

Abstract
Background: The warm‐up phenomenon observed after the second of two sequential exercise tests is characterized by an increased time to ischemia and ischemic threshold, and the latter is related to ischemic preconditioning. Previous studies have demonstrated that a single dose of glibenclamide, a cardiac ATP‐sensitive K (KATP) channel blocker, prevents ischemic preconditioning. This study aimed to investigate the effects of chronic treatment with glibenclamide during two sequential exercise tests.
Methods: Forty patients with angina pectoris were divided into three groups: 20 nondiabetics (NDM), 10 patients with diabetes in treatment with glibenclamide (DMG) and 10 diabetic patients with other treatments (DMO). All patients underwent two consecutive exercise tests.
Results: Heart rate and rate‐pressure product at 1.0 mm ST‐segment depression significantly increased during the second exercise test in NDM group (121.3 ± 16.5 vs 127.3 ± 15.3 beats/min, P < 0.001, and 216.7 + 43.1 vs 232.1 ± 43.0 beats·min−1·mmHg·102, P < 0.001), and in DMO group (114.1 ± 19.6 vs 119.6 ± 18.1 beats/min, P = 0.001, and 199.8 ± 36.6 vs 222.2 ± 29.2 beats·min−1·mmHg·102, P = 0.019), but it did not change in patients in DMG group (130.7 ± 14.5 vs 132.1 ±14.7 beats/min, P = ns, and 251.7 ± 47.2 vs 250.3 ± 42.8 beats·min−1·mmHg·102, P = ns). In the three groups, NDM, DMO, and DMG, the time to 1.0 mm ST‐segment depression during the second exercise test was greater than during the first (225.0 ± 112.5 vs 267.0 ± 122.3 seconds, P = 0.006; 187.5 ± 54.0 vs 226.5 ± 74.6 seconds, P = 0.029 and 150.0 ± 78.7 vs 186.0 ± 81.9 seconds, P < 0.001).
Conclusion: The chronic use of glibenclamide may have mediated the loss of preconditioning benefits in the warm‐up phenomenon, probably through its KATP channel‐blocker activity, but without acting upon the tolerance to exercise.
Keywords: ischemic preconditioning, diabetes mellitus, glyburide, treadmill test, sulfonylurea compounds
The warm‐up phenomenon, described in patients with more severe angina during initial effort that decreases in intensity in a subsequent exercise, is observed after the second of two sequential exercise tests 1 , 2 , 3 , 4 and characterized by an increase of both time to ischemia and ischemic threshold; and the last one is due, at least in part, to a mechanism similar to that involved in limiting experimental infarct size following a brief ischemic episode, i.e., ischemic preconditioning. 2 , 3 , 4 , 5 This hypothesis is supported by the observation that myocardial oxygen consumption is reduced during the second of two sequential exercise tests, suggesting increased metabolic efficiency, a feature of preconditioning. 2 Although the mechanisms of ischemic preconditioning are multiple and not fully established, the opening of cardiac KATP channels is believed to be crucial for the occurrence of ischemic preconditioning. 6 , 7 , 8 , 9
Non–insulin‐dependent diabetes mellitus (NIDDM) accounts for about 85% of all cases of diabetes mellitus, and has been reported to be an important risk factor for cardiovascular morbidity and mortality. 10 , 11 , 12 Up to now, oral hypoglycemic drugs, in particular sulfonylurea derivates, have represented the backbone of NIDDM therapy for the last decades. 13 , 14 The sulfonylurea derivates exert their insulinotropic effect by closing the so‐called KATP channel. 13 , 14 Interestingly, the UDPG‐study observed an increased cardiovascular mortality among patients with NIDDM treated by tolbutamide versus those treated by diet alone. 15 Moreover, smaller studies have also suggested a harmful effect of sulfonylurea derivatives on the outcome of myocardial infarction. 16 , 17 , 18
Although these two phenomena may appear unrelated, we hypothesized that blockers of KATP channels, such as the sulfonylurea drugs, can induce loss of ischemic preconditioning. Thus, in order to establish the role played by KATP channels in the warm‐up phenomenon, we assessed the effects of glibenclamide, a KATP channel blocker, in patients with stable angina and documented coronary artery disease, undergoing two sequential treadmill exercise tests.
