

Abstract
Background: The Jervell and Lange‐Nielsen syndrome (JLNS) is the autosomal recessive form of long QT syndrome (LQTS)—a familial cardiac disorder that causes syncope, seizures, and sudden death from ventricular arrhythmias, specifically torsade de pointes. JLNS is associated with sensorineural deafness and has been shown to occur with homozygous mutations in KCNQ1 or KCNE1 in JLNS families in which QTc prolongation is inherited as a dominant trait. This study investigated the molecular pathology of a family with clinical evidence of JLNS.
Methods and Results: Single‐strand conformation polymorphism, denaturing high performance liquid chromatography, and DNA sequencing analyses were used to screen for KCNQ1 mutations. Novel compound heterozygous nonsense mutations R518X/Q530X in the C‐terminus of KCNQ1 were identified in both affected dizygotic twins; both the parents and a sibling each carried only one of the mutant alleles and were asymptomatic with modestly prolonged QTc intervals (0.46, 0.50, and 0.45 seconds, respectively). These two nonsense mutations lead to premature termination of C‐terminus with truncation of the postulated assembly domain.
Conclusion: Novel compound heterozygous nonsense mutations in C‐terminus of KCNQ1 can cause JLNS. A.N.E 2003;8(3):246‐250
Keywords: JLNS, KCNQ1, long QT syndrome, compound heterozygous nonsense mutations, arrhythmia, ion channels, sensorineural deafness
The hereditary long QT syndrome (LQTS) is a familial cardiac disorder that causes syncope, seizures, and sudden death from ventricular arrhythmias, specifically torsade de pointes. 1 , 2 , 3 Both autosomal dominant LQTS (Romano‐Ward syndrome, RWS) and autosomal recessive LQTS (Jervell and Lange‐Nielsen syndrome, JLNS) have been defined. 4 , 5 , 6 Multiple heterozygous mutations in several ion channel genes (KCNQ1, HERG, SCN5A, KCNE1, and, KCNE2) have been shown to cause autosomal dominant LQTS. 7 , 8 , 9 Autosomal recessive LQTS, which is associated with deafness, has been shown to occur with homozygous mutations in KCNQ1 or KCNE1 in JLNS families in which QTc prolongation was inherited as a dominant trait. 10 , 11 , 12 , 13 Recently, an Amish JLNS family has been reported with QTc prolongation inherited as an incomplete dominant and deafness inherited as a recessive trait. 14 In this study, we report novel compound heterozygous nonsense mutations in the KCNQ1 gene of a family with clinical evidence of JLNS.
MATERIALS AND METHODS
The protocol of the study was approved by the Institutional Review Board of our Medical Center, and informed consent for genetic studies was obtained from all subjects.
Subjects
The probands are dizygotic brother (II‐2) and sister (II‐3) twins (Fig. 1a) of nonconsanguineous parents. Both twins were born congenitally deaf with QTc intervals over 0.60 seconds and required an implanted cardioverter defibrillator (ICD) for control of recurrent ventricular tachyarrhythmias. The sister (II‐3) has had >60 episodes of ventricular tachycardia/ventricular fibrillation requiring multiple ICD shocks. Her episodes are frequently accompanied by loss of consciousness. Her twin brother has used his ICD but to a much lesser extent. Both the parents (I‐1 and I‐2) and an elder sibling (II‐1) were asymptomatic for ventricular arrhythmias with modestly prolonged QTc intervals (0.46, 0.50 and 0.45 seconds, respectively).
Figure 1.
