

Abstract
Background: We conducted a study of chronic therapy with flecainide versus placebo in a small group of LQT‐3 patients with the ΔKPQ deletion to evaluate the safety and efficacy of flecainide in this genetic disorder. In vitro studies have shown that flecainide provides correction of the impaired inactivation associated with the ΔKPQ deletion.
Methods: A randomized, double‐blind, placebo‐controlled clinical trial was conducted with flecainide and placebo in six male LQT‐3 subjects with the ΔKPQ deletion.
Results: The lowest possible dose of flecainide associated with at least a 40 ms reduction in the QTc interval was determined in an initial open‐label, dose‐ranging investigation using one‐fourth or half of the recommended maximal antiarrhythmic flecainide dose. QTc reduction was achieved with a flecainide dose of 1.5 mg/kg per day in 4 subjects and with 3.0 mg/kg per day in 2 subjects. Subjects were randomized to four 6‐month alternating periods of flecainide and placebo therapy based on the open‐label dose findings. Average QTc values during placebo and flecainide therapies were 534 ms and 503 ms, respectively, with an adjusted reduction in QTc of −27.1 ms (95% confidence interval: −36.8 ms to −17.4 ms; P < 0.001) at a mean flecainide blood level of 0.11 ±0.05 μg/ml. Minimal prolongation in QRS occurred (mean: +2.5 ms), and there were no major adverse cardiac effects.
Conclusions: Chronic low‐dose flecainide significantly shortens the QTc interval in LQT‐3 subjects with the ΔKPQ mutation. No major adverse drug effects were observed with flecainide during this trial, but the sample size is not large enough to evaluate the safety of flecainide therapy in patients with this mutation.
Keywords: long‐QT syndrome, ion channels, antiarrhythmic agents
The long QT syndrome (LQTS) is a familial disorder in which affected family members have prolonged ventricular repolarization on the electrocardiogram and a propensity to syncope, polymorphic ventricular tachycardia (torsade de pointes), and sudden death. 1 Seven LQTS‐related genes have been identified involving numerous mutations. 2 , 3 , 4 One of the seven LQTS genes, SCN5A, encodes for the sodium channel, and some mutations in this gene are associated with altered inactivation of sodium entry into the myocardial cell resulting in prolonged ventricular repolarization. The functional characteristics of one particular SCN5A mutation involving a 9‐base‐pair deletion, with deletion of three amino acids (ΔKPQ) in the cytoplasmic linker region between the third and fourth domains of the sodium‐channel α‐subunit protein, have been well characterized. In brief, this mutational deletion results in dispersed and long‐lasting burst reopenings of the sodium channel with marked prolongation of the action potential duration. 5
In 1995, Schwartz, et al. demonstrated that a single oral dose of the sodium‐channel blocker, mexiletine, administered to 7 LQT‐3 patients with the ΔKPQ deletion produced significant shortening of the QTc interval within 4 hours. 6 Similar QTc shortening in LQT‐3 patients with the ΔKPQ deletion has been reported with lidocaine and tocainide. 7 In 2000, Benhorin et al. reported QT shortening with flecainide in LQT‐3 patients with the D1790G mutation involving the SCN5A gene. 8 In 2001, Windle et al. showed normalization of the QTc interval with low doses of oral flecainide administered over 48 hours in 5 patients with the ΔKPQ deletion. 9 In vitro studies have shown that flecainide has high affinity for the sodium‐channel protein and provides almost complete correction of the impaired inactivation associated with the ΔKPQ deletion. 10 However, flecainide has proarrhythmic effects in patients with organic heart disease 11 and has the potential to cause ventricular arrhythmias in patients with Brugada syndrome, a genetic disorder also involving mutations in the SCN5A gene. 12 Provocation of ventricular arrhythmias with flecainide in LQT‐3 patients with the ΔKPQ mutation is a distinct possibility. It is for this reason that we conducted a randomized, double‐blind, placebo‐controlled trial extending over 2 years with flecainide versus placebo in a small group of LQT‐3 patients with the ΔKPQ deletion to evaluate the safety and efficacy of flecainide in this genetic disorder.