METHODS
Patients
Forty patients, out of 90 patients of MASS II (Medical, Angioplasty and Surgical Study) 19 who underwent treadmill exercise test between July 1999 and July 2000 and had positive exercise test, were selected because they met this criteria: stable angina pectoris, ejection fraction >40% on echocardiography, and at least one critical stenosis (internal diameter reduction >70% by visual assessment) in the proximal two‐thirds of one major epicardial coronary artery. All patients were in sinus rhythm and without evidence of left ventricular hypertrophy or conduction defects, which could interfere with the interpretation of ST‐segment changes, and none of the patients was taking digitalis. Nitrate preparations, other than sublingual nitroglycerin and calcium entry blocking agents, were withdrawn 4 days before the study, and β‐blocking agents 5 days before.
Every patient had cineangiography in the previous year, and two independent cardiologists analyzed the results. Venous blood samples for the measurement of fasting plasma glucose levels was obtained at the day of protocol.
The patients were divided into three groups according to the presence of diabetes mellitus and the type of treatment to diabetes: 20 patients without diabetes mellitus (NDM group); 10 patients with diabetes and in use of glibenclamide in the last 6 months: 8 patients on 10 mg per day, 1 patient on 15 mg per day, and 1 patient on 20 mg per day (DMG group); and 10 patients with diabetes in use of other treatments: 5 with diet, 4 with diet and metformin therapy, and 1 with diet and insulin therapy (DMO group). All patients gave written informed consent for participation in the study, which was approved by the local Ethics Committee.
Exercise Test
All patients underwent two consecutive, computer‐assisted treadmill exercise tests, using the Bruce protocol, with a recovery period of 30 minutes between the tests to reestablish baseline electrocardiographic conditions. A standard 12‐lead electrocardiogram was obtained in standing position at baseline, at peak exercise, and at 6 minutes after the exercise. Twelve electrocardiographic leads were continuously monitored before, during, and after exercise. The level of the ST segment, 0.08 seconds after the J point, was calculated after signal averaging by means of a computer‐assisted system (FUKUDA DENSHII CO. LTD. Model ML 8000 stress test system, Tokyo, Japan) in all 12 leads. Values thus calculated were printed out, along with the heart rate, against time in trend format. This provided measurement of the ST‐segment level with an accuracy of 0.1 mm.
Criteria for interrupting the test were: (1) ST‐segment depression ≥3 mm, (2) maximal age‐related heart rate, (3) severe chest pain, (4) physical exhaustion, and the occurrence of other harmful conditions such as hypotension, severe arrhythmia, and dyspnea. Myocardial ischemia was diagnosed when a horizontal or downsloping ST‐segment depression of 1.0 mm at 80 ms from the J point was observed in at least one lead. Electrocardiographic recordings of all tests were evaluated independently in a blind fashion by two cardiologists; in case of disagreement, the matter was resolved by consensus.
The following parameters were measured: resting heart rate and blood pressure; heart rate, blood pressure, and rate‐pressure product (heart rate × systolic blood pressure) at the onset of 1.0 mm ST‐segment depression and at peak exercise; maximal ST‐segment depression; exercise duration in seconds, and time in seconds to onset of 1.0 mm ST‐segment depression.
Statistical Analysis
The paired Student's t‐test was used to compare eletrocardiographic data during repeated exercise tests in the same group. Comparisons of the continuous variables between the three groups were performed by analysis of variance, which detected significant differences; pairwise comparisons were made using the Bonferroni correction for multiple testing. The discrete variables among the three groups were analyzed using chi‐square test. Data were expressed as mean values ± 1SD; P values <0.05 were considered statistically significant.
RESULTS
Clinical Characteristics
No differences were detected among the three groups, NDM, DMO, and DMG, as to the use of antiischemic drugs and their discontinuation before the test: β‐blocking agents (85%, 90%, and 90%, P = 0.891), calcium entry blocking agents (65%, 50%, and 80%, P = 0.363), nitrate preparations (85%, 70%, and 70%, P = 0.519).
