

Abstract
Background: Although QT variables such as its interval and/or dispersion can be clinical markers of ventricular tachyarrhythmia, few data exist regarding the role of QT variables in genotyped hypertrophic cardiomyopathy (HCM). Therefore, we analyzed QT variables in genotyped subjects with or without left ventricular hypertrophy (LVH).
Methods: QT variables were analyzed in 111 mutation and 43 non‐mutation carriers who were divided into three groups: A, those without ECG abnormalities and echocardiographically determined LVH (wall thickness ≥13 mm); B, those with ECG abnormalities but LVH; and C, those with ECG abnormalities and LVH. We also examined clinical outcome of enrolled patients.
Results: Maximal LV wall thickness in group C (19.0 ± 4.3 mm, mean ±SD) was significantly greater than that in group A (9.2 ± 1.8) and group B (10.4 ± 1.8). Under these conditions, maximum QTc interval and QT dispersion were significantly longer in group C than those in group A (438 ± 38 ms vs 406 ± 30 and 64 ± 31 vs 44 ± 18, respectively; P < 0.05). QTc interval and QT dispersion in group B (436 ± 50 and 64 ± 22 ms) were also significantly greater than those in group A. During follow‐up periods, four sudden cardiac deaths and one ventricular fibrillation were observed in group C, and two nonlethal ventricular tachyarrhythmias were observed in group B.
Conclusions: Patients with HCM‐related gene mutation accompanying any ECG abnormalities frequently exhibited impaired QT variables even without LVH. We suggest that careful observation should be considered for those genotyped subjects.
Keywords: hypertrophic cardiomyopathy, QT dispersion, gene mutation
Hypertrophic cardiomyopathy (HCM) is an inherited cardiac disease that is caused by mutation of the genes, encoding sarcomeric proteins. 1 Patients with HCM are known as high risk of sudden cardiac death and/or malignant ventricular tachyarrhythmia (SCD/VT). 2 Importantly, SCD may be the first clinical manifestation. 3
QT variables such as QT intervals and QT dispersion were evaluated in patients with HCM, and QT dispersion 4 , 5 and T‐peak to T‐end interval 6 were reported as markers for SCD/VT in such patients. However, few data existed regarding relationship between the presence and absence of left ventricular hypertrophy (LVH) and those of QT variables, because the causes of LVH were complicated for analysis.
Advances in molecular genetics enable us showing whether individuals in HCM families have a gene mutation. 7 , 8 , 9 Consequently, the appearance of abnormal QT variables can be examined in genotyped subjects with or without LVH. The aim of this study was to examine relationship between LVH and abnormal QT variables in genotyped subjects and to examine impact of abnormal QT variables on clinical outcome in these subjects.
METHODS
Study Population
The study population consisted of 111 genotyped subjects from kindred with HCM associated with the beta‐myosin heavy‐chain gene mutations (Ala26Val, Ala200Thr, Met822Leu, Arg858Cys, Ser866Pro, Arg870Cys, Glu935Lys), the myosin‐binding protein‐C gene mutations (c.2067 + 1G→A, c.1777delT, Arg820Gln), the cardiac troponin T gene mutations (Val85Leu, Arg92Trp, Phe110Ile, Lys273Glu), or the cardiac troponin I gene mutation (Lys183del) identified in Kanazawa University Hospital. All mutations except Ala200Thr, Met822Leu, Arg858Cys, and Ser866Pro have been previously identified and described elsewhere. 7 , 8 , 10 , 11 , 12 , 13 , 14 , 15 Informed consent was obtained from all participants or from their guardians, in accordance with the guidelines of the Bioethical Committee on Medical Research, Kanazawa University Graduate School of Medicine. Subjects with a clinical cause of LVH such as hypertension or severe valvular heart disease were excluded from this analysis.
Groups were classified as follows: mutation carriers without any ECG abnormalities and LVH (group A), carriers with ECG abnormalities but without LVH (group B), and carriers with ECG abnormalities and LVH (group C). As controls, 43 unaffected subjects from the same kindred were selected and divided into two groups, because there were age differences between groups A and B.