METHODS
Organization of the Trial
The trial began on January 17, 2000 and enrolled LQT‐3 patients from two participating institutions (University of Rochester Medical Center and the University of Nebraska Medical Center). The protocol was approved by the institutional review boards at the two hospital centers. A Data and Safety Monitoring Board independently reviewed the ongoing results at regular intervals throughout the trial. This trial was carried out under Investigational New Drug approval by the Food and Drug Administration for oral flecainide administration in the study protocol.
Study Population
Subjects of either sex who were 8 years of age or older with genotype‐identified SCN5A‐ΔKPQ mutations were drawn from the population of subjects in the U.S. portion of the International LQTS Registry. 1 Subjects were excluded from enrollment if any of the following conditions applied: (1) not meeting eligibility criteria; (2) significant comorbidity that would preclude the subject's safe participation in the study; (3) coronary heart disease or clinical evidence of left ventricular dysfunction with an ejection fraction <0.40; (4) women of child‐bearing age not receiving appropriate contraceptive therapy; (5) weight <35 kg; (6) evidence of prior sensitivity to flecainide; (7) renal disease that might adversely effect drug excretion; (8) psychosocial problems that could compromise compliance with the protocol; (9) subjects who live at such a distance from their treating physician that travel for the frequent follow‐up visits would not be possible; (10) subjects receiving antiarrhythmic drugs other than beta blockers; or (11) subjects or parents of minors unwilling to sign the consent for participation. Subjects already taking beta blockers were allowed to continue on the drug during the trial. Subjects with an implanted pacemaker, defibrillator, or prior cervico‐thoracic sympathetic ganglionectomy were eligible for participation.
Design
After subjects provided written informed consent, a screening baseline clinical history and 12‐lead electrocardiogram were obtained and a physical examination was conducted. The study was divided into two parts:
-
1
Initial Open‐Label Flecainide Study (Phase 0): This portion of the trial involved an initial 2‐week open‐label, dose‐ranging study with initial baseline data collection including electrocardiogram and screening blood tests. Open‐label, oral dosing included 1 week of low‐dose flecainide (one‐fourth of the recommended maximal mg/kg per day dose = 1.5 mg/kg per day given in divided doses at 12‐hour intervals) followed by 1‐week washout without flecainide. If QTc shortening was less than 40 ms at the end of the first week of flecainide, subjects entered a second week of intermediate‐dose flecainide (half of the recommended maximal mg/kg per day dose = 3.0 mg/kg per day given in divided doses at 12‐hour intervals), and then a final week without any flecainide. During this 2–4‐week dose‐ranging study, appropriate electrocardiographic monitoring was performed, flecainide blood levels were obtained at anticipated trough time intervals, and subjects were monitored closely for any adverse effects. The findings from this open‐label dose‐ranging study were used to determine the appropriate dose of flecainide for use in the long‐term safety‐efficacy trial.
-
2
Long‐Term Randomized Flecainide Safety‐Efficacy Trial (Phases 1–4): Subjects were randomized to four 6‐month alternating periods (Phases 1–4) of flecainide and placebo therapy starting with either flecainide, with the oral dose determined by the open‐label dosing study or placebo. This blinded crossover design at periodic 6‐month intervals allowed each subject to serve as his or her own control. Regular scheduled follow‐up visits were set at 1 week and then monthly intervals during the first two 6‐month medication periods, and then at 1 week and at 1‐, 3‐, and 6‐month intervals during second two 6‐month medication periods. At each visit, monitoring of medication adherence and evaluation for side effects, toxicity, health status, use of nonstudy medications, and interval illness were carried out, with data recorded on standardized study forms. Each subject was specifically questioned about interim syncopal episodes, near syncope, and subjective palpitations. At each visit, a 12‐lead electrocardiogram and an 11‐minute high‐resolution digital electrocardiographic recording were obtained as well as trough flecainide blood levels. At the 1‐ and 3‐month visits of each study phase, a 24‐hour Holter electrocardiogram was recorded for safety monitoring.
