

Abstract
We describe the case of a 14‐year‐old girl with a history of syncopal episodes triggered by stress or exercise. Catecholaminergic polymorphic ventricular tachycardia was diagnosed with the aid of an implantable loop recorder. The genetic testing of the patient and her family revealed a de novo novel missense mutation (Ser4155Tyr) in the exon 90 of the ryanodine receptor gene. This mutation affects a highly conserved residue (S4155) and results to replacement of serine (S) with tyrosine (Y) leading to change in physical and chemical properties. The girl was treated with an implantable defibrillator, metoprolol and flecainide. Over 1 year of follow‐up she had no recurrence of ventricular tachycardia.
Keywords: molecular biology/genetics; basic, electrophysiology—cardiac arrest/sudden death; clinical, electrophysiology—ventricular tachycardia; clinical, implantable devices—ventricular tachycardia/fibrillation; clinical, pediatrics—implantable devices; clinical
CASE REPORT
A 14‐year‐old girl was referred for investigation of frequent syncopal episodes triggered by emotional stress (fear of dogs). Initial workup including electrocardiogram, neurological examination, blood biochemistry, echocardiogram, and tilt‐test were normal. There was no history of syncope or sudden death in the family. Resting ECG showed normal sinus rhythm with normal PR and QT intervals and no signs of Brugada syndrome. An exercise test was performed which showed ventricular ectopy with at least two different morphologies and salvos during exercise suggestive but not diagnostic of bidirectional ventricular tachycardia (Fig. 1).
Figure 1.
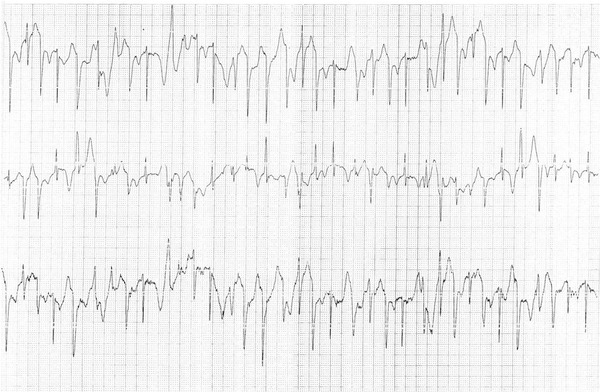
Ventricular ectopic beats and salvos with at least two different morphologies during treadmill exercise test.
Dear Dr. Luria, Would you please comment on the history and the testing so far? What are the specific concerns you have at this point of presentation?
This young girl had frequent episodes of emotional (“psychogenic”) syncope. Most frequent cause of this syndrome, usually described in medical environment, such as seeing blood or surgical procedure, is neurocardiogenic (vaso‐vagal) syncope. Any sudden strong emotional experience, such as fear of dogs, can cause similar vasovagal response. However, this patient had rapid polymorphic ventricular tachycardia (PMVT) during exercise. This is striking feature, which have to be recognized as the most apparent reason for recurrent syncopal events.
Ischemic cardiomyopathy or myocarditis usually is considered first among the reasons for PMVT in adult population. However, in a 14‐year old, with recurrent stress—induced arrhythmia and syncope, as the only clinical manifestation, and no evidence of myocardial damage (no cardiac enzyme elevation and normal myocardial function by echocardiogram)—these diagnoses are unlikely. Hereditary disorders, in this clinical setting, seem to be most probable reason for severe ventricular arrhythmia. Among these, differentiation between structural heart abnormalities and congenital defect in membrane electrical channel function (channelopathy) is of paramount importance. A normal Doppler echocardiogram rules out most structural heart disease. However some structural defects, like anomalous coronary artery origin or mild cardiomyopathy, could be missed by echocardiogram. These diseases have to be ruled out as potential cause for exercise induced PMVT. I would recommend advanced structural imaging like cardiac CT or MRI as a next step of clinical work up. At the same time, I would not expect positive findings in these studies, because syncopal PMVT as isolated clinical feature is very unusual for coronary anomaly and cardiomyopathy. After structural heart diseases are excluded, congenital channelopathies become the most important potential causes for syncope, induced by repetitive, nonsustained, rapid PMVT.