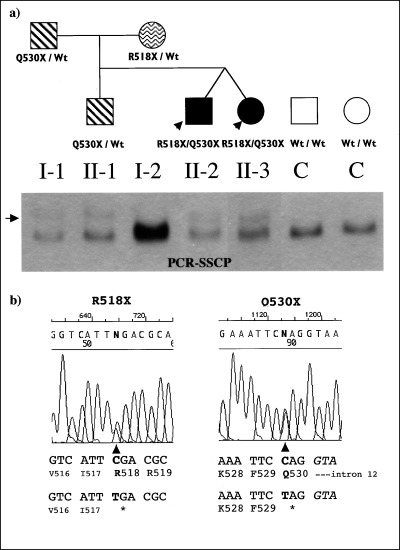
Compound heterozygous nonsense mutations in C‐terminus of KCNQ1 cause JLNS. (a), JLNS affected individuals are shown as solid circles (females) or squares (males), unaffected JLNS individuals with only one allele of the mutation as wave (R518X) or stripe (Q530X) filled circles/squares, and controls as open circles/squares. The probands of the family are indicated by arrows. The KCNQ1 genotypes of each family member are indicated. SSCP analysis on 0.5X MDE gel of genomic PCR products is presented. The control and the mother show a normal migration pattern, with R518X mutation undetectable in this assay. Q530X mutation carriers present an additional band shown by an arrow. (b), Partial KCNQ1 sequences of mutation carriers: C to T substitutions at position 1552 (R518X), and at position 1588 (Q530X) in this family.
DNA Collection and KCNQ1 Mutation Analysis
Genomic DNA was extracted from 10 ml human whole blood or cell lines derived from Epstein‐Barr virus‐transformed lymphocytes using PUREGENEr Genomic DNA Isolation Kit (Gentra System, Inc) according to standard procedure. Briefly, 20–80 ng of 1 μl genomic DNA were amplified by the polymerase chain reaction (PCR) with 22 μl PCR SuperMixr (GIBCOBRL, Inc) and 1 μl 10 pM of each primer. Fourteen exons of the KCNQ1 gene were amplified from genomic DNA by the polymerase chain reaction (PCR) primers (Table 1) with a detection rate of about 87%. 9 The thermal cycling condition of PCR reaction is denaturation at 94°C for 3 minutes and followed by 30 cycles of denaturation at 94°C (10 seconds), annealing at 58°C (20 seconds), extension at 72°C (20 seconds), and final extension at 72°C (5 minutes). The PCR products were separated by electrophoresis in 2% agarose gel with 1 X TBE buffer.
Table 1.
Oligonucleotide Primers for Detection of Mutations in the Human KCNQ1 Gene 9
| Exon | Forward Primer | Reverse Primer | Size |
|---|---|---|---|
| 2 | ATGGGCAGAGGCCGTGATGCTGAC | ATCCAGCCATGCCCTCAGATGC | 165 |
| 3 | GTTCAAACAGGTTGCAGGGTCTGA | CTTCCTGGTCTGGAAACCTGG | 256 |
| 4 | CTCTTCCCTGGGGCCCTGGC | TGCGGGGGAGCTTGTGGCACAG | 170 |
| 5 | TCAGCCCCACACCATCTCCTTC | CTGGGCCCCTACCCTAACCC | 154 |
| 6 | TCCTGGAGCCCGACACTGTGTGT | TGTCCTGCCCACTCCTCAGCCT | 238 |
| 7 | TGGCTGACCACTGTCCCTCT | CCCCAGGACCCCAGCTGTCCAA | 195 |
| 8 | GCTGGCAGTGGCCTGTGTGGA | AACAGTGACCAAAATGACAGTGAC | 191 |
| 9 | TGGCTCAGCAGGTGACAGC | TGGTGGCAGGTGGGCTACT | 185 |
| 10 | GCCTGGCAGACGATGTCCA | CAACTGCCTGAGGGGTTCT | 216 |
| 11 | CTGTCCCCACACTTTCTCCT | TGAGCTCCAGTCCCCTCCAG | 195 |
| 12 | TGGCCACTCACAATCTCCT | GCCTTGACACCCTCCACTA | 222 |
| 13 | GGCACAGGGAGGAGAAGTG | CGGCACCGCTGATCATGCA | 216 |
| 15 | GGCCCTGATTTGGGTGTTTTA | GGACGCTAACCAGAACCAC | 135 |
| 16 | CACCACTGACTCTCTCGTCT | CCATCCCCCAGCCCCATC | 297 |
Single‐Strand Conformation Polymorphism (SSCP) Analysis