Therapy
The dose of flecainide utilized in this trial was relatively low, and ranged from 25 mg to 100 mg every 12 hours depending on the initial dose‐ranging findings and patient tolerance.
Electrocardiographic Measurements
The heart rate, PR interval, and the QRS duration were taken from the automatic paper printout of the Marquette electrocardiographic (ECG) recording. The QT interval was measured manually in a blinded fashion relative to all baseline, flecainide, and placebo phases. The QT interval (ms) is the time interval from the Q wave to the end of the T wave, defined as the point of its merger with the isoelectric baseline. When a discrete U wave interrupts the T wave before the latter returns to the baseline, the end of the T wave was defined as the nadir of the curve between the T and U waves. The QT interval and its preceding RR interval were averaged from manually measured intervals on three consecutive beats in leads II, V2, and V5. Measurements in lead II were the primary measure for QT, but if lead II was not technically satisfactory in any given patient, then lead V2 or V5 was used in all QT measurements for such patients. The heart‐rate‐corrected QT interval (QTc) was calculated using the Bazett formula (QTc = QT/RR½).
Statistical Analysis
The aim of the trial was to determine if chronic flecainide therapy was associated with significant and sustained shortening of the QTc interval during two 6‐month periods of active therapy compared to two 6‐month periods of placebo therapy adjusting for baseline QTc. The trial was designed to enroll 40 LQT‐3 subjects in order to achieve 95% power to detect an average reduction in QTc of ≥40 ms at a 5% significance level. A 25% combined noncompliance, dropout, and crossover rate was assumed. Only 6 subjects were enrolled, and one of these subjects did not complete the trial. The trial ended on September 12, 2003 after the final 6‐month follow‐up visit was completed on the last enrolled subject.
The primary analysis was based on a mixed‐effects linear model of the difference between the changes in QTc, flecainide minus placebo, with adjustment for specific subject (random effect) as well as for study phase and phase‐specific baseline QTc (fixed effects). Treatment interactions and baseline values of other ECG parameters were considered and then discarded as not statistically significant. Similar analyses were performed for other ECG end points.
RESULTS
Study Population
The baseline clinical characteristics of the 6 enrolled subjects are presented in Table 1. The subjects ranged in age from 18 to 52 years, all were male, and 5 were receiving beta blockers.
Table 1.
Clinical Characteristics of the Study Population
| Characteristics | Value (Range) (N = 6) |
|---|---|
| Demographics | |
| Age (year) | 36 ± 13 (18–52) |
| Male sex (%) | 100 |
| Weight (kg) | 79 ± 15 (55–103) |
| Clinical history | |
| Syncope (%) | 33 |
| Therapy | |
| Beta‐blockers (%) | 83 |
| Pacemaker (%) | 50 |
Electrocardiographic Findings during Open‐Label, Dose‐Ranging Study
The electrocardiographic findings at baseline and during open‐label, oral flecainide dosing at 1.5 and 3.0 mg/kg per day together with flecainide blood levels are presented in Table 2. At the higher flecainide dose, there was minimal reduction in heart rate, the PR interval increased slightly, and there was widening of the QRS duration. The QT and QTc intervals progressively shortened in association with increasing flecainide dosing and blood levels.
Table 2.
Electrocardiographic Findings during Open‐Label, Dose‐Ranging Flecainide Administration
| Flecainide Dose | Number of Subjects | Heart Rate (beats/min) | PR (ms) | QRS (ms) | QT (ms) | QTc (ms) | Flecainide Blood Level (μg/ml) | |
|---|---|---|---|---|---|---|---|---|
| Baseline (no drug) | 6 | Mean | 56 | 193 | 103 | 567 | 538 | 0 |
| — | SD | 6 | 21 | 15 | 69 | 38 | — | |
| Changes from baseline while on flecainide* | ||||||||
| 1.5 mg/kg per day | 6 | Mean | 0.3 | −0.5 | −1.0 | −48 | −45 | 0.11 |
| SD | 3.2 | 17.2 | 9.6 | 28 | 25 | 0.03 | ||
| 3.0 mg/kg per day | 2† | Mean | −2.0 | 22.0 | 10.0 | −45 | −50 | 0.24 |
| SD | 1.4 | 11.3 | 14.1 | 21 | 21 | 0.14 | ||
*Values are averages of measurements taken on the third and eighth day minus baseline measurements for each subject; with means and standard deviations of these differences across subjects.