Subsequently, an electrophysiologic study was performed which revealed normal sinus node function and anterograde conduction and absence of an accessory pathway. No supraventricular or ventricular tachycardia was induced using up to three extra stimuli during ventricular stimulation at 600 ms and 400 ms drives. However, the history and stress test were suggestive of a malignant arrhythmia, therefore metoprolol 50 mg twice daily was initiated and an implantable loop recorder (REVEAL XT 9529, Medtronic Inc., Minneapolis, MN, USA) was implanted. Two months later the girl had another episode of sudden loss of consciousness after running up a flight of stairs, followed by convulsions. Despite the high beta‐blocker dose (100 mg of metoprolol daily) the loop recorder revealed a bidirectional ventricular tachycardia episode followed by a long pause (>6 seconds; Fig. 2A). Moreover, four episodes of bidirectional nonsustained ventricular tachycardia were recorded (Fig. 2B). The patient was treated with an implantable cardioverter‐defibrillator (ICD) and addition of flecainide 50 mg twice daily on top of beta‐blocker therapy to prevent defibrillator shocks. Over 12 months of follow‐up since the implantation of the ICD the patient had no recurrence of ventricular tachycardia but she received an inappropriate shock triggered by sinus tachycardia. Metoprolol 50mg twice daily was changed to bisoprolol 10 mg once daily.
Figure 2.
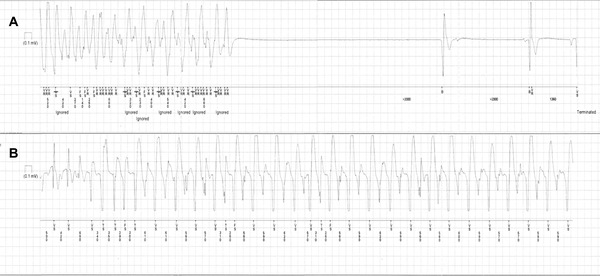
(A). Implantable loop recorder strip during an episode of loss of consciousness followed by convulsions after physical stress. A bidirectional ventricular tachycardia is recorded before the asystole which lasts for approximately 6 seconds. (B) Nonsustained bidirectional VT recorded in the loop recorder on a different occasion. The patient reported palpitations.
Dr. Luria, What are your thoughts about the utility of EP testing? Would you have considered other testing modality? Also, please comment on the management strategy. How common is recurrence of syncope while on beta blockers in this suspected condition?
Electrophysiological study (EPS) is most useful for diagnosis of supraventricular or ventricular arrhythmia, in patients with macro‐reentrant arrhythmia mechanism, such as SVT or scar related VT (for instance, in patients post myocardial infarction or ARVD). When these arrhythmias are suspected in patients with palpitations or syncope, EPS would establish exact diagnosis and provide therapeutic opportunity (ablation). However, EPS in patients without structural heart disease or even in patients with cardiomyopathy, has very small or no benefit and usually not recommended for diagnostic PMVT, as in our patient. EPS has very low sensitivity for diagnosis of sinus or atrioventricular conduction system disorder in patients with syncope and without significant conduction disease on 12‐lead ECG, and therefore, not recommended for this purpose.
Implantable loop recorder is a useful tool for diagnosis of unexplained syncope. The important disadvantage of this technology is “wait and watch” strategy. It could be dangerous, when patient has high risk for sudden cardiac death. In our patient with recurrent syncope and rapid PMVT, one should consider the risk, that next syncopal arrhythmic event can be fatal. Catecholamine‐dependent character of arrhythmia (fear, exercising), irrespective of exact diagnosis, brings about a logical assumption, that therapy with beta‐blockers can be effective in preventing of arrhythmic events. From this point of view, implantable loop recorder became reasonable approach to verify effectiveness of medical therapy and further risk stratification. ICD implantation, in a patient who is 14 years old can be a difficult decision. Patient subsequently had severe syncope with signs of cerebral anoxia (seizures) and evidence of very rapid VT recorded by the loop recorder. After such a malignant arrhythmic event on high dose of beta‐blockers, ICD implantation seems to be inevitable. Additional management strategies, as well as determination of patient prognosis, family screening and genetic consulting are dependent on exact clinical‐genetic diagnosis.