Electrocardiographic Examinations
Standard 12‐lead ECG was recorded in all subjects at paper speed of 25 mm/s with a gain of 10 mm/mV. All records were magnified by 200% and QT intervals were manually measured. In order to eliminate both interobserver variability and bias, QT intervals were measured using a digitizer in each of the 12 leads by a single observer who was blinded to all clinical findings. The QT interval was measured from the onset of the QRS complex to the end of the T wave at V5 lead. The end of the T wave was defined as the intersecting point of a tangent line on the terminal T wave and the TP baseline. 6 When a U wave was present, QT was measured to the nadir of the curve between the T and U waves. QT intervals were corrected for heart rate using Bazett's formula such as QTc = QT/RR1/2.
QT dispersion was calculated as the difference between the maximum QT and minimum QT intervals on all 12 leads of the ECG. If the height or depth of the T wave was <1.5 mm, this lead was excluded from the analysis. 16 T‐peak to T‐end interval, which represents transmural dispersion of repolarization, 17 was also measured as the interval between the peak and end of T wave at V5 lead.
In addition to QT analysis, we also defined ECG abnormalities as follows: (1) Q wave >0.04 second in duration or more than one fourth of the ensuring R wave in depth in at least two leads except in aVR 18 ; (2) LVH assessed by a Romhilt–Estes score >4 19 , 20 ; (3) ST‐segment depression of an upsloping type >0.1 mV at 0.08 second after the J point, or those of horizontal or downsloping type >0.05 mV; and (4) T‐wave inversion >0.1 mV except in aVR and V1 to V2 leads in the absence of conduction disturbance. 21
Echocardiographic Examinations
Standard transthoracic M‐mode and two‐dimensional echocardiographic studies were performed to identify and quantify morphologic features of the left ventricle. Left ventricular dimensions and the thicknesses of the septum and posterior wall were measured at the level of the tips of the mitral valve leaflet. Wall hypertrophy was defined as maximum left ventricular wall thickness ≥13 mm in adults or >95% CI of the theoretical value in children. 22 Additionally, we measured anterior and lateral walls of LV at the same level of measuring septum and posterior wall. From these measurements, we defined the maximal LV wall thickness (Fig. 1).
Figure 1.
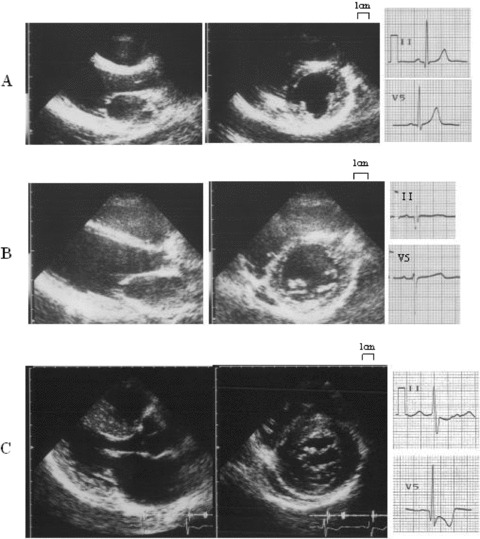
Representative cases for examination. (A) A case with neither ECG abnormalities nor LVH (group A). (B) A case with ECG abnormalities such as deep Q wave without LVH (group B). (C) A case both with ECG abnormalities such as abnormal Q waves and negative T waves and LVH (group C).
Survival Analysis
Data on survival and clinical status were collected at the time of visit of patients and were followed at this hospital and by direct telephone interview with patients, their family members, or their attending physicians at affiliated hospitals. The end points were defined as aborted cardiac arrest and SCD. Minor cardiac event such as nonlethal arrhythmia was also recorded. Enhanced antiarrhythmic treatments such as implantable cardioverter defibrillator (ICD) and amiodarone were not used until lethal arrhythmia occurred.
Statistical Analysis
Values are expressed as the mean ±SD. Comparison between the groups was performed using a one‐way analysis of variance (ANOVA) followed by Scheffe's method. Categorical data were compared using chi‐square analysis. Analysis was performed using StatView 5.0 (Abacus Concepts, Inc., Berkeley, CA, USA). A P value <0.05 was considered statistically significant.
