

Abstract
The implantable cardioverter defibrillator (ICD) is highly effective in reducing mortality due to cardiac arrhythmias in high‐risk cardiac patients. However, inappropriate therapies caused predominantly by supraventricular tachyarrhythmias (SVTs) remain a significant side effect of ICD therapy despite medical treatment, affecting 8–40% of patients. The MADIT‐RIT is a global, prospective, randomized, nonblinded, three‐arm, multicenter clinical investigation to be performed in the Unites States, Europe, Canada, Israel and Japan, and will utilize approximately 90 centers with plan to enroll 1500 patients programmed to three treatment arms. The objective of the MADIT‐RIT trial is to determine if dual‐chamber ICD or CRT‐D devices with high rate cutoff (MADIT‐RIT‐Arm B) and/or long delay in combination with detection enhancements (MADIT‐RIT‐Arm C) are associated with fewer patients experiencing inappropriate therapies than standard programming (MADIT‐RIT‐Arm A) during postimplant follow‐up of patients with indication for primary prevention device therapy. This paper describes design and analytic plan for the MADIT‐RIT trial.
Keywords: implantable defibrillators, defibrillation therapy, sudden cardiac death
The implantable cardioverter defibrillator (ICD) is highly effective in reducing mortality due to cardiac arrhythmias in high‐risk cardiac patients. 1 , 2 , 3 However, inappropriate therapies caused predominantly by supraventricular tachyarrhythmias (SVTs) remain a significant side effect of ICD therapy despite medical treatment, affecting 8–40% of patients. Inappropriate shocks can lead to pain, anxiety, depression, impaired quality of life, proarrhythmia, and poor tolerance of life‐saving ICD therapy. 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 In primary prevention cohorts, approximately 20% of ICD shock therapy is inappropriate, and in addition, inappropriate and appropriate ATP therapy can trigger ventricular tachycardia and ventricular fibrillation. 5 Moreover, recently it has been suggested that in primary prevention ICD recipients there may be an association between inappropriate shocks and increased mortality independent of interim appropriate shocks. 4 , 5 , 15
The most desirable method of limiting inappropriate therapies is accurate rhythm detection and intervention by the ICD. Intuitively, dual‐chamber detection enhancements that utilize atrial and ventricular electrograms to formulate a diagnosis were thought to be superior to single‐chamber, ventricular‐only detection enhancements. Early nonrandomized and small randomized studies failed to show clear superiority of dual‐chamber over single‐chamber detection. 14 , 15 , 16 , 17 , 18 , 19 A more recent larger randomized study suggested that dual‐chamber devices may in fact be superior at reducing inappropriate therapy for SVTs, although there was not a significant reduction in inappropriate shocks between groups. 20 Dual‐chamber ICDs provide information about the atrial rhythm during tachycardia, making it easier to determine whether the tachyarhythmia originated in the atrium or the ventricle. This is important as paroxysmal AF is the leading cause of inappropriate shocks. 5 , 21 , 23 Perhaps due to this advantage clinicians tend to implant dual chamber ICD's more frequently. Analysis of the National Cardiovascular Data Registry (NCDR) ICD Registry indicated that in a “real world” population, dual chamber ICD's are implanted twice as frequently as single chamber ICD's, despite that the addition of an atrial lead has been associated with an increased number of complications, 22 and despite the absence of a clear benefit.
Part of the inappropriate therapy problem relates to the fact that programming of primary prevention devices is not done significantly differently than secondary prevention devices. Presently, the best methodology to program a dual chamber ICD or CRT‐D device to reduce the amount of inappropriate therapy is unknown. Attempts to find optimum programming such as the PREPARE trial, 7 have demonstrated inappropriate therapy reductions when compared to historical controls, but lack the clarity of a randomized prospective trial of shock reduction strategies.
We hypothesize that increasing the rate detection to 200 bpm or significantly increasing the delay before initiating therapy in a population of primary prevention ICD and cardiac resynchronization therapy‐defibrillator (CRT‐D) recipients will significantly decrease the number of inappropriate ICD therapies without affecting morbidity or mortality when compared to standard programming, such as the one utilized in the MADIT II study. This trial, Multicenter Automatic Defibrillator Implantation Trial‐Reduce Inappropriate Therapy (MADIT‐RIT), will be the first large randomized study statistically designed to evaluate specific programming features in the implanted devices to reduce inappropriate therapy in primary prevention patients.