Comparative analysis among the three groups, NDM, DMO, and DMG, did not show differences in mean age (59.8 ± 11.6, 65.8 ± 9.5, and 61.0 ± 9.9 years, P = 0.361), male gender (85%, 70%, and 60%, P = 0.301), previous myocardial infarction (20%, 10%, and 20%, P = 0.751), hypertension (55%, 60%, and 50%, P = 0.904), smoking (35%, 10%, and 40%, P = 0.222). The mean blood glucose showed significant differences among the three groups (P < 0.001). Analysis between DMG and DMO groups did not show any significant difference (174.9 ± 44.1 and 149.7 ± 47.8 mg/dL, P = ns); but there were significant differences between DMG and NDM groups (174.9 ± 44.1 and 96.6 ± 8.6 mg/dL, P < 0.05), and between DMO and NDM groups (149.7 ± 47.8 and 96.6 ± 8.6 mg/dL, P < 0.05).
The average time since the last dose of glibenclamide in the DMG group was 14 ± 9 hours.
Exercise Tests
Comparative analysis of baseline characteristics among the three groups, NDM, DMO, and DMG, did not show differences in systolic blood pressure (153.10 ± 22.30, 162.20 ± 13.60, and 164.80 ± 21.30 mmHg, respectively, P = 0.270); but showed differences in resting heart rate (P = 0.024). Analysis between DMO and DMG groups showed significant differences (75.10 ± 19.60 and 95.00 ± 15.20 beats/min, P < 0.05), but did not show any significant difference between NDM and DMO group (79.60 ± 15.70 and 75.10 ± 19.60 beats/min, P = ns) and between NDM and DMG groups (79.60 ± 15.70 and 95.00 ± 15.20 beats/min, P = ns).
NDM Group
The main results of the two exercise tests in NDM group are summarized in Table 1. All patients achieved 1.0 mm ST‐segment depression during the two tests. Heart rate and rate‐pressure product at rest were similar during the first and the second tests.
Table 1.
Results of Two Sequential Tests in NDM Group (Mean ± SD)
| Test 1 | Test 2 | P | |
|---|---|---|---|
| Baseline | |||
| Heart rate (beats/min) | 79.6 ± 15.7 | 82.6 ± 13.1 | 0.183 |
| Systolic blood pressure (mmHg) | 153.1 ± 22.3 | 149.9 ± 19.7 | 0.381 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 121.9 ± 31.4 | 122.2 ± 25.6 | 0.959 |
| At 1.0 mm ST‐segment depression | |||
| Heart rate (beats/min) | 121.3 ± 16.5 | 127.3 ± 15.3 | <0.001 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 216.7 ± 43.1 | 232.1 ± 43.0 | <0.001 |
| Time (s) | 225.0 ± 112.5 | 267.0 ± 122.3 | 0.006 |
| Peak exercise | |||
| Heart rate (beat/min) | 133.9 ± 15.8 | 137.4 ± 14.1 | 0.017 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 251.6 ± 49.1 | 261.3 ± 48.0 | 0.047 |
| Exercise duration (s) | 337.6 ± 135.7 | 355.1 ± 136.5 | 0.009 |
| ST‐segment depression (mm) | 1.8 ± 0.5 | 1.7 ± 0.5 | 0.630 |
Both heart rate and rate‐pressure product at 1.0 mm ST‐segment depression were significantly higher during the second test than during the first (P < 0.001). Similarly, heart rate and rate‐pressure product at peak exercise were significantly higher during the second than the first test (P = 0.017 and 0.047, respectively).
Time to 1.0 mm ST‐segment depression during the second test was significantly longer than during the first test (P = 0.006), and exercise duration had the same behavior (P = 0.009).
No difference was found in maximal ST‐segment depression at peak exercise between the first and second tests.
DMO Group
The main results of the two exercise tests in the DMO group are summarized in Table 2. All patients achieved 1.0 mm ST‐segment depression during the two tests. Heart rate and rate‐pressure product at rest were similar during the first and second tests.
Table 2.
