

Abstract
Electrical remodeling in advanced stages of cardiovascular diseases creates a substrate for triggering and maintenance of arrhythmias. The electrical remodeling is a continuous process initiated already in the early stages of cardiological pathology. The aim of this opinion article was to discuss the changes in electrical properties of myocardium in left ventricular hypertrophy (LVH), with special focus on its early stage, as well as their possible reflection in the QRS amplitude of the electrocardiogram. It critically appraises the classical hypothesis related to the QRS voltage changes in LVH. The hypothesis of the relative voltage deficit is discussed in the context of supporting evidence from clinical studies, animal experiments, and simulation studies. The underlying determinants of electrical impulse propagation which may explain discrepancies between “normal” ECG findings and increased left ventricular size/mass in LVH are reviewed.
Keywords: left ventricular hypertrophy, ECG, QRS amplitude, relative voltage deficit, specific potential of myocardium
Electrical remodeling is a term comprising complex changes in active and passive electrical properties of myocardium, these changes create conditions for triggering and maintaining of arrhythmias. In a number of articles, growing evidence has been summarized from various aspects including cardiac microstructure, ion channels, energy metabolism, and gene expression. 1 , 2 , 3 , 4 , 5 , 6
Electrical remodeling is interrelated with structural remodeling in a variety of cardiac pathologies and arrhythmias usually occur as their late manifestations. However, electrical and structural remodeling is a continuous process beginning already in the early stages of cardiac pathology. Naturally, the question arises — whether and how is remodeling manifested in the electrocardiogram prior to the manifestation of arrhythmias. The identification of such early changes in the ECG would be of utmost diagnostic and prognostic importance.
The pertinent literature on the electrical remodeling and arrhythmias deals mostly with the role of repolarization changes (the prolongation of AP duration, ion channels, etc.). However, electrical and structural remodeling affects also depolarization.
In left ventricular hypertrophy (LVH), the classical expectation of the clinical ECG diagnostics is the increased QRS amplitude in defined leads (so‐called voltage criteria). It is supported by the assumption that excitation of the larger and thicker muscle mass results in larger and longer living activation boundaries, which in turn, result in the more than usual preponderance of the leftward and posteriorly oriented electrical forces. In other words, the increase in the muscle mass of the left ventricle exaggerates the normal preponderance of the left ventricular potential. However, the increased QRS voltage is seen only in a minority of LVH cases in both clinical and animal studies and consequently voltage criteria suffer from a high number of false negative results and low sensitivity.
In 1976, Mashima 7 presented a concept of “ideal” hypertrophy causing the enlargement of the QRS amplitude. The assumptions characterizing the ideal hypertrophy model are: (i) the hypertrophy is diffuse and symmetrical, (ii) the sequence of electrical activation is unaltered, and (iii) the strength of the double layer and the velocity of the activation wave are the same as normal. The assumption on “ideal hypertrophy” can be illustrated by the results of the computer simulation study by Szathmary et al. 8 This computer model, using identical electrical characteristics for individual elements of working ventricular myocardium shows an increase of maximum spatial vector magnitude with the increasing extent of the activation front.
Mashima points out that discrepancies in actual cases indicate deviations from the ideal state. He assumes that myocardial alteration other than hypertrophy, as well as individual variations in the body build and surrounding tissue obscure the genuine effect of hypertrophy. As regards the strength of double layer he considers its alteration by myocardial edema and “other pathological processes.”
In our previous articles, we have demonstrated a novel approach to so‐called false negative ECG results in LVH and we formulated a hypothesis that these false negative results might reflect changes in electrical properties of myocardium in the LVH development. 9 , 10 Using Mashima's term of “deviations from the ideal state” we assume that these deviations are caused by changes in electrical properties of the myocardium, attributable to hypertrophy already in the early stages of LVH progress. We termed this deviations “the relative voltage deficit” since the QRS voltage is lower than expected according to the classical expectations.
The aim of this opinion article was to provide an overview of changes in electrical properties of the myocardium in LVH with the focus on changes of depolarization in the early stage of LVH and to review the evidence that links altered electrical properties of myocardium to changes in QRS voltage.
THE HYPOTHESIS ON THE RELATIVE VOLTAGE DEFICIT
Our hypothesis on the relative voltage deficit assumes that: (i) a unit of pathologically changed myocardium in LVH is a less efficient generator of cardioelectric field as compared to a unit of healthy myocardium, (ii) the relative voltage deficit starts already in the early stage of LVH development and varies with the progress of LVH — it is diminished in the stage of compensated LVH and is enhanced again in the stage of developing heart failure, and (iii) the relative voltage deficit is caused by altered active and passive electrical properties of myocardium due to electrical and structural remodeling. In terms of the spatial angle theory, the relative voltage deficit represents the changes in nonspatial determinants influencing the QRS voltage. The specific potential of myocardium (SP), calculated as the ratio of the QRS voltage and left ventricular mass (LVM), has been introduced as a measure for the relative voltage deficit. 10
Supporting Evidence for the Relative Voltage Deficit in LVH
In our previous articles, we have demonstrated that the ventricular mass is not always the major determinant of QRS voltage in LVH. 9 , 10 , 11
In clinical studies, we showed lower SP values in hypertensive patients as compared to healthy subjects contrasting with higher QRSmax values in hypertensive patients. 12 During a screening program, lower values of both QRSmax and SP were recorded in hypertensive subjects with newly diagnosed hypertension as compared to healthy subjects. 13 A decrease in QRS voltage was also observed in girls in the first 21 months of intensive training of competitive sport. 14
We have shown a decrease in both absolute and relative values of QRS amplitude in the initial stage of experimental models of LVH due to volume and pressure overload, respectively. 15 , 16 Similar decrease in QRS amplitude was observed also in the experimental model of exercise‐induced LVH in swimming normotensive rats. 17 We have concluded that the decrease in the QRS amplitude could be an early sign of hypertrophic rebuilding of the myocardium, reflecting changes in its electrical properties.
Treatment with antihypertensive drugs provides another model to study the relationship between QRS amplitude and LVM. Antihypertensive drugs reduce increased blood pressure, leading consequently to the regression of LVH by different mechanisms. We study changes in QRS amplitude in relation to LVM changes in spontaneously hypertensive rats treated by lacidipine (calcium antagonist) and enalapril (angiotensin converting enzyme inhibitor). 18 While systolic blood pressure and LVM decreased significantly during the follow‐up period, no significant changes were observed in the QRS amplitude. On the other hand, the values of the specific potential of myocardium increased during the treatment, in the case of enalapril significantly — e.g., the relative voltage deficit reduced.
Recently, the problem of the effect of anabolics on myocardium is of increasing interest, especially in relation to the increasing incidence of sudden death in athletes. We studied the relation between the QRS amplitude and LVM in rats exposed to regular training, combined with the effect of nandrolon. 19 We found an increase in LVM in all groups under study with a maximum increase in groups after nandrolon administration, but the changes in QRS amplitude were not proportional to the increase of LVM. The maximum discrepancies were observed in groups with nandrolon administration. The values of QRS amplitude increased in the group of swimming rats with nandrolon administration, accordingly, the SP values increase, what was associated with a decrease in physical performance.
ELECTRICAL IMPULSE GENERATION AND PROPAGATION IN LVH
Figure 1 summarizes the major factors involved in electrical impulse generation and propagation at the subcellullar, cellular and tissue levels in a form of a mind map to illustrate the complexity and variety of factors involved. The structure of this mind map was adopted from the review on the basic mechanisms of cardiac impulse propagation in relation to arrhythmias by Kleber and Rudy. 1
Figure 1.