OBJECTIVES
The purpose of the MADIT‐RIT trial is to compare the occurrence of inappropriate therapy using high rate cutoff and/or very long delays in primary prevention patients receiving an ICD or CRT‐D device against standard programming. Inappropriate therapy is defined as the highest energy therapy delivered (ATP or shock) for nonventricular tachyarrhythmias or for inappropriate sensing. The objective of the MADIT‐RIT trial is to determine if ICD or CRT‐D devices with high rate cutoff (MADIT‐RIT‐Arm B) and/or long delay in combination with detection enhancements (MADIT‐RIT‐Arm C) are associated with fewer patients experiencing inappropriate therapies than standard programming (MADIT‐RIT‐Arm A) during postimplant follow‐up of patients with indication for primary prevention device therapy.
DESIGN
MADIT‐RIT is a global, prospective, randomized, nonblinded, three‐arm, multi‐center clinical investigation to be performed in the United States, Europe, Canada, Israel and Japan, and will utilize approximately 90 centers, depending on enrollment rate. An average of 120–130 patients per month is expected to result in 1500 patients programmed to treatment arm in approximately 12 months. The ongoing recruitment rate will be closely monitored.
The trial is expected to end in approximately 36 months with six months for full start‐up, 10–14 months for recruitment, 9–12 months to complete follow‐up of the last patient programmed to treatment arm, and six months for data analysis. Slower recruitment will prolong study duration.
Patients with an approved primary prevention indication for ICD or CRT‐D device will be randomized and programmed within one calendar month of implant to one of three treatment arms in a 1:1:1 ratio (Fig. 1). Optimal pharmacological therapy as per established guidelines for the underlying cardiac condition will be required for patients in all three treatment arms. Patients without successful device implantation will be withdrawn from the study.
Figure 1.
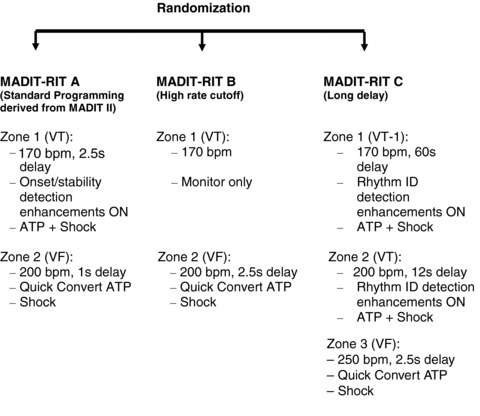
Three arms of MADIT‐RIT.
ELIGIBILITY
The MADIT‐RIT trial will utilize the current approved guidelines for indications and contraindications of ICD and CRT‐D devices for primary prevention therapy. Only Boston Scientific (St. Paul, MN, USA) ICD (TELIGEN) and CRT‐D (COGNIS) devices and future generations of Boston Scientific ICD and CRT‐D devices that are commercially available in the United States for U.S. sites, Europe for European Sites, Canada for Canadian sites, and Israel for Israeli sites, will be used in this trial.
Patients who meet all of the following criteria at enrollment may be given consideration for inclusion in this clinical investigation:
-
1
Primary prevention patient with ischemic or nonischemic heart disease who meets current. guidelines for dual‐chamber ICD or CRT‐D device therapy.
-
2
Patient in sinus rhythm at the time of the implant.
-
3
Patient on stable optimal pharmacologic therapy for their cardiac condition.
-
4
Patient ≥21 years of age, or legal representative, willing and capable of giving informed consent.
The ICD is indicated for primary prevention patients with:
-
1
Prior MI and LVEF ≤ 30%, or
-
2
Ischemic or nonischemic dilated cardiomyopathy, LVEF ≤ 35%, and NYHA class II/III, or
-
3
Prior MI and LVEF ≤ 35% and documented, nonsustained VT and inducible VT at EP testing.
The CRT‐D is indicated for patients with:
-
1
NYHA class II and III, and
-
2
LVEF ≤ 35%, and
-
3
QRS width ≥ 120 ms, and
-
4
Remains symptomatic despite stable, optimal heart failure drug therapy.
Should the indications for ICD/CRT‐D implantation change during the MADIT‐RIT trial, patients who meet the new criteria will be eligible to participate. Please note that for the purposes of the MADIT‐RIT trial, CRT‐D patients with an NYHA Class IV indication will be excluded.