SSCP and DNA sequencing analyses were used to screen for KCNQ1 mutations. After amplification, 5 μl PCR product was mixed with 5 μl formamide buffer (95% formamide, 10 mM NaOH, 0.25% bromphenol blue, 0.25% xylene cyanol). The mixture was denatured at 94°C for 3 minutes, then cooled rapidly on ice and held for 5 minutes. For each sample, 5 μl of the denatured mixture was loaded onto 0.5X MDEr gel (FMC Bioproducts, Inc) supported with GelBond PAGr film (FMC Bioproducts, Inc). MDEr gel electrophoresis was performed in 0.6X TBE running buffer at 6 W for 14 hours at room temperature. The SSCP bands were detected by the silver‐stained method. The gel was fixed in a solution containing 50% ethanol and 5% acetic acid for 40 minutes. After brief water rinsing, the gel was soaked in 600 ml of the silver stain solution (containing 0.6 g silver nitrate, 900 μl 37% formaldehyde) for 30 minutes, then soaked in 600 ml of the prechilled developing solution (containing 18 g sodium carbonate anhydrous, 900 μl 37% formaldehyde, 76 μl sodium thiosulfate) until the SSCP bands appear. The stained gel was postfixed in 10% acetic acid for 10 minutes and air‐dried.
DHPLC (Denaturing High Performance Liquid Chromatography) Analysis
The same PCR was performed for dHPLC analysis as for SSCP assay. However, to induce the formation of the heteroduplex from the genetic variant and the normal DNA, a post‐PCR re‐annealing step was added by an incubation of the PCR products for 3 minutes at 95°C, followed by the gradual re‐annealing from 94°C to 65°C (decreasing 0.6°C/min) for about 50 minutes. The quality of PCR products was checked on the agarose gel prior to dHPLC analysis. The dHPLC analysis was performed with the ProStarr Helix system (Varian). The melting temperature predictions for each amplicon were made by using the Stanford DHPLC Melt Program (http://www.insertions.stanford.edu/melt.html). Chromatogram data were analyzed based on visual inspection and comparison with normal controls is included in each run. Heterozygous profiles were detected as distinct elution peaks from homozygous wild‐type peaks.
DNA Sequencing
Sequencing of the purified products was performed using the BigDye Terminatorr cycle sequencing kit by the University of Rochester Core Nucleic Acid Laboratory.
RESULTS
SSCP assay was performed as the initial LQTS gene mutation screening for this family. Similar aberrant SSCP conformers of the DNA fragment spanning the whole KCNQ1 exon 12 and its intron‐exon boundaries, were identified in the affected dizygotic twins II‐2 and II‐3, the father (I‐1) and a brother (II‐1) (Fig. 1); these SSCP anomalies were not observed in DNA samples from the mother (I‐2) (Fig. 1), and from more than 130 control subjects (data not shown). Direct sequence analysis of the SSCP‐positive PCR products revealed the presence of novel compound heterozygous nonsense mutations R518X/Q530X in the C‐terminus of KCNQ1 in both affected dizygotic twins. The father and a sibling (II‐1) carried only one mutant allele Q530X. The mother (I‐2) carried the other mutant allele R518X. These two nonsense mutations lead to premature termination of the C‐terminus, not observed in 50 control subjects (data not shown).
While our SSCP assay only picked up Q530X, our dHPLC method detected both mutations (Fig. 2)
Figure 2.
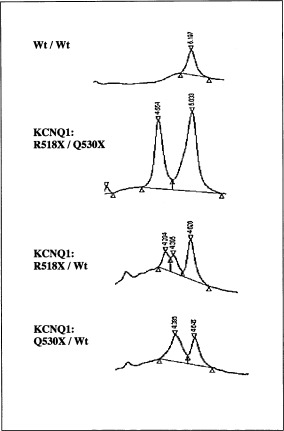
DHPLC mutation analysis of KCNQ1 exon12. DHPLC assay confirms the mutation (Q530X) detected by SSCP and identifies the mutation (R518X) missed by SSCP.