Results of Two Sequential Tests in DMO Group (Mean ± SD)
| Test 1 | Test 2 | P | |
|---|---|---|---|
| Baseline | |||
| Heart rate (beats/min) | 75.1 ± 19.6 | 77.9 ± 18.0 | 0.122 |
| Systolic blood pressure (mmHg) | 162.2 ± 13.6 | 159.3 ± 13.6 | 0.306 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 121.4 ± 32.4 | 123.7 ± 29.9 | 0.441 |
| At 1.0 mm ST‐segment depression | |||
| Heart rate (beats/min) | 114.1 ± 19.6 | 119.6 ± 18.1 | 0.001 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 199.8 ± 36.6 | 222.2 ± 29.2 | 0.019 |
| Time (s) | 187.5 ± 54.0 | 226.5 ± 74.6 | 0.029 |
| Peak exercise | |||
| Heart rate (beats/min) | 125.1 ± 19.8 | 130.3 ± 17.6 | 0.007 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 232.8 ± 27.0 | 247.1 ± 23.9 | 0.006 |
| Exercise duration (s) | 327.1 ± 72.8 | 358.3 ± 103.3 | 0.046 |
| ST‐segment depression (mm) | 1.8 ± 0.6 | 1.6 ± 0.6 | 0.199 |
Heart rate and rate‐pressure product at 1.0 mm ST‐segment depression were significantly higher during the second test than during the first test (P = 0.001 and P = 0.019, respectively). Similarly, heart rate and rate‐pressure product at peak exercise were significantly higher during the second than during the first test (P = 0.007 and 0.006, respectively).
Time to 1.0 mm ST‐segment depression during the second test was significantly longer than during the first test (P = 0.029), and exercise duration had the same behavior (P = 0.046).
There was no difference in maximal ST‐segment depression at peak exercise between the first and second tests.
DMG Group
The main results of the two exercise tests in the DMG group are summarized in Table 3. All patients achieved 1.0 mm ST‐segment depression during the two tests. Heart rate and rate‐pressure product at rest were similar during the first and second tests.
Table 3.
Results of Two Sequential Tests in DMG Group (Mean ± SD)
| Test 1 | Test 2 | P | |
|---|---|---|---|
| Baseline | |||
| Heart rate (beats/min) | 95.0 ± 15.2 | 98.3 ± 12.8 | 0.154 |
| Systolic blood pressure (mmHg) | 164.8 ± 21.3 | 164.6 ± 25.6 | 0.973 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 157.6 ± 36.0 | 161.5 ± 31.8 | 0.450 |
| At 1.0 mm ST‐segment depression | |||
| Heart rate (beats/min) | 130.7 ± 14.5 | 132.1 ± 14.7 | 0.298 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 251.7 ± 47.2 | 250.3 ± 42.8 | 0.838 |
| Time (s) | 150.0 ± 78.7 | 186.0 ± 81.9 | <0.001 |
| Peak exercise | |||
| Heart rate (beats/min) | 142.9 ± 14.8 | 144.2 ± 14.2 | 0.064 |
| Rate‐pressure product (beats·min−1·mmHg·102) | 292.0 ± 43.4 | 286.9 ± 32.7 | 0.480 |
| Exercise duration (s) | 282.4 ± 89.9 | 301.1 ± 101.0 | 0.057 |
| ST‐segment depression (mm) | 2.0 ± 0.7 | 1.9 ± 0.7 | 0.423 |
Heart rate and rate‐pressure product at 1.0 mm ST‐segment depression did not show any differences between the first and second tests. Similarly, heart rate and rate‐pressure product at peak exercise did not reveal any differences from the first to second test.
Time to 1.0 mm ST‐segment depression during the second test was significantly longer than during the first test (P < 0.001), exercise duration was longer in the second test than in the first, but this difference was not statistically significant (P = 0.057).
There was no difference in maximal ST‐segment depression at peak exercise between the first and second tests.
Figures 1 and 2 display heart rate and rate‐pressure product behavior in the three groups, NDM, DMO, and DMG.
Figure 1.
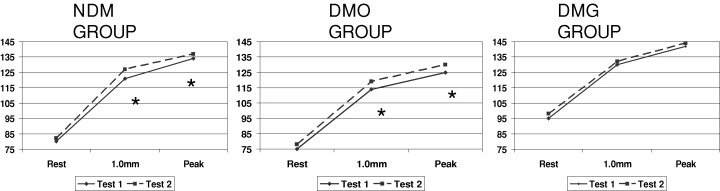
Mean values of heart rate (beats/min) at rest, at 1.0 mm ST‐segment depression, and at peak of exercise. *P < 0.05 (exercise test 1 vs 2).