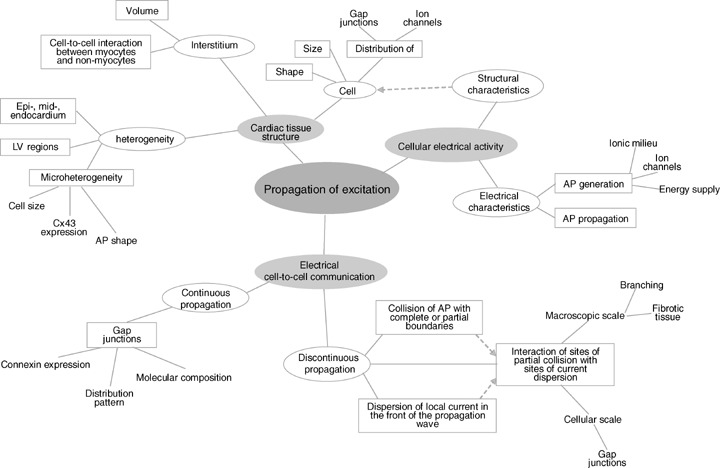
Mind map: Factors involved in the impulse propagation which have been reported to develop changes in the early stage of left ventricular hypertrophy.
Structural Remodeling
The increase in size of individual cardiomyocytes is the basic characteristics in LVH and a number of studies on humans as well as on variety of experimental animal models have been published. Table 1 illustrates a selection of findings with a focus on the early stage of LVH in SHR which could be related to the findings of decreased QRS amplitude in our animal studies.
Table 1.
Structural Characteristics of Myocytes in Left Ventricular Hypertrophy (with a Focus on the Changes of Cardiomyocytes at the Early Stage of LVH in SHR)
| Reference | LVH Model | Finding |
|---|---|---|
| Sawada et al. 20 | Human | Concentric hypertrophy: increased diameter of cardiomyocytes, the length/width ratio decreased Excentric hypertrophy: thickened and prolonged cardiomyocytes Increase number of series branches per cell Thicker and longer series and lateral branches |
| Yamamoto et al. 21 | Human | Thicker and longer series and lateral branches Increase number of series branches per cell — generation of new branches? |
| Kawamura et al. 22 | SHR 3,5,9,11,15,21,52 w | 5 week: Increase in cardiomyocyte diameter in posterior papillary muscle, differences between the size of cardiomyocytes in left ventricular lateral wall and posterior papillary muscle 11 week: Increase in diameter in left ventricular lateral wall |
| McCrossan et al. 23 | SHR 20 w | 20 week: Increased volume, smaller length/width ratio Regional differences in length, width, and volume between epicardial and endocardial regions |
| Aiello et al. 24 | SHR 16–20 w | 16–20 weeks: Increased length as compared to WKY and Wistar rats Cell width and cross‐sectional area in SHR greater as compared to Wistar rats, no difference as compared to WKY rats |
| Okabe et al. 25 | SHR 15 w | 15 week: Disproportionate hypertrophy (a greater increase in diameter than in length) Proliferation of the lateral branches |
| Cagalinec et al. 26 | SHR 12,20 w | 12, 20 week: Increase in diameter, length, volume Disproportionate hypertrophy |
| Rithalia et al. 27 | SHR 20 w | 20 week: Increased length, the regional differences in degree of hypertrophy — myocyte length greater in subendocardial than subepicardial myocytes |
Changes in diameter, length, branching and number of connected cardiomyocytes to an individual cardiomyocyte can consequently effect the impulse propagation. Applying the cable theory, the increase in the cable diameter decreases the resistance and increases conductivity. Joyner 28 show the effect of length of cells on the velocity of action potential propagation. Spach et al. 2 in their simulation study demonstrate the effect of cell size and distribution pattern of gap junctions on cell‐to‐cell propagation delay and upstroke velocity of the action potential, and show that the cell size has even a significantly larger effect than gap junction pattern. The contribution of branching to slow conduction is demonstrated by Kucera and Rudy 29 in their experimental and theoretical model. To summarize, there is evidence on significant changes in size and shape of cardiomyocytes in the early stage of LVH that could themselves affect the excitation propagation.
Electrical Characteristics of Myocardium in LVH
Resting membrane potential and action potential
With respect to active electrical properties of myocardium, the main changes in LVH are observed during the repolarization phase of action potential (AP), especially the prolongation of AP duration. The findings on the depolarization phase of AP are not that explicit. On one hand, no differences in resting membrane potential, upstroke velocity or amplitude of the action potential are reported, on the other hand however, significant changes are documented. Conduction velocity in hypertrophied myocardium is changed, as has been shown in animal experiments and in clinical studies, however there is no consistent pattern in these changes. The findings on the electrical characteristics of hypertrophied myocardium are presented in Table 2. The reasons for differences can include differences in the experimental model of LVH, species and preparation used, variations in the severity of hypertrophy, methodological aspects, etc., but there is a solid base of evidence on the changes of the electrical characteristics of hypertrophied myocardium.
Table 2.
Electrical Characteristics of Myocardium in Left Ventricular Hypertrophy. In the Case of Action Potential only Characteristics Related to Depolarization are Reported
| Reference | LVH Model | Finding |
|---|---|---|
| Hicks et al. 30 | Perinephritic hypertension in New Zealand rabbits | No differences in resting membrane potential, upstroke velocity or amplitude of the action potential as compared to control animals |
| Keung and Aronson 31 | Renal hypertension in rat | No difference in action potential |
| Gulch et al. 32 | Coarctation of renal artery in rat | No differences in transmembrane resting potential, upstroke velocity or amplitude of the action potential |
| Scamps et al. 33 | Stenosis of abdominal aorta in rat | No difference in current density |
| Winterton et al. 34 | Aortic constriction in guinea pig | No changes in AP in hypertrophied hearts 50 and 150 days after the operation, a significant conduction delay in 150‐day hypertrophied hearts |
| Brooksby 35 | SHR | No difference in resting membrane difference |
| Kleiman and Houser 36 | Right ventricular hypertrophy in cat | More depolarized resting membrane potential |
| Ryder et al. 37 | Infrarenal aortic constriction in guinea pig | More depolarized resting membrane potential |
| Nordin et al. 38 | Banded ascending aorta in guinea pigs | Lower resting potential |
| Yokoshiki et al. 39 | SHR 12 w | Greater membrane capacitance in SHR, normalized to the control level during regression of LVH produced by captopril treatment |
| Aiello et al. 24 | SHR 16–20 w | Higher membrane capacitance in SHR compared to Wistar rats |
| Cerbai et al. 40 | SHR 12, 72 w | Increase in membrane capacitance |
| Botchway et al. 41 | Aortic constriction in guinea pig | Decreased conduction velocity |
| McIntyre and Fry 42 | Human | Conduction velocity decreased progressively as cell diameter increased both in septal and papillary muscle preparations |
| Cooklin et al. 43 , 44 | Constriction of thoracic aorta in guinea pig | Declined conduction velocity in the later stage (150 days) of LVH in guinea‐pig with constriction of thoracic aorta, with a conduction velocity increase in the earlier stage of LVH (50 days post‐operation), conduction velocity increased |
| Wiegerinck et al. 45 | Pressue‐volume overload in rabbits | Longitudinal and transversal subepicardial conduction velocities higher in animals with heart failure than in controls, transmural conduction velocity unchanged |
Moreover, it has been shown that changes in passive electrical properties — the process of uncoupling — may itself induce changes in action potential characteristics (duration). Thomas et al. 46 did not find differences in action potential amplitude in cultured neonatal ventricular myocytes between heterozygote connexin43 (Cx43) knockout mice Cx43+/– and homozygote wild type mice Cx43+/+, however, they observed an increased maximal upstroke velocity of the transmembrane action potential and a shortening of action potential duration in Cx43± . They concluded that the heterozygote mutation of Cx43 produces a complex phenotype in synthetic strands that is characterized by both changes in cell‐to‐cell coupling and ion channel function, with possible involvement of the ion channels remodeling. Similarly it was shown that a transient change in resistance results in changes in action potential upstroke for poor coupling dynamic model. 47
Gap Junction Remodeling
The low‐resistance gap junctions are aggregates of intercellular channels that in heart enable the functional connections between cardiac cells and facilitate the transmission of electrical current between myocytes. According to current knowledge, they are considered to be crucial for the propagation of the cardiac impulse, although there are also some questions of their role. 48 In advanced stages of cardiac pathology, connexin expression and intracellular coupling are diminished, and gap junction channels become redistributed, these changes have been strongly implicated in the pathogenesis of lethal ventricular arrhythmias. In the case of LVH, studies on humans, experimental animal models and cultured cardiomyocytes report alterations in the distribution and expression of Cx43, the predominant isoform in the adult ventricles. As it is seen in Table 3, there is a growing evidence of significant changes in density and distribution of gap junctions, as well as to Cx43 expression and redistribution in LVH. The changes are observed already in early stages of developing hypertrophy, creating a solid basis for changes in impulse propagation. Similarly as it was already mentioned, inconsistency in the gap junction remodeling data may be related to the different experimental procedures used to induce hypertrophy, interspecies differences, methodological differences, and/or rapidity and severity of the hypertrophic response and sampling period. However, there is robust evidence on the changes of gap junction and Cx43 in LVH, including the early stages of the remodeling process.