EXCLUSIONS
Patients who meet any one of the following criteria at enrollment will be excluded from this clinical investigation:
-
•
Patient with an implanted pacemaker or CRT‐P
-
•
Patient with an existing ICD or CRT‐D device components
-
•
Patient with a history of VT or VF
-
•
Patient with permanent or chronic AF within the past three calendar months before enrollment
-
•
Patient with coronary artery bypass graft surgery or percutaneous coronary intervention within the past three calendar months prior to enrollment
-
•
Patient with enzyme‐positive myocardial infarction within the past three calendar months prior to enrollment
-
•
Patient with angiographic evidence of coronary disease who are candidates for coronary revascularization and are likely to undergo coronary artery bypass graft surgery or percutaneous coronary intervention in the foreseeable future
-
•
Patient with second or third degree heart block
-
•
Patient in NYHA Class IV
-
•
Patient who is pregnant or plans to become pregnant during the course of the trial
-
•
Patient with irreversible brain damage from preexisting cerebral disease
-
•
Patient with presence of any disease, other than the patient's cardiac disease, associated with a reduced likelihood of survival for the duration of the trial, e.g., cancer, uremia, liver failure, etc.
-
•
Patient with chronic renal disease with BUN ≥ 50 mg/dL or creatinine ≥2.5 mg/dL
-
•
Patient participating in any other clinical trial
-
•
Patient unwilling or unable to cooperate with the protocol
-
•
Patient who lives at such a distance from the clinic that travel for follow‐up visits would be unusually difficult
-
•
Patient who does not anticipate being a resident of the area for the scheduled duration of the trial
-
•
Patient unwilling to sign the consent for participation
-
•
Patients whose physician does not allow participation
CONSENT
After a patient has been determined to meet eligibility criteria and not to have any exclusions from the MADIT‐RIT study the Enrolling Center Investigation team will present in an oral and written fashion the nature and requirements of the study and follow‐up, including the risks of any procedures and participation. All materials will be approved by the local IRB and the Sponsor and Data Coordinating Center. After addressing all questions and concerns and an appropriate time period to consider the option of participating the patient can sign the consent form. The patient will receive a copy of the signed consent form and will also be maintained in the center's records.
BASELINE EVALUATION
Before device implantation and randomization the enrolled subjects will have a history and physical, medication list, 12‐lead ECG, and quality of life form completed.
DEVICE IMPLANT
After informed consent has been obtained the patient will undergo ICD or CRT‐D implantation within 14 days. Patients meeting appropriate indications for an ICD or for a CRT‐D device will be implanted with either a dual chamber, commercially available TELIGEN (ICD) or COGNIS (CRT‐D) (or future generation of Boston Scientific ICD and CRT‐D device). A dual‐chamber ICD will be used to allow similar detection programming to the CRT‐D devices and to optimize the ability to determine the type of arrhythmia triggering device delivered therapy. Patients should meet all FDA and other applicable criteria for the dual chamber ICD or CRT‐D implant. Routine clinical methods for device implantation and testing (including defibrillation threshold testing as per the device FDA approval labeling) will be followed.
RANDOMIZATION
Randomization will be performed after successful ICD or CRT‐D implantation, ideally within 24‐hours after implant and before hospital discharge but can be performed up to ≤1 calendar month after implant. Prior to randomization the patient may be programmed according to physician preference. The Enrolling Center team will enter the type of device (ICD or CRT‐D) and presence or absence of a prior history of self‐terminating paroxysmal atrial fibrillation; randomization will be stratified by each of these three criteria. Neither the patient nor the study personnel will be blinded to the programming assignment (Fig. 2).
Figure 2.
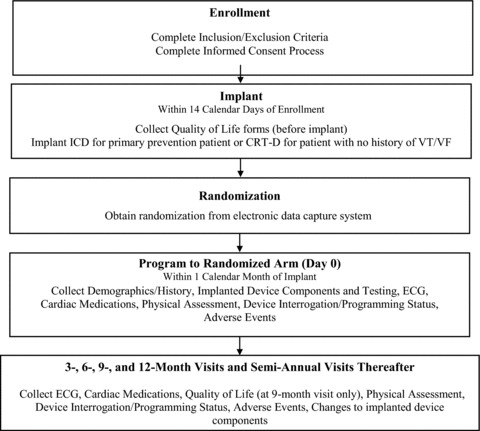
Study time line and patient flow.
DEVICE PROGRAMMING
Bradycardia Pacing
Dual chamber ICD devices will be programmed using the AV Search + Algorithm for optimization of the pacing AV delay and AV Search Hysteresis ON to prevent unnecessary RV pacing.
CRT‐D device programming should include the SMARTDelay algorithm for optimization of AV delay and VRR and BiV Trigger ON to maximize biventricular pacing.