†Limited to the two subjects with <40 ms changes from baseline QTc on the eighth day of the 1.5 mg/kg per day dose.
Long‐Term Randomized Flecainide/Placebo Trial
Five of 6 LQT‐3 subjects completed all four phases of the 24‐month randomized trial. One subject, aged 52 years with baseline QRS duration of 128 ms, developed electrocardiographic evidence of ST segment elevation in leads V1–V3 during the trial. Drug unblinding revealed that the subject was on flecainide (50 mg) every 12 hours with a flecainide blood level of 0.14 μg/ml. Flecainide was stopped and the subject was discontinued from the trial. An ST segment pattern similar to the one observed during the flecainide therapy was found on an electrocardiogram that had been recorded a few years prior to the start of trial. The Data Safety Monitoring Board concluded that the ST segment findings were not solely due to flecainide. Another subject developed a 3‐second sinus pause while on blinded drug therapy. Unblinding revealed placebo therapy, and a dual‐chamber pacemaker was implanted and programmed to the AAI mode. The subject was continued in the trial after the pacemaker was implanted. No other adverse cardiac effects were noted and no ventricular tachyarrhythmias were uncovered during numerous 24‐hour Holter recordings obtained during the course of the trial.
The mean values of the electrocardiographic measurements and of the flecainide blood levels during baseline, placebo, and flecainide portions of the trial are presented in Table 3. Flecainide treatment significantly shortened the adjusted QTc interval when compared to placebo by 27.1 ms (P < 0.001), and the adjusted QRS duration increased by 2.5 ms (P = 0.04). PR interval and heart rate were not significantly different during flecainide and placebo therapies (P > 0.05). Twenty‐four‐hour Holter monitoring showed no significant change in mean heart rate between placebo and flecainide therapies.
Table 3.
Findings during Randomized Placebo and Flecainide Therapy
| Treatment Groups | Number of Observations | Heart Rate (beats/min) | PR (ms) | QRS (ms) | QT (ms) | QTc (ms) | Flecainide Blood Level (μg/ml) |
|---|---|---|---|---|---|---|---|
| Raw data* | |||||||
| Placebo | 61 | 57 | 187 | 97 | 553 | 534 | 0 |
| Flecainide | 56 | 56 | 196 | 100 | 523 | 503 | 0.11 ± 0.05 |
| Changes from phase‐specific baseline† | |||||||
| Placebo | 61 | +0.8 | +6.8 | +1.5 | +3.5 | +4.8 | 0 |
| 95% CI | (−0.6, 2.1) | (−3.8, 17.5) | (−0.4, 3.5) | (−7.5, 14.5) | (−6.8, 16.3) | ||
| P value | 0.27 | 0.20 | 0.13 | 0.53 | 0.41 | ||
| Flecainide | 54 | +0.3 | +10.3 | +4.0 | −33.8 | −22.4 | 0.11 ± 0.05 |
| 95% CI | (−1.1, 1.8) | (−0.7, 21.2) | (2.0, 6.0) | (−45.3, −22.4) | (−34.2, −10.5) | ||
| P value | 0.65 | 0.07 | <0.001 | <0.001 | <0.001 | ||
| Difference | −0.4 | +3.4 | +2.5 | −37.3 | −27.1 | ||
| 95% CI | (−2.2, 1.3) | (−6.7, 13.6) | (0.2, 4.9) | (−52.7, −21.8) | (−36.8, −17.4) | ||
| P value | 0.62 | 0.50 | 0.04 | <0.001 | <0.001 | ||
*Data for the end points represent mean values.