Table 3.
Gap Junction Remodeling in Left Ventricular Hypertrophy
| Reference | LVH Model | Finding |
|---|---|---|
| Kostin et al. 49 | Human aortic stenosis | 44.3% increase in Cx43 protein, augmented number of GJs per 100 μm2 intercalated disc, increased GJ surface density in compensated hypertrophy |
| Dimished and heterogenous Cx43 distribution in decompensated hypertrophy | ||
| Peters et al. 50 | Human aortic stenosis | Cx43 gap junction expression per myocyte not significantly different from normal, reduced by 40% per unit volume of myocyte |
| Dupont et al. 51 | Human cardiomyopathy with end‐stage heart failure | Reduced level of Cx43, spatially heterogeneous |
| Emdad et al. 52 | Aortic‐band rats 4–12 months after operation | Dispersion of punctuate Cx43 labeling over the entire cell surface, significantly decreased proportion of Cx43 label at the intercalated disk, reduction of Cx43 gap junctions in the intercalated disc center Cx43‐containing gap junctions displaced from the usual locations to form side‐to‐side contacts distant from the disk, also appearing as annual profiles |
| Wang and Gerdes 53 | Aortic stenosis in guinea pigs 4, 6 months after operation | No change at the compensated hypertrophy stage, a decrease in Cx43 per LV myocyte by 37% at the congestive heart failure stage |
| Peters et al. 54 | Renovascular hypertension in guinea pigs | In early phases — a substantially increased connexin43 gap junction expression compared to controls, both when measured per cell (increased by 45%) and per unit volume of myocyte (increase by 30%) |
| Okabe et al. 25 | SHR 15 w | Diminution of the number of step‐to‐step and side‐to‐side junctions |
| Tribulova et al. 55 | SHR 13 w | No differences in the overall density of myocardial gap junctions as compared to control WKY rats, a significant decrease in phosphorylated isoform of Cx43, a higher number of lateral, side‐to‐side connections |
| Bacharova et al. 56 | SHR 20 w | Lower values of Cx43 of about 40% as compared to Wistar normotensive rats, corresponding to lower values of QRSmax and SP of about 37 and 50%, respectively |
| Itoh et al. 57 | Arteriovenous shunt in rabbits 12 weeks after operation | Significant decrease in Cx43 mRNA expression 12 weeks after the shunt operation |
| Goldfine et al. 58 | Ao regurgitation in New Zealand white rabbits 1 month, ≥2.5 year after operation | Initial tendency of Cx43 expression in AR animals to be less than that of age‐matched controls (1 month), and its increase as in the ≥ 2.5 year AR animals |
| Formigli et al. 59 | Volume overload in pig 6, 24, 48, 96, 168 hours 2, 3 months | Initial increase during the acute (compensated) hypertrophic response (6 hour after surgery) and decrease with the progression of hypertrophy (from 168 hours up to 3 months) |
| van Veen et al. 60 | Transgenic hypertrophic mouse model of forced retinoic acid signaling 4–6 month old | Heterogeneous reduction of expression in left ventricle. The remodeling accompanied by a reduction in conduction velocity |
| Chu et al. 61 | Transgenic mice over expressing a constitutively active for of calcineurin | Cx43 protein markedly down‐regulated, dephosphorylated and redistributed from the intercalated discs to the lateral borders |
| Sarkar et al. 62 | Transgenic mice with cardiac‐specific overexpression of myotrophin 4, 8 month old | A reduction in Cx43 mRNA expression at 4 weeks of age, with an even further reduction at 9 months compared to nontransgenic |
| Darrow et al. 63 | Cultured neonatal rat ventricular myocyte, exposed to cAMP | Two‐fold increase in the total content of connexin43, an increase in the number of gap junctions after exposure of neonatal rat ventricular myocyte cultures to cAMP, significant increase in conduction velocity |
| Dodge et al. 64 | Cultured neonatal rat ventricular myocyte, exposed to angiotensin II | An increase in Cx43 content and in the number of gap junction profile in cultured neonatal rat ventricular myocytes exposed to angiotensin II |
| Zhuang et al. 65 | Cultured neonatal rat ventricular myocyte, strech model | Upregulation of intercellular junction proteins, paralleled by increase in propagation velocity. No changes in upstroke velocity of the action potential or cell size |
| Uzzaman et al. 66 | RVH, monocrotaline induced pulmonary hypertension | Thirty‐five percent decrease in gap junction area per intercalated disk after 4 weeks of right ventricular hypertrophy caused by monocrotaline‐induced pulmonary hypertension in the rat, concomitant with a 30% decrease in longitudinal conduction velocity |
| Yao et al. 67 | Cx43‐deficient mice | Clusters of cells expressing normal levels of Cx43 randomly interspersed among Cx43‐deficient myocytes |
| Gutstein et al. 68 | Cx43‐deficient mice | Heterogeneous Cx43 expression resulting in conduction defects |
| Wiegerinck et al. 45 | Pressure‐volume overload in rabbit | Reduction of connexin43 content in mid‐myocardium but unchanged in subepicardium, longitudinal and transversal subepicardial conduction velocities higher in animals with heart failure |
The Effect of Cx43 Reduction
The specific effect of Cx43 reduction on conductivity is studied mainly using genetically engineered murine models of Cx43 knockout mice or of conditionally knockout mice expressing progressively decreasing levels of Cx43.
Beauchamp et al. 69 studied intercellular electrical conductance and electrical propagation in synthesized strands and pairs of ventricular myocytes from germline Cx43 knockout mice. They observed reduction in intercellular conductance by 32% in Cx43+/− cell pairs and by 96% in Cx43–/– cell pairs, the propagation was very slow and highly discontinuous in Cx43–/– strands. In right ventricular hypertrophy in the rat, Uzzaman et al. 66 find a 35% decrease in gap junction area per intercalated disk after 4 weeks, associated with a 30% decrease in longitudinal conduction velocity.
On the other hand Thomas et al. 46 did not find a detectable difference in propagation velocity in synthetic strand of cultured neonatal ventricular myocytes from Cx43+/+ wild type mice and Cx43+/− heterozygote mice in their experimental and simulation study. Their computer simulation shows relatively small dependence of propagation velocity on gap junction coupling, and they suggest the role of ion channel function and cell‐to‐cell coupling. Jongsma and Wilders 70 using a computer modeling showed that a reduction in total gap junction content by as much as 40% without changes in the size of the gap junction plaques may by itself have only moderate effect on conduction velocity. Their results point out to the role of the cytoplazmatic resistivity and cellular geometry, a conclusion advocated also by Spach et al. 2 , 3 , 71
In intact ventricles of Cx43–/– mice, Gutstein et al. 72 showed a decrease in propagation velocity to 50% in both longitudinal and transverse directions, with highly irregular epicardial conduction patterns. Ventricular conduction was reduced by 38% in Cx43+/− mice compared with wild type. 73 In conditionally knockout mice, the decrease in Cx43 to 59% of control values did not affect significantly the conduction velocity, the further decrease to 18% slowed the conduction velocity to 50% of control hearts. 74 Similarly, van Rijen et al. 75 reported that heterozygous expression of Cx43 in adult mice with inducible deletion of Cx43 do not affect ventricular conduction velocity, but up to 95% decrease of Cx43 protein reduces conduction velocity and increases dispersion of conduction.