The genetic testing revealed a novel mutation in the ryanodine 2 receptor (RyR2) gene (Fig. 3). The RyR2 exons 3, 8, 14–15, 37, 44–50, 83, 83–105, and flanking introns were assessed. A heterozygous missense mutation S4155Y (or g741775C>A) in exon 90 was identified. The mutation S4155Y affects a highly conserved residue (S4155) and results to replacement of serine (S) with tyrosine (Y) leading to change in physical and chemical properties such as the hydrophobicity and mass (Gratham's distance 144 [0–215]). According to Uniprot Database (http://www.uniprot.org) the amino acid S4155 is located in an intracellular loop where the transmembrane parts M2 (amino acids 3978–3995) and M3 (amino acids 4233–4256) bind together. S4155 is located in the carboxy‐terminal region of the molecule in domain III (channel region) which is a known “hot spot” for mutations.
Figure 3.
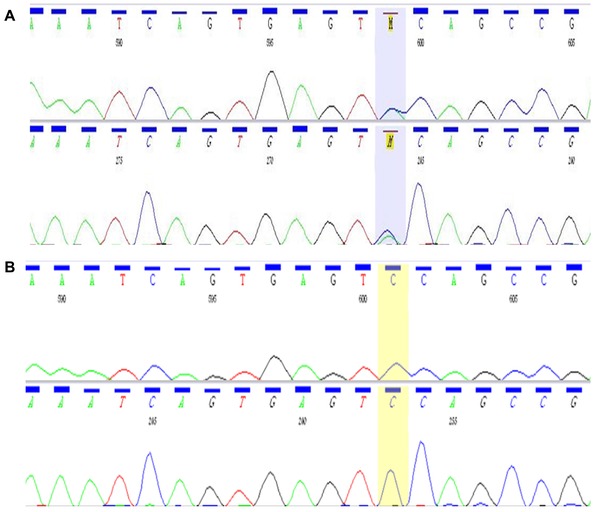
Automated DNA sequencing with the ABI Prism dye‐terminator cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). A. Patient and B. Normal subject.
To assess the effect of the replacement of S with Y, in silico analysis was performed using three available software applications: Polyphen‐2 (Polymorphism Phenotyping) (v2.2.2, http://genetics.bwh.harvard.edu/pph2/Harvard Medical School), Sorting Intolerant From Tolerant (SIFT) (Ensembl 63, http://sift.jcvi.org/J. Craig Venter Institute, San Diego, CA, USA) and PMut (http://mmb.pcb.ub.es/PMut/Molecular Modelling & Bioinformatics Unit IRB‐PCB & University of Barcelona). Polyphen‐2 indicated a harmful replacement (value = 0.999, range 0–1). SIFT indicated that this replacement affects the function of the protein (value = 0.00/values < 0.05 = harmful mutations) but this prediction is of low reliability. PMut predicted that this mutation may be pathogenetic with a reliability value of 6 (range 0–9).
All other family members (parents and a younger sister) were asymptomatic. Both parents underwent treadmill exercise test which was normal. Genetic testing of the parents and sister showed that this mutation was not present in any other member of the family. A pedigree is presented in Figure 4.
Figure 4.