VT/VF Detection and Therapy Programming
Ventricular tachycardia and fibrillation detection and therapy programming will be according to randomization arm, and will be equivalent for ICD and for CRT‐D devices. Arm A, the conventional detection arm, will use two‐zone detection at 170 bpm for VT and 200 bpm for VF (Fig. 1). MADIT‐RIT Arm B will use a monitor‐only zone between 170 bpm and 200 bpm and a VF detection zone at 200 bpm (Fig. 1). MADIT‐RIT Arm C patients will be programmed to a three‐zone detection at 170 bpm for VT and 200 bpm for fast VT and 250 bpm for VF. In addition they will have a long delay of 60 seconds in the 170 to 199 bpm range (Fig. 1).
OPTIMAL PHARMACOLOGIC THERAPY
Patient eligibility for MADIT‐RIT consists of primary prevention patients with ischemic or nonischemic heart failure and low ejection fraction. The patients should be treated with stable, optimal pharmacologic therapy including beta‐blockers, angiotensin converting enzyme inhibitors (or angiotensin receptor blocking agents), diuretic agents and aldosterone antagonists, according to appropriate guidelines and continue to have low ejection fraction despite appropriate therapy.
FOLLOW‐UP
After randomization and programming to appropriate randomized setting (within 1 month after implantation), patients will be followed in the enrolling centers at 3‐month intervals for the first year and then at 6‐month intervals thereafter. At each follow‐up visit a list of medications, an interim history focusing on occurrence of adverse events, a physical exam, ECG and device interrogation will be recorded. The device interrogation will be saved to disc in duplicate with a copy kept at the enrolling center and one sent to the Electrogram Core Lab. Device shocks or other events may prompt an interim visit during which the same data as for a scheduled follow‐up will be collected including the device interrogation disc downloads.
END POINTS
The primary end point in MADIT‐RIT is occurrence of a first inappropriate therapy (either ATP or shock). Inappropriate therapy is defined as therapy for a nonventricular arrhythmia, i.e., a supraventricular arrhythmia or an abnormal sensing episode. If both ATP and a shock occur during an episode, as adjudicated by the electrogram core lab, the episode is classified as an inappropriate shock based on the highest energy delivered during the episode. The enrolling center PI, investigators, and research coordinators will ascertain the occurrence of device therapy and classify it as appropriate or inappropriate at scheduled follow‐up or after a patient‐reported event results in an interim outpatient visit and device interrogation. The final analysis of the electrogram core lab shall be the official determination of whether an episode is appropriate or inappropriate (See Core Laboratories). The secondary end point is all‐cause mortality, although the trial is not powered to detect a difference in mortality between arms. Mortality events will be reported to the sponsor (Boston Scientific, Inc., St Paul, MN) within the required timeframe. Tertiary end points include comparison between Arms B and C, for which the trial is not powered for comparison. Occurrence of appropriate therapies and shocks, recurrent inappropriate therapy, quality of life, stroke, syncope, untreated VT are among other tertiary end points (Table 1).
Table 1.
End points of the MADIT‐RIT Trial
| Primary end point | |
| First inappropriate therapy | |
| Secondary end point | |
| All‐cause mortality | |
| Tertiary end point | |
| First inappropriate therapy episodes in MADIT‐RIT‐B vs MADIT‐RIT‐C | |
| Quality of life in Arms A‐C | |
| Florida patient acceptance and shock anxiety surveys Arms A‐C | |
| Recurrent inappropriate therapy in Arms A‐C | |
| Occurrence of events dependent upon history of paroxysmal AF | |
| Inappropriate therapy event rates in ischemic vs nonischemic heart in Arms A‐C | |
| Total shocks delivered in each of the three arms of the trial | |
| Total energy delivered in each of the three arms of the trial | |
| Frequency of syncope in each of the three arms of the trial | |
| Stroke rate in each of the three arms of the trial | |
| Relative health care costs in each of the three arms of the trial | |
| Frequency of arrhythmic hospitalizations in each of the three arms of the trial | |
| Frequency of heart failure hospitalizations in each of the three arms of the trial | |
| Differences in inappropriate and appropriate therapy in ICD vs CRT‐D device | |
| Frequency of untreated sustained VT in each of the three arms of the trial |
CORE LABORATORIES AND COMMITTEES
Electrogram and Device Interrogation Core Laboratory
This lab consists of a director and co‐director and their appointed staff. The core lab will review all device interrogations using electronic media downloaded from device interrogations at the enrolling centers. The enrolling center's interpretation and classification will be available to the core lab but they will form a final opinion of the nature of the arrhythmia resulting in device firing (ATP or shock). The core lab will review all interrogation to capture and adjudicate any events not reported by the enrolling center. The events will be classified by definitions similar to those used for MADIT II and as per the protocol. For example, the definition of VT or VF is based on the electrogram morphology and rate and not device classification of arrhthmia as VT or VF (Fig. 3).