The reduction in total Cx43 levels in itself may have only moderate effect on the conduction velocity. However, in conjunction with other hypertrophy‐induced changes of active and passive electrical properties of myocardium it can contribute to alterations in conduction of electrical impulses, e.g., a decrease in gap junction conductance by lowering pH and Ca2+ ions concentration is observed. 76 The more, in a pathological heart, Cx43 expression is heterogeneous, i.e., some regions have a virtually normal density of Cx43 expression, while others lack Cx43 almost completely. In conjunction with other disease‐related changes this gives rise to discontinuities in conduction. It should be also mentioned that measures of total connexin levels do not provide information on the quantity of functional (open) channels; hence, a reduction of Cx43 may not, per se, be detrimental. (For details see reviews. 1 , 70 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 )
With respect to QRS amplitude, articles studying the effect of decrease in Cx43 on conduction in relation to electrocardiogram are focused on arrhythmias and accordingly evaluate parameters of arrhythmia. However, some of them present also a “representative” electrocardiogram where QRS changes can be seen.
Thomas et al. 73 found no consistent changes in the voltage of the QRS complexes in the heterozygote Cx43+/− mice, but the pattern of the QRS complex, presented in their article in the figure 3, shows a pattern of intraventricular conduction disorder. On the other hand, Morley et al. 86 showed progressive diminution in the amplitude of the QRS complex, indicative of altered ventricular depolarization in genetically engineered mice with cardiac‐restricted knockout of connexin43 (OCKO), although action potential amplitude and maximum upstroke velocities were not significantly different in adult OCKO and controls. Slowing of conduction velocity was diminished by about 50% in hearts from 8‐ to 16‐week‐old OCKO mice. Similarly, Danik et al. 74 observed a gradual decrease in QRS amplitude, which closely parallel the loss of Cx43 expression. Except of decrease in their amplitude, the QRS complexes do not show any additional changes in their morphology. The loss of Cx43 is associated with conduction velocity slowing, but not with changes in QRS duration. Ventricular contractility and other invasive hemodynamic parameters in the OCKO mice did not differ as compared to controls. The animals did not exhibit pericardial effusion, anasarca, or gross ventricular dilatation — i.e., factors which are frequently discussed as possible causes of the low sensitivity of voltage criteria in LVH clinical diagnostics. Curiously, the authors found this result interesting, but it was not further discussed in their article.
Ion Channel Remodeling
Changes in AP shape and duration result from alteration in the functional expression of depolarizing and repolarizing currents. In this article, we focus on the depolarizing currents influencing the action potential generation: the fast sodium current I Na, L‐type calcium current I Ca(L) and Na+–Ca2+ exchanger I Na/Ca. The fast I Na is thought to be responsible for the rapid upstroke of the action potential in ventricular myocytes, the I Ca(L) is the primary source of Ca2+ entry, triggering release of Ca2+ from the sarcoplasmatic reticulum. The changes in Na+–Ca2+ exchange (NCX) activity influence both contractile behavior and electrical events, the alterations in NCX could also alter the action potential configuration. 87 , Table 4 reviews the findings on ion channel changes in LVH and demonstrates that the ion channel involvement in hypertrophic remodeling is present already in the early stages of LVH. The ion channel remodeling and mechanisms involved in this process are reviewed in details by Richard et al., 109 Tomaselli and Marban, 4 Hill, 110 Nattel and Li, 5 Shaw and Rudy, 111 Sipido et al. 87
Table 4.
Ion Channel Remodeling in Left Ventricular Hypertrophy
| Reference | LVH Model | Finding |
|---|---|---|
| Chorvatova et al. 88 | Aorta banding in guinea pig | Decrease in I Na density, slowing in the time course of fast inactivation of I Na |
| Ahmed et al. 89 | Aorta banding in guinea pig | Increase in I Na |
| Beuckelmann et al. 90 | Human cardiomyopathy with terminal heart failure undergoing transplantation | No difference in unstimulated I Ca(L) densities |
| Mewes and Ravens 91 | Human | No difference in I Ca(L) densities |
| Li and Jiang 92 | SHR 16–20 w | Increase in I Na and I Ca,L amplitude, the current density similar to that of Wistar rat |
| Scamps et al. 33 | Stenosis of abdominal aorta in rat | No difference in I Ca(L) current density |
| Gomez et al. 93 | Stenosis of abdominal aorta in rat | No difference in I Ca(L) current density |
| Momtaz et al. 94 | DOCA‐pellet implantation in uninephrectomized saline‐drinking rat | The I Ca(L) current density remains unaltered in either state of hypertrophy |
| Brooksby et al. 35 | SHR | No difference in the magnitude, time course or voltage dependence of I Ca(L) current |
| Cerbai et al. 40 | SHR 12, 72 w | No significant modification of I Ca (L) density |
| Ouadid et al. 95 | Human | Lower I Ca(L) density |
| Ming et al. 96 | Ascending aortic banding in guinea pig | A decrease in I Ca(L) current density |
| Santos et al. 97 | Hypertrophied tissue surviving a healed MI in rat | A decrease in I Ca(L) current density |
| Nuss and Houser 98 | Pressure overload of the right ventricle in cats | A decrease in I Ca(L) current density |
| Xiao and McArdle 99 | SHR 10 w | An increase in I Ca(L) |
| Keung 100 | Goldblatt renovascular hypertensive rats | An increase in I Ca(L) current density |
| Ryder et al. 37 | Abdominal aortic coarctation in guinea pig | An increase in I Ca(L) current density |
| Wang et al. 101 | Thoracic aortic banding in mouse | An increase in I Ca(L) current density |
| Chorvatova et al. 102 | Isoprenaline induced hypertrophy in rat | Steady state I (Na/Ca) density significantly increased |
| Meszaros et al. 103 | Isoprenaline induced hypertrophy in rat | An increase in the density of caffeine‐induced I Na/Ca |
| Ito et al. 104 | Aortic stenosis in mouse | An increase in protein levels of I Na/Ca |
| David‐Dufilho et al. 105 | SHR 3–4 w | Increased NCX activity in sarcolemmal vesicles of 3–4‐week‐old SHR, before the increase in blood pressure |
| Nakanishi et al. 106 | SHR 22 w | An increase in Na+‐dependent Ca2+ uptake |
| Wagner et al. 107 | Syrian cardiomyopathic hamsters | Na+–Ca2+ exchange in heart is increased by 400% in 30‐day old animals, while at 360 days cardiac Na+–Ca2+ exchange is decreased by 50% |
| Andrawis et al. 108 | Renovascular hypertension in rat | The 50% decrease of sarcolemmal vesicle uptake rate |
I Na= Fast sodium current; I Ca(L)= L‐type calcium current; I Na/Ca= Na/Ca exchange current; MI = myocardial infarction.
The factors involved in the structural, gap junction and ion channel remodeling are mutually interrelated, as is schematically illustrated in Figure 2. Their interplay can have significant relevance to conduction under the conditions of hypertrophied left ventricle. Kucera et al. 112 demonstrated that conduction is influenced by cleft currents and potentials that result from localization of sodium channel to clefts. Under conditions of substantially reduced gap junctional conductance, interaction between I Na and cleft potentials results in enhancement of conduction when there are narrow intercellular clefts. Shaw and Rudy 111 explore the ionic mechanisms and functional role of the fast sodium current, I Na, and the L‐type calcium current, I Ca(L) during conduction slowing using a multicellular theoretical fiber. They showed that the functional status in propagation of the action potential is changing when intercellular coupling is reduced. Thomas et al. 46 observed an increased upstroke velocity in cultured myocytes with reduced Cx43, possibly resulting from upregulation of sodium current.
Figure 2.