Figure 2.
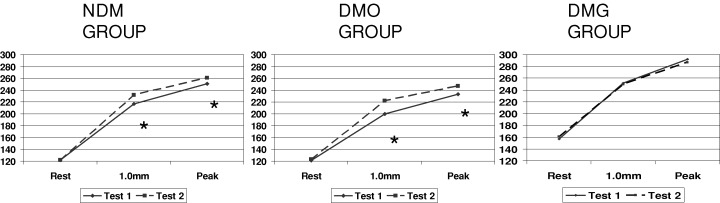
Mean values of rate‐pressure product (beats·min−1·mmHg·102) at rest, at 1.0 mm ST‐segment depression, and at peak of exercise. *P < 0.05 (exercise test 1 vs 2).
DISCUSSION
This study was carried out in patients with stable angina who were separated according to the presence or absence of diabetes mellitus, and to its treatment, either with glibenclamide or others; patients underwent two consecutive exercise tests, which showed: (1) an improvement of the ischemic threshold during the second exercise test in the NDM and DMO groups, but no such improvement in the DMG group and (2) enhanced exercise tolerance during the second exercise test in the three groups.
The warm‐up phenomenon was associated with a reduction in myocardial oxygen consumption, thus suggesting that the same mechanism involved in limiting experimental infarct size following a brief ischemic episode, i.e. ischemic preconditioning, 2 , 3 , 4 might have occurred. Of note, the time course between the first and second test, 30 minutes, is consistent with that of ischemic preconditioning, as it lasts no longer than 60—120 minutes. 3 , 4 , 20 , 21
In this study, the variables analyzed, heart rate and rate‐pressure product at 1.0 mm ST‐segment depression and at peak of exercise, represent objective, non‐invasive indexes of the ischemic threshold, i.e., of oxygen consumption at the onset of ischemia. 22 The other variables, such as the time to 1.0 mm ST‐segment depression and the duration of exercise, are due to a training effect caused by peripheral mechanisms, 23 , 24 and may be influenced by the subjective attitude of the patient.
Thus, we can conclude that the chronic use of glibenclamide abolished the improvement of ischemic threshold during the second exercise test, but did not interfere with the tolerance to exercise. 3 , 4 In particular, these findings seem to confirm that at least one component of the warm‐up phenomenon is due to ischemic preconditioning, and indicated that KATP channels are involved in such component.
It has been postulated that the opening of the KATP channels is responsible for the shortening of action potential duration observed during ischemia and the consequent reduction in Ca2+ influx that may lead to reduced myocardial contractility, vasodilatation, and sparing of ATP. 25 , 26 Then, the blockers of KATP channels can induce loss of ischemic preconditioning. 25
Two previous studies demonstrated that only one dose of glibenclamide in nondiabetic patients 27 and in diabetic patients who were not using glibenclamide 28 could abolish the ischemic preconditioning in two sequential stress tests. Although the dose used in these studies was similar to the dose used in the clinical practice, we cannot extrapolate these results to patients in chronic use of glibenclamide.
Apparently, the plasma concentrations of these drugs are sufficiently high to close KATP channels in the pancreas, but there are no evidences of this same effect in cardiovascular channels. In vitro, glibenclamide concentrations as low as 0.01 μmol/L (≈5 ng/mL) were able to reduce the vascular relaxation induced by the KATP channel opening drug pinacidil. 29 In patients with NIDDM, treated with glibenclamide, plasma concentrations may reach levels above 300 ng/mL. Although the free drug concentrations are probably much lower because sulfonylurea derivates are extensively protein‐bound, 13 the free plasma concentration may still be sufficient to interact with cardiovascular KATP channels.