Figure 3.
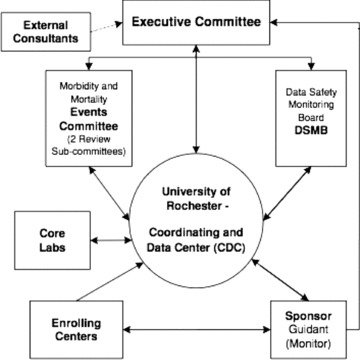
MADIT‐RIT organizational structure.
Mortality Review Committee
The three‐member Mortality Committee will review the enrolling center's classification and form as well as all available medical record source documents using a modified Hinkle‐Thaler definition to arrive at a consensus interpretation of the cause of death as cardiac (sudden or nonsudden) or noncardiac. 24 , 25 , 26 , 27 , 28
DATA MONITORING AND QUALITY CONTROL
The Coordination and Data Center (CDC) will monitor the study data that will be entered in a common relational database. A Web‐enabled portal provides access to the system. The system is configured to have data entered electronically at the investigational sites by authorized personnel, with at‐source real‐time data editing and correction. The underlying foundation of the system provides the capability to define data editing rules for ensuring the quality and consistency of the clinical data, a query management process to track data discrepancies and their resolutions, and source monitoring and verification of the clinical data. The web‐enabled system provides audit trail, security mechanisms, and electronic signature capabilities that meet FDA 21 CFR Part 11 requirements regarding electronic records and electronic signatures. This database permits data expansion, easy updating, and rapid retrieval; it has simplified report‐generating routines and audit trail component.
All submitted data will be subject to an extensive computer edit‐checking process for completeness, internal consistency, identification of numerical values outside specified rational limits, invalid codes, subject identification errors, and date errors. The system enables the CDC to monitor data status with feedback provided to the enrolling centers if data is due, overdue, backlogged or erred.
Boston Scientific personnel will conduct regular monitoring visits throughout the study. Visits will be made to all enrolling centers to ensure adherence to the protocol and operational procedures as well as maintenance of the highest‐quality data with data checked per source documentation at each enrolling center.
The device interrogation data, syncope and mortality data, and adverse event reports will be continuously monitored by the Data Safety and Monitoring Board. All data will be managed by the CDC throughout the trial and the Boston Scientific study leadership team will be blinded to the study treatment arm assignment throughout the trial and not reported via the progress reports to the IRBs and enrolling centers. The syncope and mortality review committee will be blinded to the randomized treatment arm.
STATISTICAL DESIGN AND ANALYSIS
This study will be carried out as if two trials conducted simultaneously, one evaluating treatment B (high rate cutoff) relative to A (conventional) and the other evaluating treatment C (long delay) relative to A. The primary end point (after programming) is the time to first episode of inappropriate therapy (highest‐energy therapy delivered, classified as ATP or shock). The null hypothesis in each trial (B vs A and C vs A—hereafter T vs A with T being either B or C) is that of no difference in risk of first inappriopriate therapy between arms T and A. The alternative of interest, in each trial, is that arm T has reduced risk relative to A. Nevertheless, we allow for a two‐sided alternative in each trial, with a 5% signficance level in each. The two trials are in parallel, with inference to be made in each, and hence no adjustment for multiplicity is deemed appropriate. Specifically, for T vs A, we hypothesize that, after programming of a dual‐chamber ICD or CRT‐D according to T or A, the ratio of ongoing risks of an inappropriate therapy episode, in patients alive and without prior inappropriate therapy, will be less than unity.
We focus on power to detect a value 0.5 for the ratio of risks, but consider values in the range 0.67–0.33 as well. The hypotheses are common to the two trials. These hypotheses are only relevant if mortality is unaffected by B or C relative to A, which will be assumed for the primary analysis, as there is insufficient power to detect significant differences.
