. Genotype‐phenotype relationship in congenital long QT syndrome. J Electrocardiol 2005;38:64–68. [DOI] [PubMed] [Google Scholar]
- 92. Tamargo J, Caballero R, Gomez R, et al. Pharmacology of cardiac potassium channels. Cardiovasc Res 2004;62:9–33. [DOI] [PubMed] [Google Scholar]
- 93. Stengl M, Volders PGA, Thomsen MB, et al. Accumulation of slowly activating delayed rectifier potassium currents (IKs) in canine ventricular myocytes. J Physiol 2003;551:777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Boulet IR, Raes AL, et al. Functional effects of a KCNQ1 mutation associated with the long QT syndrome. Cardiovasc Res 2006;70:466–474. [DOI] [PubMed] [Google Scholar]
- 95. Kääb S, Hinterseer M, Näbauer M, et al. Sotalol testing unmasks altered repolarization in patients with suspected acquired long‐QT‐syndrome‐a case‐control pilot study using i.v. sotalol. Eur Heart J 2003;24:649–657. [DOI] [PubMed] [Google Scholar]
- 96. Ehrlich JR, Pourrier M, Weerapura M, et al. KvLQT1 modulates the distribution and biophysical properties of HERG. A novel alpha‐subunit interaction between delayed rectifier currents. J Biol Chem 2004;279:1233–1241. [DOI] [PubMed] [Google Scholar]