In our study, the median time between the last dose of glibenclamide and the first stress test was 14 hours, this drug has elimination half time of about 10 hours and an effective duration of action of up to 24 hours. 14
The results of our study suggest that diabetic patients exposed to a long‐term treatment with glibenclamide could not be functionally protected by ischemic preconditioning at the time of acute myocardial infarction, and this finding may be helpful in explaining the excess mortality from cardiovascular causes observed in diabetic patients on sulfonylureas in the UGPD 15 and BARI 30 trials, and the worse outcome of patients on sulfonylureas at the time of acute myocardial infarction. 16 , 17 , 18
This study has some limitations. First, the differences in heart rate at rest among the groups may have influenced the results. Although the resting heart rate was significantly different between DMO and DMG groups, there were no significant differences between NDM and DMG groups, and between DMO and NDM groups. Second, we cannot rule out that glibenclamide may have prevented the improvement in ischemic threshold during the second test through reduction of ischemic vasodilatation by blockade of KATP channels in vascular smooth muscle; 31 however, if this were the case, we should have seen more severe ischemic changes in the DMG group during the first exercise test.
Finally, these evidences suggest that glibenclamide should be used carefully in patients with coronary artery disease and diabetes mellitus, or if possible be shifted to insulin or a new generation sulfonylurea with a specific effect on pancreatic KATP channels.
REFERENCES
- 1. Williams DO, Bass TA, Gerwirtz H, et al Adaptation to the stress of tachycardia in patients with coronary artery disease; insight into the mechanism of the warm‐up phenomenon. Circulation 1985;71: 687–692. [DOI] [PubMed] [Google Scholar]
- 2. Okazaki Y, Kodama K, Sato H, et al Attenuation of increased regional myocardial oxygen consumption during exercise as a major cause of warm‐up phenomenon. J Am Coll Cardiol 1993;21: 1597–1604. [DOI] [PubMed] [Google Scholar]
- 3. Stewart RAH, Simmonds MB, Williams MJA. Time course of “warm‐up” in stable angina. Am J Cardiol 1995;76: 70–73. [DOI] [PubMed] [Google Scholar]
- 4. Tomai F, Crea F, Danesi A, et al Mechanisms of the warm‐up phenomenon. Eur Heart J 1996;17: 1022–1027. [DOI] [PubMed] [Google Scholar]
- 5. Gross GJ, Auchapach JA. Blockade of ATP‐sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res 1992;70: 223–233. [DOI] [PubMed] [Google Scholar]
- 6. Parrat JR, Kane KA. KATP channels in ischemic preconditioning. Cardiovasc Res 1994;28: 783–787. [DOI] [PubMed] [Google Scholar]
- 7. Speechly‐Dick ME, Grover GJ, Yellon DM. Does ischemic preconditioning in the human involve protein kinase C and the ATP‐dependent K+ channel? Studies of contractile function after simulated ischemia in an atrial in vitro model. Circ Res 1995;77: 1030–1035. [DOI] [PubMed] [Google Scholar]
- 8. Cleveland JC, Meldrum DR, Cain BS, et al Oral sulfonylurea hypoglicemic agents prevent ischemic preconditioning in human myocardium. Two paradoxes revisited. Circulation 1997;96: 29–32. [DOI] [PubMed] [Google Scholar]
- 9. Tomai F, Crea F, Gaspardone A, et al Ischemic preconditioning during coronary angioplasty is prevented by glibenclamide, a selective ATP‐sensitive K+ channel blocker. Circulation 1994;90: 700–705. [DOI] [PubMed] [Google Scholar]
- 10. Abbot RD, Brand FN, Kannel WB. Epidemiology of some peripheral arterial findings in diabetic men and women: Experiences from the Framingham study. Am J Med 1990;88: 376–381. [DOI] [PubMed] [Google Scholar]
- 11. Fuller JH, Shipley MJ, Rose G, et al Mortality from coronary heart disease and stroke in relation to degree of glycaemia: The Whitehall study. BMJ 1983;287: 867–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. HilIer R, Sperduto RD, Podgor MJ, et al Diabetic retinopathy and cardiovascular disease in type II diabetics. The Framingham heart study and Framingham eye study. Am J Epidemiol 1988;128: 402–409. [DOI] [PubMed] [Google Scholar]