The primary analysis will be based on a proportional‐hazards analysis for risk of a first inappropriate therapy episode, recognizing mortality as a competing (and possibly related) risk 24 ; as a consequence, follow‐up for inappropriate therapy will be censored upon death. The analysis will be stratified by enrolling center, by history of paroxysmal AF at baseline and by device type (ICD or CRT‐D). To achieve 90% power at a hazard ratio of 0.5, representing a 50% reduction in risk, a total of 88 events are required in two arms being compared, both in B and A together and in C and A together. The hazard ratios considered here are assumed to include any effects of crossovers.
The total sample size of 1500 patients (with successful implantations), with approximately 500 in each arm, is expected to generate sufficient numbers of end point events, in arms B + A and in C + A, to provide 90% power to detect a 50% reduction in risk, in each trial. This estimate is based on recruitment requiring 12 months, with 9 months additional follow‐up, resulting in 15 months average follow‐up per patient, and—based on data from MADIT‐II and an assumption that nonischemic patients will have slightly less mortality—we estimate that 6–7% of arm A patients will die within one year and an additional 1–2% of the patients will be lost to active participation. This leads us to estimate a loss of 10–12% of the patients in each arm during the study. Based on data from MADIT‐II and from SCD‐HeFT, we expect 10% of arm A patients to have at least one end point event within one year, implying a risk of 0.88% per patient per month (pppm). Under null hypothesis conditions, the same risk is expected in arm T, while under target alternative conditions the risk would be reduced in half, namely 0.44% pppm. These conditions will likely lead to accumulations of at least 88 end points in B + A and in C + A. However, if after a minimum of 9 months of follow‐up for all surviving and active patients, these numbers have not been accumulated, the DSMB will report this fact to the PI (without further details on end points by arm), and the trial will be continued an additional 3 months, providing a minimum of 12 months follow‐up. By that time, it is highly probable (under conditions stated above) that there will be at least 75 events accumulated in each trial, a sufficient number to provide 85% power to detect a 50% reduction in risk; even 66 events will provide 80% power (Table 2).
Table 2.
Power to Detect a Reduction in Risk of Inappropriate Therapy for a Particular Treatment Arm versus Control
| Hazard Ratio | Power (in %) | |
|---|---|---|
| 88 End points | 75 End points | |
| 1.00 | 2.5 | 2.5 |
| 0.67 | 47 | 41 |
| 0.60 | 69 | 60 |
| 0.50 | 90 | 85 |
| 0.40 | 99 | 98 |
| 0.33 | >99 | >99 |
Power is for a test with two‐sided significance level of 5%, as a function of the hazard ratio and the number of accumulated end points in arms T + A together.
Validity of proportional‐hazards modeling will be evaluated; if found invalid, an alternative analysis using piecewise‐constant hazards will be carried out. (Kaplan–Meier time‐to‐event curves are not appropriate due to the necessity of censoring follow‐up at death. 24
As a tertiary analysis, any difference between arms B and C will be analyzed similarly. However, there will likely be insufficient power to detect any meaningful difference.
Analysis of Mortality
One source of information on mortality is from interrogation of implanted devices (CRT‐Ds or ICDs). If no therapy was offered by the device prior to death, determination as to whether such therapy may have been offered had the patient been in a different arm of the study can be sought. Although any occurrence of such an untoward incident would provide cause for concern, such incidences are unlikely to be sufficiently frequent to provide statistical evidence for or against differences in mortality risk. The DSMB will have the option of recommending termination of the trial should such evidence develop during the trial, however.
The secondary end point of all‐cause mortality will be analyzed similarly to the primary end point, along with Kaplan–Meier mortality curves. With 6–7% mortality expected at 12 months, a total of 135 deaths among the 1500 patients may be expected. This is too few to provide even 50% power, with a one‐sided significance level of 10%, and with pooling arms B and C together to compare with arm A.
Statistical tests of equivalence in mortality are similarly underpowered.
PROTECTION OF HUMAN SUBJECTS
The local enrolling center Principal Investigators and their associated enrollment teams are responsible for the initial evaluation, discussion of the study, obtaining consent, and all follow‐up of the study subjects. The enrolling teams have been instructed to follow all applicable FDA guidelines for clinical research, the MADIT‐RIT protocol, the “Declaration of Helsinki: Recommendations Guiding Medical Doctors in Biomedical Research Involving Human Patients,” and their own local Institutional Review Boards regulations. At each encounter with a potential or enrolled study subject the Investigator must be responsible for protecting the subject and determining if further participation in the clinical trial is appropriate for that individual patient. The Investigators, Research Coordinators and the Sponsor's team are charged with maintaining the confidentiality of patient and clinical trial information.
Drs. Schuger and Daubert functioned as coprimary authors.