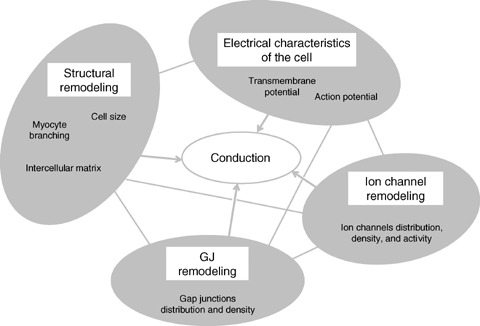
Schematic presentation of interrelations among changes in structural and electrical characteristics in hypertrophied myocardium.
These results suggest that the passive myocardial structure has a major effect on the role of membrane currents in propagation of the action potential. The nonmembrane change (i.e., decreased intercellular coupling) can cause the membrane to switch to a different process (calcium instead of sodium current) as the major mechanism for supporting conduction. It follows, that the functional role of excitation currents (i.e., I Na and I Ca(L)) can be determined to a significant extent by passive structural factors external to the membrane and not only by intrinsic membrane factors. These processes are further modified by a broad spectrum of changes observed in LVH, such as ion concentration (e.g., Na+, Ca2+), gene expression, metabolic changes, energy metabolism, and many others.
Microscopic Heterogeneity
Microscopic heterogeneity in the size of hypertrophied cardiomyocytes, Cx43 reduction, gap junction location, action potential characteristics and membrane currents might be additional factors resulting in alteration of the normal ordered pattern of the microconduction pathway. Kawamura et al. 22 described differences in cell diameter between lateral wall and papillary muscle. In SHR, it was shown that cell enlargement is not uniform across the SHR left ventricular wall. Significant changes in myocyte length, width and volume are seen in the epicardial and endocardial regions, and not in the mid‐myocardial region. 23 With respect to connexin expression, single cells and more complex higher‐order clusters of cells that express normal levels of Cx43 are randomly interspersed among Cx43‐deficient myocytes in the C‐CKO hearts, 111 creating microscopic cellular heterogeneity, even when the activation front is not deformed. Similarly, Gutstein et al. 68 reported that heterogeneous Cx43 expression results in conduction defects in chimeric mice. In a rabbit model with pressure‐volume overload, longitudinal and transversal subepicardial conduction velocities were higher in animals with heart failure and Cx43 content was reduced in mid‐myocardium but unchanged in subepicardium. 45 Regional differences in action potential characteristics and membrane currents in left ventricle were also observed. 113 , 114
CONCLUSION
The broad spectrum of growing evidence on structural and electrical remodeling and on their numerous interrelations indicates that the deviations from the original Mashima's “ideal” hypertrophy could be caused also by the changes in electrical properties of myocardium due to structural and functional hypertrophic remodeling. Myocardial remodeling in LVH is multifactorial and complex. This process involves changes in the structure and function of myocardium, including myocyte hypertrophy and apoptosis, changes in electrical and contractile phenotype, and alterations in the quantity and composition of the extracellular matrix. These complex changes are interrelated and the relative contribution of individual components to the QRS amplitude induced by LVH changes over the time.
In this review we showed that electrical properties of hypertrophied myocardium are changed already in the early stages of LVH, and that they have to be taken into account in the interpretation of the so‐called negative ECG results. Understanding of these processes will contribute to the improvement in clinical diagnostic and prognostic possibilities of electrocardiogram focusing on the unique information provided by ECG — on the information about the electrical properties of myocardium — and could bring answers to many clinical questions including the arrhythmogenic effect of drugs.
The details of mechanisms linking the changes in electrical properties of myocardium to the decrease in QRS amplitude in LVH development have to be established. The meaning of increased QRS voltage in LVH has to be also re‐evaluated, as well as the discrepancies/agreements in changes in QRS voltage and in LVM during the regression of LVH due to treatment.
Our approach to the false negative ECG results is based on the complex understanding of the changes of electrical properties of myocardium in LVH. The concept of the relative voltage deficit in LVH, and the specific potential of myocardium:
-
•
Distinguishes the anatomical and electrical information in LVH diagnostics and utilizes both.
-
•
Considers the so‐called false negative results as true results, showing the deviation from “ideal” hypertrophy caused by electrical remodeling.
-
•
Considers nonlinear changes of structural and electrical characteristics and their interplay in different stages of LVH development in terms of LVH stages described by Meerson 115 and Fizel et al. 116
This approach stresses the need to differentiate between the anatomical size and the function of the heart as a source of cardioelectric field — an approach repeatedly emphasized by electrocardiologists. 117 , 118 , 119 The unique information and the added value of ECG in LVH diagnostics is in providing the information on the electrical properties of the heart and not in the imperfect estimation of the LVM. Answering the question from the title of this review it can be concluded that there is growing evidence that the structural and electrical remodeling in LVH could create a substrate for the relative voltage deficit (i.e., the deviations from “ideal” state), which can be manifested in electrocardiogram as a decrease in QRS amplitude or as a lack of its increase, what is till now earmarked as “false negative ECG result.”
Supported by the grant VEGA 1/3406/06 from The Science Grant Agency, Slovak Republic.
REFERENCES
- 1. Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev 2004;84:431–488. [DOI] [PubMed] [Google Scholar]
- 2. Spach MS, Heidlage JF, Dolber PC, et al Electrophysiological effects of remodeling cardiac gap junctions and cell size. Experimental and model studies of normal cardiac growth. Circ Res 2000;86:302–311. [DOI] [PubMed] [Google Scholar]
- 3. Spach MS, Barr RC. Effects of cardiac microstructure on propagating electrical waveforms. Circ Res 2000;86:e23–e28. [DOI] [PubMed] [Google Scholar]
- 4. Tomaselli FG, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res 1999;42:270–283. [DOI] [PubMed] [Google Scholar]
- 5. Nattel S, Li D. Ionic remodeling in the heart. Pathophysioogical significance and new therapeutic opportunities for atrial fibrillation. Circ Res 2000;87:440–447. [DOI] [PubMed] [Google Scholar]
- 6. Akazawa H, Komuro I. Roles of cardiac transcription factors in cardiac hypertrophy. Circ Res 2003;92:1079–1088. [DOI] [PubMed] [Google Scholar]
- 7. Mashima S. Theoretical considerations on the electrocardiogram of ventricular hypertrophy. J Electrocardiology 1976;9:133–138. [DOI] [PubMed] [Google Scholar]
- 8. Szathmary V, Ruttkay‐Nedecky I, Osvald R. Computer simulation of propagated activation in different types of left ventricular enlargement. Comput Methods Programs Biomed 1994;44:85–91. [DOI] [PubMed] [Google Scholar]
- 9. Bacharova L. Effect of left ventricular hypertrophy on the cardiac electrical field: The concept of the specific potential of myocardium. Exp Clin Cardiol 1998;3:128–133. [Google Scholar]
- 10. Bacharova L, Kyselovic J. Electrocardiographic diagnosis of left ventricular hypertrophy: Is the method obsolete or should the hypothesis be reconsidered? Medical Hypotheses 2001;57:487–490. [DOI] [PubMed] [Google Scholar]
- 11. Bacharova L. Evidence‐based medicine: A lesson for electrocardiography? Arq Bras Cardiol 2003;81:102–110. [DOI] [PubMed] [Google Scholar]
- 12. Bacharova L, Melotova J, Sedlakova K. The “specific potential” as a parameter of myocardial changes in left ventricular hypertrophy In Abel H. (ed): Electrocardiology 1988. Amsterdam , Elsevier Science Publisher, Excerpta Medica, 1989, pp. 195–198. [Google Scholar]
- 13. Bacharova L, Baum OV, Muromtseva GA, et al The relation between QRS amplitude and left ventricular mass in patients with mild hypertension identified at screening . Anatolian Journal of Cardiology 2007; 7(Suppl 1): in press. [PubMed] [Google Scholar]
- 14. Bacharova L, Tibenska M, Kucerova D, et al Decrease in QRS amplitude in juvenile female competitive athletes during the initial twenty‐one months of intensive training. Cardiology Journal 2007; 14: in press. [PubMed]
- 15. Bacharova L, Bernadic M, Fizelova A. Electrocardiographic manifestation of experimental left ventricular hypertrophy In Jagielski J, Gornicki M. (eds): Electrocardiology 91. Singapore , World Scientific Publ Co, 1992, pp. 29–32. [Google Scholar]