†All values are means, adjusted for specific subject (random effect) as well as for study phase and phase‐specific baseline value (fixed effect). Placebo changes are those while on placebo with phase‐specific baseline value subtracted; flecainide changes are those recorded while on flecainide, also with phase‐specific baseline value subtracted. The difference between the changes, flecainide minus placebo, is the primary measure of treatment effect.
The relationship between QTc and flecainide blood levels throughout the randomized portion of the trial is shown in Figure 1. The QTc declined with increasing flecainide blood levels that range from 0.02 to 0.21 μg/ml. A graphic presentation of the QTc values in one subject during the four phases of the flecainide trial are presented in Figure 2, with representative electrocardiographic tracings during placebo and flecainide therapy shown in Figure 3 for that subject.
Figure 1.
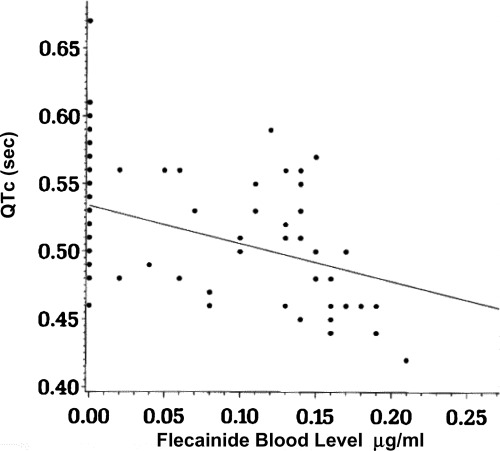
QTc versus flecainide blood levels during the randomized placebo or flecainide therapy. The linear regression equation relating these two parameters (126 observations) is QTc = 0.53–0.28(flecainide concentration), with R2= 0.16.
Figure 2.
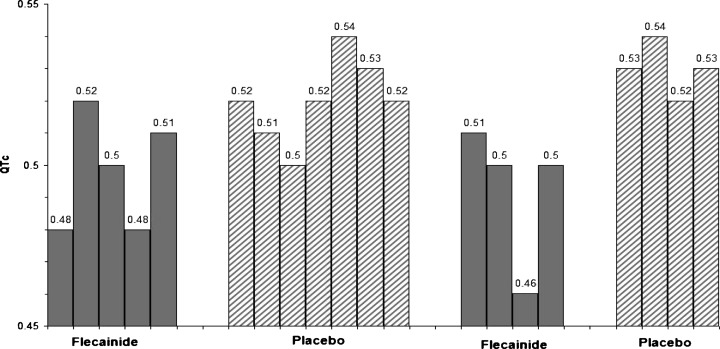
QTc in seconds at multiple times during each of four 6‐month periods of randomized therapy with placebo (striped bars; average QTc = 0.52s) or flecainide (gray bars; average QTc = 0.49s) in one LQT‐3 subject.
Figure 3.

Electrocardiographic tracings during selected phases of placebo and flecainide therapy in the same LQT‐3 subject as presented in Figure 2.
DISCUSSION
This randomized, double‐blind, placebo‐controlled trial shows that chronic low‐dose flecainide significantly shortens the QTc interval in LQT‐3 subjects with the ΔKPQ mutation in the SCN5A sodium‐channel gene. At an average flecainide blood level of 0.11 μg/ml, the QTc shortened by 27.1 ms when compared to placebo therapy, and there were no major adverse cardiac effects. This low flecainide blood level was associated with a 2.5 ms increase in QRS duration.
The findings in this trial support the prior, preliminary finding of QTc reduction with short‐term (48 hour) flecainide therapy that was reported in 1998 in 5 subjects with LQT‐3 (ΔKPQ mutation). 9 The current trial provides substantial evidence that chronic flecainide therapy has a sustained effect when the drug is administered over several months. However, caution must be exercised when administering this medication even in low doses to subjects with the ΔKPQ mutation. One subject in our study developed minor ST segment changes suggesting a possible Brugada‐like pattern at low blood levels of flecainide, and the flecainide was promptly stopped in this patient. Although the observed electrocardiogram (ECG) pattern was similar to one previously recorded in this patient a few years before the start of the trial, we simply do not know what the effect of flecainide therapy would have been had we continued the administration of this medication.