- 13. Groop LC. Sulfonylureas in NIDDM. Diabetes Care 1992;15: 737–754. [DOI] [PubMed] [Google Scholar]
- 14. Gerich JE. Oral hypoglicemic agents. N Engl J Med 1989;321: 1231–1245. [DOI] [PubMed] [Google Scholar]
- 15. University Group Diabetes Program . A study of the effects of hypoglycemic agents on vascular complications in patients with adult‐onset diabetes mellitus: II. Mortality results. Diabetes 1970: 19(2):785–830. [PubMed] [Google Scholar]
- 16. Klimt CR, Canner PHPL, Jacobs DC, et al The prognostic importance of plasma glucose levels and of the use of oral hypoglycemic drugs after myocardial infarction in men. Diabetes 1977;26: 453–465. [PubMed] [Google Scholar]
- 17. Soler NG, Bennet MA, Malins JM. Coronary care for myocardial infarction in diabetics. Lancet 1974;I: 475–477. [DOI] [PubMed] [Google Scholar]
- 18. Rytter L, Troelsen S, Beck‐Nielsen H. Prevalence and mortality of acute myocardial infarction in patients with diabetes. Diabetes Care 1985;8: 230–234. [DOI] [PubMed] [Google Scholar]
- 19. The Medicine, Angioplasty, or surgery Study (MASS‐II): A Randomized controlled clinical trial of 3 therapeutic strategies for multi‐vessel coronary artery disease: 1‐year results. J Am Coll Cardiol 2004;43: 1743–1751. [DOI] [PubMed] [Google Scholar]
- 20. Murry CE, Richard VJ, Jennings RB, et al Myocardial protection is lost before cantractile function recovers from ischemic preconditioning. Am J Physiol 1991;260: H796–H804. [DOI] [PubMed] [Google Scholar]
- 21. Li YW, Whittaker P, Kloner RA. The transient nature of the effect of ischemic preconditioning on myocardial infarct size and ventricular arrhythmia. Am Heart J 1992;123: 346–353. [DOI] [PubMed] [Google Scholar]
- 22. Gobel FL, Nordstrom LA, Nelson RR, et al The rate‐pressure product as an index of myocardial oxygen consumption during exercise in patients with angina pectoris. Circulation 1978;57: 549–556. [DOI] [PubMed] [Google Scholar]
- 23. Casaburi R. Principles of exercise testing. Chest 1992;101: 263–267. [PubMed] [Google Scholar]
- 24. Waters DD, McCans JL, Crean PA. Serial exercise testing in patients with effort angina: Variable tolerance, fixed threshold. J Am Coll Cardiol 1985;6: 1011–1015. [DOI] [PubMed] [Google Scholar]
- 25. Gross GJ, Auchampach JA. Blockade of ATP‐sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res 1992;70: 223–233. [DOI] [PubMed] [Google Scholar]
- 26. Hearse DJ. Activation of ATP‐sensitive potassium channels: A novel pharmacological approach to myocardial protection? Cardiovasc Res 1995;30: 1–17. [PubMed] [Google Scholar]
- 27. Toami F, Danesi A, Ghini AS, et al Effects of KATP channel blockade by glibenclamida on the warm‐up phenomenon. Eur Heart J 1999;20: 196–202. [DOI] [PubMed] [Google Scholar]
- 28. Övünç K. Effects of glibenclamide, a KATP channel blocker, on warm‐up phenomenon in type II diabetic patients with chronic stable angina pectoris. Clin Cardiol 2000;23: 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meisheri KD, Khan SA, Martin JL. Vascular pharmacology of ATP‐sensitive K‐channels: Interactions between glyburide and K‐channel‐openers. J Vasc Res 1993;30: 2–12. [DOI] [PubMed] [Google Scholar]
- 30. Detre KM, Lombardero MS, Brooks MM, et al for the Bypass Angioplasty Revascularization Investigation Investigators. The effect of previous coronary‐artery bypass surgery on the prognosis of patients who have acute myocardial infarction. N Engl J Med 2000;342: 989–997. [DOI] [PubMed] [Google Scholar]
- 31. Daut J, Maier‐Rudolph W, Von Beckerath N, et al Hypoxic dilation of coronary arteries is mediated by ATP‐sensitive potassium channels. Science 1990;247: 1341–1344. [DOI] [PubMed] [Google Scholar]