- 16. Bacharova L, Kyselovic J, Klimas J. The initial stage of left ventricular hypertrophy in spontaneously hypertensive rats is manifested by a decrease in the QRS amplitude/left ventricular mass ratio. Clin Exp Hypertens 2004;26:557–567. [DOI] [PubMed] [Google Scholar]
- 17. Bacharova L. Michalak K, Kyselovic J, et al The relation between QRS amplitude and left ventricular mass in the initial stage of exercise‐induced left ventricular hypertrophy in rats. Clin Exp Hypertens 2005;27:533–541. [DOI] [PubMed] [Google Scholar]
- 18. Bacharova L, Kyselovic J, Klimas J, et al Changes in QRS amplitude to left ventricular mass relation in rats treated by antihypertensive drugs In Hiraoka M, Ogawa S, Kodama I, Inoue H, Kasanuki H, Katoh T. (eds): Advances in Electrocardiology 2004. Singapore , World Scientific, 2005, pp. 636–639. [Google Scholar]
- 19. Michalak K, Klimas J, Krenek P, et al ECG signs of left ventricular hypertrophy in rats exposed to traning combined with the effect of anabolic steroids. J Mol Cell Cardiol 2002;34:A87 (Abstract). [Google Scholar]
- 20. Sawada K, Kawamura K. Architecture of myocardial cells in human cardiac ventricles with concentric and eccentric hypertrophy as demonstrated by quantitative scanning electron microscopy. Heart Vessels 1991;6:129–142. [DOI] [PubMed] [Google Scholar]
- 21. Yamamoto S, James TN, Sawada K, et al Generation of new intercellular junctions between cardiocytes. A possible mechanism compensating for mechanical overload in the hypertrophied human adult myocardium. Circ Res 1996;78:362–370. [DOI] [PubMed] [Google Scholar]
- 22. Kawamura K, Kashii C, Imamura K. Ultrastructural changes in hypertrophied myocardium of spontaneously hypertensive rats. Jpn Cicr J 1976;40:1119–1145. [DOI] [PubMed] [Google Scholar]
- 23. McCrossan ZA, Billeter R, White E. Transmural changes in size, contractile and electrical properties of SHR left ventricular myocytes during compensated hypertrophy. Cardiovasc Res 2004;63:283–292. [DOI] [PubMed] [Google Scholar]
- 24. Aiello EA, Villa‐Abrille MC, Escudero EM, et al Myocardial hypertrophy of normotensive Wistar‐Kyoto rats. Am J Physiol Heart Circ Physiol 2004;286:H1229–H1235. [DOI] [PubMed] [Google Scholar]
- 25. Okabe M, Kawamura K, Terasaki F, et al Remodeling of cardiomyocytes and their branches in juvenile, adult, and senescent spontaneously hypertensive rats and Wistar Kyoto rats: Comparative morphometric analyses by scanning electron microscopy. Heart Vessels 1999;14:15–28. [DOI] [PubMed] [Google Scholar]
- 26. Cagalinec M, Kyselovic J, Blaskova E, et al Comparative study of the effects of lacidipine and enalapril on the left ventricular cardiomyocyte remodeling in spontaneously hypertensive rats. J Cardiovasc Pharmacol 2006;47:561–570. [DOI] [PubMed] [Google Scholar]
- 27. Rithalia A, Hopkins PM, Harrison SM. Effects of halothane on action potential configuration in subendocardial and sub‐epicardial myocytes from normotensive and hypertensive rat left ventricle. Br J Anaesth 2001;90:501–503. [DOI] [PubMed] [Google Scholar]
- 28. Joyner RW. Effects of the discrete pattern of electrical coupling on propagation through an electrical syncytium. Circ Res 1982;50:192–200. [DOI] [PubMed] [Google Scholar]
- 29. Kucera JP, Rudy Y. Mechanistic insight into very slow conduction in branching cardiac tissue. A model study. Circ Res 2001;89:799–806. [DOI] [PubMed] [Google Scholar]
- 30. Hicks MN, McIntosh MA, Kane KA, et al The electrophysiology of rabbit hearts with left ventricular hypertrophy under normal and ischemic conditions. Cardiovasc Res 1995;30:181–186. [PubMed] [Google Scholar]
- 31. Keung ECH, Aronson RS. Non‐uniform electrophysiological properties and electrotonic interaction in hypertrophied rat myocardium. Circ Res 1981;49:150–158. [DOI] [PubMed] [Google Scholar]
- 32. Gulch RW, Bauman NR, Jacob R. Analysis of myocardial action potential in left ventricular hypertrophy of Goldblatt rats. Basic Res Cardiol 1979;74:69–82. [DOI] [PubMed] [Google Scholar]
- 33. Scamps F, Mayoux E, Charlemagne D, et al Calcium current in single cells isolated from normal and hypertrophied rat heart. Cir Res 1990;67:199–208. [DOI] [PubMed] [Google Scholar]
- 34. Winterton SJ, Turner MA, O'Gorman DJ, et al Hypertrophy causes delayed conduction in human and guinea pog myocardium: Accentuation during ischemic perfusion. Cardiovasc Res 1994;28:47–54. [DOI] [PubMed] [Google Scholar]
- 35. Brooksby P, Levi AJ, Jones JV. The electrophysiological characteristics of hypertrophied ventricular myocytes from spontaneously hypertensive tar. J Hypertens 1993;11:611–622. [DOI] [PubMed] [Google Scholar]
- 36. Kleiman RB, Houser SR. Outward currents in normal and hypertrophied feline ventricular myocytes. Am J Physiol 1989;256:H1450‐H1461. [DOI] [PubMed] [Google Scholar]
- 37. Ryder KO, Bryant SM, Hart G. Membrane current changes in left ventricular myocytes isolated from guinea pig after abdominal aortic coarctation. Cardiovasc Res 1993;27:1278–1287. [DOI] [PubMed] [Google Scholar]
- 38. Nordin C, Siri F, Aronson RS. Electrophysiologic characteristics of single myocytes isolated from hypertrophied guinea‐pig hearts. J Mol Cell Cardiol 1989;21:729–739. [DOI] [PubMed] [Google Scholar]
- 39. Yokoshiki H, Kohya T, Tomita F, et al Restoration of action potential duration and transient outward current by regression of left ventricular hypertrophy. J Mol Cell Cardiol 1997;29:1331–1339. [DOI] [PubMed] [Google Scholar]
- 40. Cerbai E, Barbieri M, Li Q, et al Ionic basis of action potential prolongation of hypertrophied cardiac myocytes isolated from hypertensive rats of different ages. Cardiovasc Res 1994;28:1180–1187. [DOI] [PubMed] [Google Scholar]
- 41. Botchway AN, Turner MA, Sheridan DJ, et al Electrophysiological effects accompanying regression of left ventricular hypertrophy. Cardiovasc Res 2003;60:510–517. [DOI] [PubMed] [Google Scholar]
- 42. McIntyte H, Fry CH. Abnormal action potential conduction in isolated human hypertrophied left ventricular myocardium. J Cardiovasc Electrophysiol 1997;8:887–894. [DOI] [PubMed] [Google Scholar]
- 43. Cooklin M, Wallis WRJ, Sheridan DJ, et al Changes in cell‐to‐cell electrical coupling associated with left ventricular hypertrophy. Circ Res 1997;80:765–771. [DOI] [PubMed] [Google Scholar]
- 44. Cooklin M, Wallis WRJ, Sheridan DJ, et al Conduction velocity and gap junction resistance in hypertrophied, hypoxic guinea‐pig left ventricular myocardium. Exp Physiol 1998;83:763–770. [DOI] [PubMed] [Google Scholar]
- 45. Wiegerinck RF, Verkerk AO, Belterman CN, et al Larger cell size in rabbits with heart failure increases myocardial conduction velocity and QRS duration. Circulation 2006;113:806–813. [DOI] [PubMed] [Google Scholar]
- 46. Thomas SP, Kucera JP, Bircher‐Lehmann L, et al Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res 2003;92:1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henriquez AP, Vogel R, Muller‐Borer BJ, et al Influence of dynamic gap junction resistance on impulse propagation in ventricular myocardium: A computer simulation study. Biophys J 2001;81:2112–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Speralakis N. An electric field mechanism for transmission of excitation between myocardial cells. Circ Res 2002;91:985–987. [DOI] [PubMed] [Google Scholar]
- 49. Kostin S, Dammer S, Hein S, et al Connexin43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res 2004;62:426–436. [DOI] [PubMed] [Google Scholar]
- 50. Peters NS, Green CR, Poole‐Wilson PA, et al Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation 1993;88:864–875. [DOI] [PubMed] [Google Scholar]
- 51. Dupont E, Matsushita T, Kaba RA, et al Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol 2001;33:359–371. [DOI] [PubMed] [Google Scholar]
- 52. Emdad L, Uzzaman M, Takagishi Y, et al Gap junction remodeling in hypertrophied left ventricles of aortic‐banded rats: Prevention by angiotensin II type 1 receptor blockade. J Mol Cell Cardiol 2001;33:219–231. [DOI] [PubMed] [Google Scholar]
- 53. Wang X, Gerdes AM. Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: III. Intercalated disc remodeling. J Mol Cell Cardiol 1999;31:333–343. [DOI] [PubMed] [Google Scholar]
- 54. Peters NS. New insight into myocardial arrhythmogenesis: Distribution of gap‐junctional coupling in normal, ischemic and hypertrophied human hearts. Clin Sci (Lond) 1996;90:447–452. [DOI] [PubMed] [Google Scholar]
- 55. Tribulova N, Okruhlicova L, Imanaga I, et al Factors involved in the susceptibility of spontaneously hypertensive rats to low K± induced arrhythmias. Gen Physiol Biophys 2003;22:369–382. [PubMed] [Google Scholar]
- 56. Bacharova L, Plandorova J, Klimas J, et al Decreased QRS amplitude in the stage of developing left ventricular hypertrophy is associated by decreased expression of connexin43. The 33th International Congress on Electrocardiology, ICE 2006, Cologne, Germany , June 2006. Abstractband PS 16, p. 13.