Mutations in different regions of the SCN5A gene are present in LQT‐3 2 , 8 and in Brugada syndrome, 13 and overlap has been reported in these two conditions with a few specific SCN5A mutations. 14 , 15 It is possible that patients with the ΔKPQ mutation could progress from just QTc prolongation to an intraventricular conduction disturbance and then onto a Brugada‐type electrocardiographic pattern with age. Since flecainide challenge can induce ST segment elevation of the Brugada type in some LQT‐3 subjects, 16 it is important to obtain ongoing follow‐up electrocardiographic monitoring when administering flecainide to subjects with the ΔKPQ form of LQT‐3.
The weaker sodium‐channel blocking agent, mexiletine, has also been shown to shorten the QTc interval in subjects with the ΔKPQ mutation. Acute oral loading with mexiletine shortened the QTc interval in 8 LQT‐3 subjects, 7 with ΔKPQ mutation. 6 It is interesting that mexiletine does not block open sodium channels, but it may promote or stabilize closed/inactivated channels when inactivation is not entirely crippled. 17 The effects of chronic, long‐term mexiletine therapy on the QTc interval have not been reported.
Benhorin et al., reported the effectiveness of open‐label oral flecainide in shortening the QTc in 8 asymptomatic subjects with the D1790G mutation involving the SCN5A gene. 8 The QTc‐shortening effects were maintained during 4–12 months of therapy without adverse effects. Although the D1790G mutation has a clinical phenotype similar to the ΔKPQ mutation, the electrophysiologic mechanisms are quite different in the two SCN5A mutations. Flecainide corrects the impaired inactivation associated with the ΔKPQ mutation. 10 The D1790G mutation changes the manner by which the altered sodium channel interacts with sodium‐channel blocking agents, particularly with flecainide, with the mutation conferring a potent use‐dependent block with flecainide. 18 , 19 Thus, it is not surprising that the clinical efficacy of flecainide is similar in subjects with the ΔKPQ and the D1790G mutations, although the mechanism of this drug in these two sodium‐channel mutations is quite different in expressed channels. Of interest, flecainide was associated with increased heart rate in those with the D1790G mutation, but no change in heart rate was observed with flecainide in subjects with the ΔKPQ mutation.
The current trial enrolled fewer patients than planned, and all the subjects were males. The small sample size and the absence of female subjects compromise the generalizability of the findings. The sample size is not large enough to evaluate the long‐term safety of flecainide in patients with the ΔKPQ mutation. Nevertheless, the study provides encouraging evidence in support of mutation‐specific therapy in LQTS. At the present time, flecainide still should be considered an investigational drug in LQT‐3 patients with the ΔKPQ mutation. If flecainide is administered to high‐risk patients with this mutation, careful follow‐up with frequent ECG monitoring is essential. Furthermore, the lowest possible flecainide dose should be used, and follow‐up blood levels should be obtained with the aim to keep the flecainide blood level less than 0.25 μg/ml. A larger clinical trial with flecainide in patients with this mutation is needed before this therapy can be recommended for the treatment of patients with this genetic disorder.
Acknowledgments
Acknowledgments: The authors thank the members of the Data and Safety Monitoring Board (Drs. Raymond L. Woosley, Nancy Cox, Frank I. Marcus, and an anonymous member) for their valuable assistance and Dr. Peter M. Spooner for his intellectual input and commitment to this research project.
This study was supported in part by research grant HL58731 and by General Clinical Research Center grant 5M01RR00044 from the National Center for Research Resources from the National Institutes of Health, Bethesda, MD.