MADIT‐RIT is sponsored by a Research Grant to The University of Rochester Medical Center from Cardiac Pacemakers, Inc, DBA‐Boston Scientific Cardiac Rhythm Management (CRM), 4100 Hamline Ave. N., St. Paul, MN 55112, Tel: 1800 CARDIAC.
Disclosures: Claudio Schuger: Claudio Schuger receives research support from Boston Scientific, St. Jude, Medtronic, Biosense‐Webster; and honoraria for advisory board participation from Premier. James Daubert receives research support from Boston Scientific, St. Jude, Medtronic, Biosense‐Webster; institutional fellowship support from Boston Scientific, St. Jude, Medtronic, Biosense‐Webster and Bard; and honoraria for advisory board participation or lectures from Biosense‐Webster, Sanofi‐Aventis, Boston Scientific, St. Jude, Premier and Sorin Medical. Mary Brown, Jackson Hall and Helmut Klein receives research support from University of Rochester Medical Center. David Cannom and Brian Olshansky receives research support from The Hospital of the Good Samaritan. N.A. Mark Estes III and David Wilber receives research support from New England Medical Center.
Wojciech Zareba received research support from Boston Scientific
Arthur J. Moss receives research support from Boston Scientific and honoraria for lectures from Boston Scientific, Medtronic, and St. Jude.
REFERENCES
- 1. Moss AJ, Hall WJ, Cannom DS, et al Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. Multicenter Automatic Defibrillator Implantation Trial Investigators. N Engl J Med. 1996;335:1933–1940. [DOI] [PubMed] [Google Scholar]
- 2. Moss AJ, Zareba W, Hall WJ, et al Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. New Engl J Med. 2002;346:877–883. [DOI] [PubMed] [Google Scholar]
- 3. Bardy GH, Lee KL, Mark DB, et al Amiodarone or an implantable cardioverter‐defibrillator for congestive heart failure. N Engl J Med. 2005;352:225–237. [DOI] [PubMed] [Google Scholar]
- 4. Poole JE, Johnson GW, Hellkamp AS, et al Prognosticimportance of defibrillator shocks in patients with heart failure. N EnglJ Med 2008;359:1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daubert JP, Zareba W, Cannom DS, et al MADIT II Investigators. Inappropriate implantablecardioverter‐defibrillator shocks in MADIT II: Frequency, mechanisms, predictors, and survival impact. J Am Coll Cardiol 2008;51:1357–1365. [DOI] [PubMed] [Google Scholar]
- 6. Ellenbogen KA, Levine JH, Berger RD, et al Defibrillatorsin Non‐Ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) Investigators. Are implantable cardioverter defibrillator shocks a surrogate for sudden cardiac death in patients with nonischemic cardiomyopathy? Circulation 2006;113:776–782. [DOI] [PubMed] [Google Scholar]
- 7. Wilkoff BL, Williamson BD, Stern RS, et al PREPARE Study Investigators. Strategic programming of detection and therapy parameters in implantablecardioverter‐defibrillators reduces shocks in primary prevention patients: Results from the PREPARE (Primary Prevention Parameters Evaluation) study. J Am Coll Cardiol 2008;52:541–550. [DOI] [PubMed] [Google Scholar]
- 8. Grimm W, Flores BF, Marchlinski FE. Electrocardiographically documented unnecessary, spontaneous shocks in 241 patients with implantable cardioverter defibrillators. Pacing Clin Electrophysiol 1992;15:1667–1673. [DOI] [PubMed] [Google Scholar]
- 9. Schmitt C, Montero M, Melichercik J. Significance of supraventricular tachyarrhythmias in patients with implanted pacing cardioverter defibrillators. Pacing Clin Electrophysiol 1994;17:295–302. [DOI] [PubMed] [Google Scholar]
- 10. Theuns DA, Klootwijk AP, Simoons ML, et al Clinical variables predicting inappropriate use of implantable cardioverter‐defibrillator in patients with coronary heart disease or nonischemic dilated cardiomyopathy. Am J Cardiol 2005;95:271–274. [DOI] [PubMed] [Google Scholar]
- 11. Schron EB, Exner DV, Yao Q, et al Quality of life in the antiarrhythmics versus implantable defibrillators trial: Impact of therapy and influence of adverse symptoms and defibrillator shocks. Circulation 2002;105:589–594. [DOI] [PubMed] [Google Scholar]