- 57. Itoh M, Takeishi Y, Nakada S, et al Long‐term treatment with angiotensine II type receptor antagonist, CV‐11974, restores beta‐catenin mRNA expression in volume‐overloaded rabbit hearts. Heart Vessels 2002;17:36–41. [DOI] [PubMed] [Google Scholar]
- 58. Goldfine SM, Walcott B, Brink PR, et al Myocardial connexin43 expression in left ventricular hypertrophy resulting from aortic regurgitation. Cardiovasc Pathol 1999;8:1–6. [DOI] [PubMed] [Google Scholar]
- 59. Formigli L, Ibba‐Manneschi L, Perna AM, et al Altered Cx43 expression during myocardial adaptation to acute and chronic volume overload. Histol Histopathol 2003;18:359–369. [DOI] [PubMed] [Google Scholar]
- 60. Van Veen TAB, Van Rijen HVM, Wiegerinck RF, et al Remodeling of gap junctions in mouse hearts hypertrophied by forced retinoic acid signaling. J Mol Cell Cardiol 2002;34:1411–1423. [DOI] [PubMed] [Google Scholar]
- 61. Chu G, Carr AN, Young KB, et al Enhanced myocyte contractility and Ca2+ handling in a calcineurin transgenic model of heart failure. Cardiovasc Res 2002;54:105–116. [DOI] [PubMed] [Google Scholar]
- 62. Sarkar S, Leaman DW, Gupta S, et al Cardiac overexpression of myotrophin triggers myocardial hypertrophy and heart failure in transgenic mice. J Biol Chem 2004;279:20422–20434. [DOI] [PubMed] [Google Scholar]
- 63. Darow BJ, Fast VG, Kleber AG, et al Functional and structural assessment of intrecellular communication: Increased conduction velocity and enhanced connexin expression in dibutyryl cAMP‐treated cultured cardiac myocytes. Circ Res 1996;79:174–183. [DOI] [PubMed] [Google Scholar]
- 64. Dodge SM, Beardslee MA, Darrow BJ, et al Effect of angiotensin II on expression of the gap junction channel protein connexin43 in neonatal rat ventricular myocytes. J Am Coll Cardiol 1998;32:800–807. [DOI] [PubMed] [Google Scholar]
- 65. Zhuang J, Yamada KA, Saffitz JE, et al Pulsatile stretch remodels cell‐to‐cell communication in cultured myocytes. Circ Res 2000;87:316–322. [DOI] [PubMed] [Google Scholar]
- 66. Uzzaman M, Honjo H, Takagishi Y, et al Remodeling of gap‐junctional coupling in hypertrophied right ventricles of rats with monocroatline‐induced pulmonary hypertension. Circ Res 2000;86:871–878. [DOI] [PubMed] [Google Scholar]
- 67. Yao J‐A, Gutstein DE, Liu F, et al Cell coupling between ventricular myocyte pairs from connexin43‐deficient murine hearts. Circ Res 2003;93:736–743. [DOI] [PubMed] [Google Scholar]
- 68. Gutstein DE, Morley GE, Vaidaya D, et al Heterogeneous expression of gap junction channels in the heart lead to conduction defects and ventricular dysfunction. Circulation 2001;104:1194–1199. [DOI] [PubMed] [Google Scholar]
- 69. Beauchamp P, Choby C, Desplantez T, et al Electrical propagation in synthetic ventricular myocyte strands from germline connexin43 knockout mice. Circ Res 2004;95:170–178. [DOI] [PubMed] [Google Scholar]
- 70. Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease. Circ Res 2000;86:1193–1197. [DOI] [PubMed] [Google Scholar]
- 71. Spach MS. Anisotropy of cardiac tissue: A major determinant of conduction? J Cardiovasc Electrophysiol 1999;10:887–890. [DOI] [PubMed] [Google Scholar]
- 72. Gutstein DE, Morley GE, Tammadon H, et al Conduction slowing and sudden arrhythmic death in mice with cardiac‐restricted inactivation of connexin43. Circ Res 2001;88:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thomas SA, Schuessler RB, Berul CL, et al Disparate effect of deficient expression if connexin43 on atrial and ventricular conduction. Evidence for chamber‐specific molecular determinants of conduction. Circulation 1998;97:686–691. [DOI] [PubMed] [Google Scholar]
- 74. Danik SB, Liu F, Zhang J, et al Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res 2004;95:1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Van Rijen HVM, Eckardt D, Degen J, et al Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 2004;109:1048–1055. [DOI] [PubMed] [Google Scholar]
- 76. Firek L, Weingart R. Modification of gap junction conductance by divalent cations and protons in neonatal rat heart cells. J Mol Cell Cardiol 1995;27:1633–1643. [DOI] [PubMed] [Google Scholar]
- 77. Saffitz JE. Regulation of intracellular coupling in acute and chronic heart disease. Braz J Med Biol Res 2000;33:407–413. [DOI] [PubMed] [Google Scholar]
- 78. Harris AL. Emerging issues of connexdin channels: Biophysics fills the gap. Q Rev Biophysw 2001;35:325–472. [DOI] [PubMed] [Google Scholar]
- 79. Jalife J, Morley GE, Vaidya D. Connexins and impulse propagation in the mouse heart. J Cardiovasc Electrophysiol 1999;10:1649–1663. [DOI] [PubMed] [Google Scholar]
- 80. Lo CW. Role of gap junctions in cardiac conduction and development. Insight from the connexin knockout mice. Circ Res 2000;87:346–348. [DOI] [PubMed] [Google Scholar]
- 81. Peters NS, Wit AL. Myocardial architecture and ventricular arrhythomogenesis. Circulation 1998;97:1746–1754. [DOI] [PubMed] [Google Scholar]
- 82. Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res 2004;62:309–322. [DOI] [PubMed] [Google Scholar]
- 83. Severs NJ. Gap junction remodeling and cardiac arrhythmogenesis: Cause or coincidence? J Cell Mol Med 2001;5:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Teunissen BEJ, Jongsma HJ, Bierhuizen MFA. Regulation of myocardial connexins dusring hypertophic remodeling. Eur Heart J 2004;25:1979–1989. [DOI] [PubMed] [Google Scholar]
- 85. Kanno S, Saffitz JE. The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc Pathol 2001;10:169–177. [DOI] [PubMed] [Google Scholar]
- 86. Morley GE, Danik S, Bernstein, et al Reduced intercellular coupling leads to paradoxical propagation across the Purkinje‐ventricular junction and aberrant myocardial activation. PNAS 2005;102:4126–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sipido KR, Volders PGA, Vos MA, et al Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: A new target for therapy? Cardiovasc Res 2002;53:782–805. [DOI] [PubMed] [Google Scholar]
- 88. Chorvatova A, Snowdon R, Hart G, et al Effects of pressure overload‐induced hypertrophy on TTX‐sensitive inward currents in guinea pig left ventricle. Mol Cell Biochem 2004;261:217–226. [DOI] [PubMed] [Google Scholar]