[ Michel Mirowski with his Daughters Used with permission of Ariella M. Rosengard, MD. This photograph may not be reproduced, stored, or transmitted in any form or by any means without the prior permission in writing from Dr. Rosengard. ]
REFERENCES
- 1. Moss AJ, Schwartz PJ, Crampton RS, et al The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 1991;84: 1136–1144. [DOI] [PubMed] [Google Scholar]
- 2. Splawski I, Shen J, Timothy KW, et al Spectrum of mutations in long‐QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000;102: 1178–1185. [DOI] [PubMed] [Google Scholar]
- 3. Plaster NM, Tawil R, Tristani‐Firouzi M, et al Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 2001;105: 511–519. [DOI] [PubMed] [Google Scholar]
- 4. Mohler PJ, Schott JJ, Gramolini AO, et al Ankyrin‐B mutation causes type 4 long‐QT cardiac arrhythmia and sudden cardiac death. Nature 2003;421: 634–639. [DOI] [PubMed] [Google Scholar]
- 5. Dumaine R, Wang Q, Keating MT, et al Multiple mechanisms of Na+ channel‐linked long‐QT syndrome. Circ Res 1996;78: 916–924. [DOI] [PubMed] [Google Scholar]
- 6. Schwartz PJ, Priori SG, Locati EH, et al Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene‐specific therapy (see comments). Circulation 1995;92: 3381–3386. [DOI] [PubMed] [Google Scholar]
- 7. Rosero S, Zareba W, Robinson JL, et al Gene‐specific therapy for long QT syndrome: QT shortening with lidocaine and tocainide in patients with mutation of the sodium channel gene. Ann Noninvasive Electrocardiol 1997;2: 274–278. [Google Scholar]
- 8. Benhorin J, Taub R, Goldmit M, et al Effects of flecainide in patients with new SCN5A mutation: Mutation‐specific therapy for long‐QT syndrome? Circulation 2000;101: 1698–1706. [DOI] [PubMed] [Google Scholar]
- 9. Windle JR, Geletka RC, Moss AJ, et al Normalization of ventricular repolarization with flecainide in long qt syndrome patients with scn5a:deltakpq mutation. Ann Noninvasive Electrocardiol 2001;6: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nagatomo T, January CT, Makielski JC. Preferential block of late sodium current in the LQT3 DeltaKPQ mutant by the class I(C) antiarrhythmic flecainide. Mol Pharmacol 2000;57: 101–107. [PubMed] [Google Scholar]
- 11. Echt DS, Liebson PR, Mitchell LB, et al Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991;324: 781–788. [DOI] [PubMed] [Google Scholar]
- 12. Antzelevitch C. The Brugada syndrome: Ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol 2001;12: 268–272. [DOI] [PubMed] [Google Scholar]
- 13. Chen Q, Kirsch GE, Zhang D, et al Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392: 293–296. [DOI] [PubMed] [Google Scholar]
- 14. Bezzina C, Veldkamp MW, Van Den Berg MP, et al A single Na(+) channel mutation causing both long‐QT and Brugada syndromes. Circ Res 1999;85: 1206–1213. [DOI] [PubMed] [Google Scholar]
- 15. Veldkamp MW, Viswanathan PC, Bezzina C, et al Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ Res 2000;86: E91–E97. [DOI] [PubMed] [Google Scholar]
- 16. Priori SG, Napolitano C, Schwartz PJ, et al The elusive link between LQT3 and Brugada syndrome: The role of flecainide challenge. Circulation 2000;102: 945–947. [DOI] [PubMed] [Google Scholar]
- 17. Bennett PB. Long QT syndrome: Biophysical and pharmacologic mechanisms in LQT3. J Cardiovasc Electrophysiol 2000;11: 819–822. [DOI] [PubMed] [Google Scholar]
- 18. Abriel H, Wehrens XH, Benhorin J, et al Molecular pharmacology of the sodium channel mutation D1790G linked to the long‐QT syndrome. Circulation 2000;102: 921–925. [DOI] [PubMed] [Google Scholar]
- 19. Wehrens XH, Abriel H, Cabo C, et al Arrhythmogenic mechanism of an LQT‐3 mutation of the human heart Na(+) channel alpha‐subunit: A computational analysis. Circulation 2000;102: 584–590. [DOI] [PubMed] [Google Scholar]