DISCUSSION
Homozygous mutations in KCNQ1 or KCNE1 that result in JLNS have been reported in several consanguineous JLNS families in which QTc prolongation was inherited as a dominant trait. 10 , 11 , 12 , 13 Recently, an Amish JLNS family has been reported with QTc prolongation inherited as an incomplete dominant and deafness inherited as a recessive trait. 14 The JLNS mutations spread over the KCNQ1 or KCNE1 genes and no hot spots have been identified. All LQTS genes identified to date belong to the family of voltage‐gated ion channels. These channels have similar structural characteristics: the functional channel is a tetramer formed by assembly of four α‐subunits. K+ channel genes encode only one of these α‐subunits, while in SCN5A all four α‐subunits are encoded by a single gene. Each α‐subunit consists of six α‐helical transmembrane segments (S1–S6). The S4 segment contains several positively charged amino acids and is involved in the voltage‐dependent activation. The linker between S5 and S6 as well as the S5 and S6 segments participates in the formation of the ion channel pore (Fig. 3). In addition, KCNQ1 is associated with the auxiliary subunit minK 15 , 16 that has only a single transmembrane segment (Fig. 3). In vitro biochemical and functional studies indicate that the KCNQ1 C‐terminus functions as an assembly domain for KCNQ1 subunits. 11 Both of the two mutations identified in this study result in truncation of this postulated assembly domain. Each of them individually has been associated with RWS. 9 In our case, when one of the mutant alleles was present, the carriers were asymptomatic with regard to the cardiac phenotypes but with modestly prolonged QTc intervals (I‐1 for 0.46 seconds, I‐2 for 0.50 seconds, and II‐1 for 0.45 seconds, respectively); the combination of two mutant alleles resulted in the more severe phenotype of deafness, syncope, and more prolonged QTc intervals (II‐2 and II‐3: >0.6 seconds).
Figure 3.
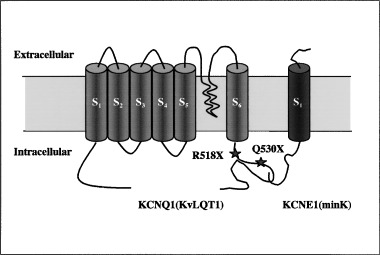
KCNQ1 mutations in JLNS patients. The positions of the mutations within the KCNQ1 channel identified in the family are illustrated.
Our findings provide the first description that novel compound heterozygous nonsense mutations in C‐terminus of KCNQ1 cause JLNS. While we prepared this article, two cases of other compound heterozygous mutations of KCNQ1 in JLNS were reported by Towbin and his colleagues. 17 In one family, a missense mutation G268D at exon 6 and an incomplete‐penetrance nonsense mutation at exon 3 together resulted in JLNS. The other family carried a missense mutation D202N and frameshift mutation at Nt585. 17 It was reported in one study that 33% of clinically unaffected family members in kindreds containing a variety of LQTS mutations were found to be gene carriers. 18 An unpublished study identified that out of 275 probands in LQTS kindreds, nine carried two mutations, either in the same LQTS gene or in different LQTS genes (Splawski, personal communication). In those families, the phenotype of individuals carrying two mutations in LQTS genes was more severe than individuals carrying one. Thus, it is not unreasonable to hypothesize that different compound heterozygous mutations of KCNQ1 may be more common than previously thought in JLNS. Our case of compound heterozygous nonsense mutations suggests a loss‐of‐function as the molecular pathogenic mechanism in this disease.
It is unknown why the SSCP screening missed the R518X mutation even with repeated efforts although it is known that the sensitivity of SSCP can vary significantly depending on various parameters, including sequence. 19 However, this mutation could be readily detected by dHPLC, giving another example of the higher sensitivity of the new method.