- 12. Namerow PB, Firth B, Heywood GM, et al Quality of life six months after CABG surgery in patients randomized to ICD versus no ICD therapy: Findings from the CABG Patch Trial. PACE 1999;22:1305–1313. [DOI] [PubMed] [Google Scholar]
- 13. Irvine J, Dorian P, Baker BM, et al Quality of life in the Canadian Implantable Defibrillator Study (CIDS). Am Heart J 2002;144:282–289. [DOI] [PubMed] [Google Scholar]
- 14. Klein RC, Raitt MH, Wilkoff BL, et al Analysis of implantable cardioverter defibrillator therapy in the Antiarrhythmics Versus Implantable Defibrillators (AVID) Trial. J Cardiovasc Electrophysiol 2003;14:940–948. [DOI] [PubMed] [Google Scholar]
- 15. Van Rees JB, Borleffs CJW, De Bie MK, et al Inappropriate implantable cardioverter‐defibrillator shocks: Incidence, predictors, and impact on mortality. J Am Coll Cardiol 2011;57 556–562 [DOI] [PubMed] [Google Scholar]
- 16. Kuhlkamp V, Dornberger V, Mewis C, et al Clinical experience with the new detection algorithms for atrial fibrillation of a defibrillator with dual chamber sensing and pacing. J Cardiovasc Electrophysiol 1999;10:905–915. [DOI] [PubMed] [Google Scholar]
- 17. Deisenhofer I, Kolb C, Ndrepepa G, et al Do current dual chamber cardioverter defibrillators have advantages over conventional single chamber cardioverter defibrillators in reducing inappropriate therapies? A randomized, prospective study. J Cardiovasc Electrophysiol 2001;12: 134–142. [DOI] [PubMed] [Google Scholar]
- 18. Theuns DA, Klootwijk AP, Goedhart DM, et al Prevention of inappropriate therapy in implantable cardioverter‐defibrillators: Results of a prospective, randomized study of tachyarrhythmia detection algorithms. J Am College Cardiol 2004;44:2362–2367. [DOI] [PubMed] [Google Scholar]
- 19. Olshansky B, Day JD, Moore S, et al Is dual‐chamber programming inferior to single‐chamber programming in an implantable cardioverter‐defibrillator? Results of the INTRINSIC RV (Inhibition of Unnecessary RV Pacing With AVSH in ICDs) Study. Circulation 2007;115:9–16. [DOI] [PubMed] [Google Scholar]
- 20. Friedman PA, McClelland RL, Bamlet WR, et al Dual‐chamber versus single‐chamber detection enhancements for implantable defibrillator rhythm diagnosis: The detect supraventricular tachycardia study. Circulation 2006;113:2871–2879. [DOI] [PubMed] [Google Scholar]
- 21. Rienstra M, Smit MD, Nieuwland W, et al Persistent atrial fibrillation is associated with appropriate shocks and heart failure in patients with left ventricular dysfunction treated with an implantable cardiovertr defibrillator. Am Heart J 2007;143:120–126. [DOI] [PubMed] [Google Scholar]
- 22. Dewland TA, Pellegrini CN, Wang Y, et al Dual‐chamber implantable cardioverter‐defibrillator selection is associated with increased complication rates and mortality among patients enrolled in the NCDR Implantable Cardioverter‐Defibrillator Registry. J Am Coll Cardiol 2011;58:1007–1013 [DOI] [PubMed] [Google Scholar]
- 23. Spragg DD, Berger R. How to avoid inappropriate shocks. Heart Rhythm 2008;5:762–765. [DOI] [PubMed] [Google Scholar]
- 24. Lawless JF. Statistical Models and Methods for Lifetime Data (Chapter 9), 2nd edition, 2003. Hoboken , NJ , John Wiley. [Google Scholar]
- 25. Muhlbaier LH. HIPAA Training Handbook for Researchers, Opus Communications, Inc; Marblehead , MA , 2002. [Google Scholar]
- 26. Kim SG, Fogoros RN, Furman S, et al Standardized reporting of ICD patient outcome: The report of a North American Society of Pacing and Electrophysiology Policy Conference, February 9–10, 1993. Pacing Clin Electrophysiol 193;16:1358–1362. [DOI] [PubMed] [Google Scholar]
- 27. Epstein AE, Carlson MD, Fogoros RN, et al Classification of death in antiarrhythmia trials. J Am Coll Cardiol 1996;27:433–442. [DOI] [PubMed] [Google Scholar]
- 28. Hinkle LE, Jr. , Thaler HT. Clinical classification of cardiac deaths. Circulation 1982;65:457–464. [DOI] [PubMed] [Google Scholar]