- 89. Ahmmed GU, Dong PH, Song G, et al Changes in Ca2+ cycling proteins underlie cardiac action potential prolongation in apressure‐overload guinea pig model with cardiac hypertrophy and failure. Circ Res 2000;86:558–570. [DOI] [PubMed] [Google Scholar]
- 90. Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 1992;85:1046–1055. [DOI] [PubMed] [Google Scholar]
- 91. Mewes T, Ravens U. L‐type calcium currents of human myocytes from ventricle of non‐failing and failing hearts and from atrium. J Mol Cell Cardiol 1994;26:1307–1320. [DOI] [PubMed] [Google Scholar]
- 92. Li X, Jiang W. Electrical remodelling of membrane ionic channels of hypertrophied ventricular myocytes from spontaneously hypertensive rats. Chin Med J 2000;113:584–587. [PubMed] [Google Scholar]
- 93. Gomez AM, Benitah JP, Henzel D, et al Modulation of electrical heterogeneity by compensated hypertrophy in rat left ventricle. Am J Physiol 1997;272:H1078–1086. [DOI] [PubMed] [Google Scholar]
- 94. Momtaz A, Coulombe A, Richter P, et al Action potential and plateau ionic currents in moderately and severely DOCA‐salt hypertrophied rat hearts. J Mol Cell Cardiol 1996;28:2511–2522. [DOI] [PubMed] [Google Scholar]
- 95. Ouadid H, Albat B, Nargeot J. Calcium currents in diseased human cardiac cells. J Cardiovasc Pharmacol 1995;25:282–291. [DOI] [PubMed] [Google Scholar]
- 96. Ming Z, Nordin C, Siri F, et al Reduced calcium current density in single myocytes isolated from hypertrophied failing guinea pig hearts. J Mol Cell Cardiol 1994;26:1133–1143. [DOI] [PubMed] [Google Scholar]
- 97. Santos PE, Barcellos LC, Mill JG, et al Ventricular action potential and L‐type calcium channel in infarct‐induced hypertrophy in rats. J Cardiovasc Electrophysiol 1995;6:1004–1014. [DOI] [PubMed] [Google Scholar]
- 98. Nuss HB, Houser SR. Voltage dependence of contraction and calcium current in severely hypertrophied feline ventricular myocytes. J Mol Cell Cardiol 1991;23:717–726. [DOI] [PubMed] [Google Scholar]
- 99. Xiao YF, McArdle JJ. Elevated density and altered pharmacologic properties of myocardial calcium current of the spontaneously hypertensive rat. J Hypertens 1994;12:783–790. [PubMed] [Google Scholar]
- 100. Keung EC. Calcium current is increased in isolated adult myocytes from hypertrophied rat myocardium. Circ Res 1989;64:753–763. [DOI] [PubMed] [Google Scholar]
- 101. Wang Z, Kutschke W, Richardson KE, et al Electrical remodeling in pressure‐overload cardiac hypertrophy. Role of calcineurin. Circulation 2001;104:1657–1663. [DOI] [PubMed] [Google Scholar]
- 102. Chorvatova A, Hart G, Hussain M. Na+/Ca2+ exchange current (I(Na/Ca)) and sarcoplasmatic reticulum Ca2+ release in catecholamine‐induced cardiac hypertrophy. Cardivasc Res 2004;61:278–287. [DOI] [PubMed] [Google Scholar]
- 103. Meszaros J, Khananshvili D, Hart G. Mechanisms underlying delayed afterdepolarizations in hypertrophied left ventricular myocytes of rats. Am J Physiol Heart Circ Physiol 2001;281:H903–H914. [DOI] [PubMed] [Google Scholar]
- 104. Ito K, Yan X, Tajima M, et al Contractile reserve and intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing hearts. Circ Res 2000;87:588–595. [DOI] [PubMed] [Google Scholar]
- 105. David‐Dufilho M, Pernollet MG, LeQuan Sang H, et al Active Na+ and Ca+ transport, Na+‐Ca2+ exchange, and intracellular Na+ and Ca2+ content in young spontaneously hypertensive rats. J Cardiovasc Pharmacol 1986;8(Suppl.8):S130–S135. [DOI] [PubMed] [Google Scholar]
- 106. Nakanishi H, Makino N, Matsui H, et al Sarcolemmal Ca2+ transposrt activities in cardiac hypertrophy caused by pressure overload. Am J Physiol 1989;257:H349–H356. [DOI] [PubMed] [Google Scholar]
- 107. Wagner JA, Weisman HF, Snowman AM, et al Alterations in calcium antagonists receptors and sodium‐calcium exchange in cardiomyopathic hamster tissues. Curc Res 1989;65:205–214. [DOI] [PubMed] [Google Scholar]
- 108. Andrawis NS, Kuo TH, Giacomelli F, et al Altered calcium regulation in the cardiac plasma membrane in experimental renal hypertension. J Mol Cell Cardiol 1988;20:625–634. [DOI] [PubMed] [Google Scholar]
- 109. Richard S, Leclercq F, Lemaire S, et al Ca2+ currents in compensated hypertrophy and heart failure. Cardiovasc Res 1998;37:300–311. [DOI] [PubMed] [Google Scholar]
- 110. Hill JA. Electrical remodeling in cardiac hypertrophy. Trends Cardiovasc Med 2003;13:316–322. [DOI] [PubMed] [Google Scholar]
- 111. Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of sodium and L‐type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res 1997;81:727–741. [DOI] [PubMed] [Google Scholar]
- 112. Kucera JP, Rohr S, Rudy Y. Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ Res 2002;91:1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Main MC, Bryant SM, Hart G. Regional differences in action potential characteristics and membrane currents of guinea‐pig left ventricular myocytes. Exp Physiol 1998;83:747–761. [DOI] [PubMed] [Google Scholar]
- 114. Belardinelli L, Antzelevitch C. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J Cardiovasc Electrophysiol 2006;17(Suppl 1):S79–S85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Meerson FZ. The myocardium in hyperfunction, hypertrophy and heart failure. Circ Res 1969;25(Suppl. II):II‐1–II‐163. [PubMed] [Google Scholar]
- 116. Fizel A, Fizelova A, Turcany. The relations between ultrastructural and metabolic changes of the myocardium of the hypertrophied and failing heart [Vztah medzi ultrastrukturalnymi a metabolickymi zmenami myokardu hypertrofovaneho a zlyhavajuceho srdca]. Folia Fac Med Univ Comeniane Bratisl 1984;22:9–119 (in Slovak, abstract in English). [Google Scholar]
- 117. Stern S. Electrocardiogram: Still the cardiologist's best friend. Circulation 2006;113:753–756. [DOI] [PubMed] [Google Scholar]
- 118. Stern S. The year 2005 in electrocardiology. ANE 2006;11:187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Mirvis DM. What's wrong with electrocardiography? J Electrocardiol 1998;31:313–316. [DOI] [PubMed] [Google Scholar]