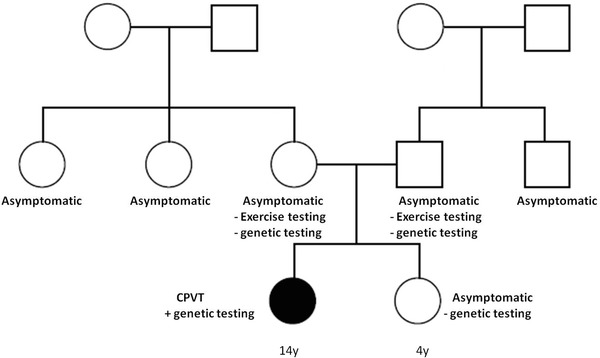
Pedigree of the patient's family. The phenotype of CPVT was not present in any other member of the family and the genetic testing of the parents and sister was negative.
DISCUSSION
More than 20 mutations in exon 90 have been identified so far and exon 90 is among the 16 out of 105 exons of the RyR2 gene that host >65% of total known mutations.1 No more than 15% of the mutations identified in RyR2 have been studied in vitro. However, cosegregation with the disease and absence in normal subjects are suggestive of pathogenicity of a mutation. In cases where CPVT is strongly suspected there is a 95% estimated probability that the identification of a rare missense mutation would have a causal relationship with the patient's CPVT rather than being a rare amino acid substitution.1
Recurrence of VT or sudden death have been described despite of ICD and β‐blocker treatment in patients with CPVT. However, it is still unclear whether specific mutations or other factors are related to increased risk of sudden death.2 Sympathetic denervation may provide an additional therapeutic option for patients with drug refractory arrhythmias and multiple defibrillator shocks.3
Dr. Luria, thank you for your thoughts so far. Please comment on the genetic findings here. Does localization of genotype mutation help in risk stratification of patients with CPVT?
Genetic diseases of cell membrane channels (channelopathies) are very important potential causes for PMVT that can result in syncope and sudden cardiac death, especially in young age. The most frequent is Long QT syndromes, caused by abnormal function of sodium or potassium channels of cellular membrane. PMVT in this diseases could be catecholamine‐induced (especially long QT1 and 2) and usually has specific appearance (“torsades de pointes”), at least in part of ECG leads. In our patient QT is normal, and PMVT had different, bidirectional appearance. Therefore, two other groups of channelopathies need to be taken into consideration.
Andersen‐Tawil syndrome can manifest with bi‐directional VT and no or borderline QT prolongation. Other clinical features of this disease are periodic paralysis and craniofacial dysmorphism. However, only in two‐thirds of cases complete clinical presentation is apparent. Mutation in KCNJ2 gene is responsible for this disease, causing reduction of IK1 membrane current at the end of repolarization and during diastole.
Exercise induced polymorphic VT (usually bidirectional) is the key clinical manifestation of another group of channelopathies, related to intracellular calcium handling and known as catecholamine‐sensitive polymorphic ventricular tachycardia (CPVT). These patients have frequent exercise induced syncope and high risk of sudden cardiac death in young age. Biological basis of this disease in the vast majority of cases is malfunction of ryanodine 2 receptors (RyR2), responsible for calcium release from sarcoplasmic reticulum into cytoplasm during systole. Mutation in RyR2 gene causes diastolic leakage of calcium cytosol and diastolic depolarization (after‐depolarizations) of cell's membrane, triggering PMVT. In rare cases (about 3%), CPVT is a result of mutation in calsequestrin protein, related to RYR2 molecular complex.
About 20% of RyR2 mutations arise de novo, as in our case. Genetic analysis affords a diagnosis of patients with PMVT related to congenital channelopathy and it contributes to risk stratification in patients with long QT syndrome, but not yet in patients with CPVT.
Acknowledgments
LM is supported by grants from the European Heart Rhythm Association and the Hellenic Cardiological Society.
REFERENCES
- 1. Medeiros‐Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2‐encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise‐induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol 2009;54(22):2065–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sy RW, Gollob MH, Klein GJ, et al. Arrhythmia characterization and long‐term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2011;8(6):864–871. [DOI] [PubMed] [Google Scholar]
- 3. Watanabe H, Knollmann BC. Mechanism underlying catecholaminergic polymorphic ventricular tachycardia and approaches to therapy. J Electrocardiol 2011;44(6):650–655. [DOI] [PubMed] [Google Scholar]