This work is supported in part by Grants from HHMI (Research Support Program, M.Q.), the United States Public Health Service (HL51618, A.J.M), and General Clinical Research Center of URMC (M.Q.).
REFERENCES
- 1. Ackerman MJ. The long QT syndrome. Pediatr Rev 1998;19: 232–238. [DOI] [PubMed] [Google Scholar]
- 2. Moss A, Robinson J. The cardiology patient page: The long‐QT syndrome. Circulation 2002;105: 784–786. [Google Scholar]
- 3. Schwartz PJ, Moss AJ, Vincent GM, et al Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88(2):782–784. [DOI] [PubMed] [Google Scholar]
- 4. Romano C, Gemme G, Pongiglione R. Arithmie cardiache rare dell'eta pediatrica Clin Pediatr 1963;45: 656–683. [PubMed] [Google Scholar]
- 5. Ward OC. A new familial cardiac syndrome in children. J. Israel. Med. Assoc. 1964;54: 103–106. [PubMed] [Google Scholar]
- 6. Jervell A, Lange‐Nielsen F. Congenital deaf‐mutism, function heart disease with prolongation of the Q‐T interval and sudden death. Am Heart J 1957;54: 59–68. [DOI] [PubMed] [Google Scholar]
- 7. Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 2001;104: 569–580. [DOI] [PubMed] [Google Scholar]
- 8. Towbin JA, Wang Z, Li H. Genotype and severity of long QT syndrome. Drug Metab Dispos 2001;29: 574–579. [PubMed] [Google Scholar]
- 9. Splawski I, Shen J, Timothy KW, et al Spectrum of mutations in long‐QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000;102: 1178–1185. [DOI] [PubMed] [Google Scholar]
- 10. Ilhan A, Tuncer C, Komsuoglu SS, et al Jervell and Lange‐Nielsen syndrome: Neurologic and cardiologic evaluation. Pediatr Neurol 1999;21: 809–813. [DOI] [PubMed] [Google Scholar]
- 11. Schmitt N, Schwarz M, Peretz A, et al A recessive C‐terminal Jervell and Lange‐Nielsen mutation of the KCNQ1 channel impairs subunit assembly. Embo J 2000;19: 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schulze‐Bahr E, Haverkamp W, Wedekind H, et al Autosomal recessive long‐QT syndrome (Jervell Lange‐Nielsen syndrome) is genetically heterogeneous. Hum Genet 1997;100: 573–576. [DOI] [PubMed] [Google Scholar]
- 13. Tranebjaerg L, Bathen J, Tyson J, et al Jervell and Lange‐Nielsen syndrome: A Norwegian perspective. Am J Med Genet 1999;89: 137–146. [PubMed] [Google Scholar]
- 14. Chen Q, Zhang D, Gingell RL, et al Homozygous deletion in KVLQT1 associated with Jervell and Lange‐Nielsen syndrome. Circulation 1999;99: 1344–1347. [DOI] [PubMed] [Google Scholar]
- 15. Sanguinetti MC, Curran ME, Zou A, et al Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996;384: 80–83. [DOI] [PubMed] [Google Scholar]
- 16. Barhanin J, Lesage F, Guillemare E, et al K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current [see comments]. Nature 1996;384: 78–80. [DOI] [PubMed] [Google Scholar]
- 17. Wang Z, Li H, Moss AJ, et al Compound heterozygous mutations in KvLQT1 cause Jervell and Lange‐Nielsen syndrome. Mol Genet Metab 2002;75: 308–316. [DOI] [PubMed] [Google Scholar]
- 18. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long‐QT syndrome: clinical impact. Circulation 1999;99: 529–533. [DOI] [PubMed] [Google Scholar]
- 19. Liu Q, Feng J, Sommer SS. Bi‐directional dideoxy fingerprinting (Bi‐ddF): a rapid method for quantitative detection of mutations in genomic regions of 300‐600 bp. Hum Mol Genet 1996;5: 107–114. [DOI] [PubMed] [Google Scholar]