RESULTS
Baseline Characteristics
Baseline characteristics of the study subjects are summarized in Table 1. Twenty‐two subjects were assigned to group A (Fig. 1A), 12 to group B (Fig. 1B), and 77 to group C (Fig. 1C). Mean age of group A (27 ± 22) was significant younger than those in group B (40 ± 24) and group C (50 ± 17). There were no differences in prevalence of the gene mutations between the three groups. Maximal wall thickness and interventricular septal wall thickness of group C (18.9 ± 4.5 mm and 17.9 ± 4.7) were significantly greater than those in groups A (9.1 ± 1.8 and 8.7 ± 1.9) and B (10.3 ± 1.9 and 9.8 ± 2.1). There were no differences in maximal wall thickness and interventricular septal wall thickness between groups A and B. Left ventricular dimensions and left ventricular fractional shortening were not different among the three groups.
Table 1.
Baseline Characteristics
| Group A | Control A | Group B | Control B | Group C | |
|---|---|---|---|---|---|
| Number | 22 | 27 | 12 | 16 | 77 |
| Male/female | 10/12 | 15/12 | 5/7 | 5/11 | 42/35 |
| Age (year) | 26.8 ± 22.0 | 28.5 ± 16.2 | 39.7 ± 23.8 | 40.0 ± 17.9 | 50.4 ± 17.2 |
| Gene | |||||
| MYH7 | 4 | – | 1 | – | 11 |
| MYBPC3 | 8 | – | 1 | – | 23 |
| TNNT2 | 3 | – | 3 | – | 15 |
| TNNI3 | 7 | – | 7 | – | 28 |
| Echocardiogram | |||||
| MaxWT (mm) | 9.1 ± 1.8 | 9.2 ± 1.4 | 10.3 ± 1.9 | 9.5 ± 1.2 | 18.9 ± 4.5* |
| IVST (mm) | 8.7 ± 1.9 | 9.0 ± 1.7 | 9.8 ± 2.1 | 9.1 ± 1.2 | 17.9 ± 4.7* |
| PWT (mm) | 8.7 ± 1.9 | 8.7 ± 1.4 | 9.7 ± 1.8 | 8.9 ± 1.3 | 11.3 ± 2.0 |
| LVDd (mm) | 42.3 ± 8.4 | 46.1 ± 4.0 | 43.9 ± 3.7 | 45.7 ± 3.5 | 44.6 ± 5.9 |
| LVDs (mm) | 27.6 ± 8.8 | 28.7 ± 3.8 | 26.4 ± 6.3 | 27.6 ± 3.5 | 28.1 ± 7.2 |
| FS (%) | 35.5 ± 7.3 | 37.7 ± 6.1 | 39.7 ± 10.8 | 39.6 ± 6.3 | 37.7 ± 9.4 |
| LAD (mm) | 31.6 ± 9.0 | 32.2 ± 5.0 | 35.4 ± 7.2 | 31.6 ± 5.1 | 38.3 ± 7.4 |
*P < 0.001 for comparison of group A, group B, control A and control B.
MaxWT = maximum wall thickness; IVST = interventricular septal wall thickness; PWT = left ventricular posterior wall thickness; LVDd = left ventricular end‐diastolic dimensions; LVDs = left ventricular end‐systolic dimensions; FS = left ventricular fractional shortening; LAD = left atrial dimensions.
QT Variables in the Study Groups
QT variables in the study groups are summarized in Table 2. Maximum QTc interval and QT dispersion were significantly longer in group C in comparison with those in group A (Figs. 2). Interestingly, QT dispersion in group B was also significantly increased over that in group A and that in age‐matched controls (control B). In contrast, QT variables in group A were not different from those in age‐matched controls (control A). As for T‐peak to T‐end interval, there was no significant difference among these groups (Table 2).
Table 2.
QT Variables in Each Group
| Group A | Control A | Group B | Control B | Group C | |
|---|---|---|---|---|---|
| RR (ms) | 880 ± 189 | 899 ± 153 | 935 ± 152 | 922 ± 127 | 959 ± 146 |
| MaxQT (ms) | 379 ± 41 | 387 ± 36 | 403 ± 40 | 386 ± 26 | 427 ± 47** |
| MinQT (ms) | 338 ± 37 | 341 ± 27 | 342 ± 33 | 340 ± 23 | 366 ± 43** |
| QT dispersion (ms) | 42 ± 19 | 46 ± 16 | 61 ± 22* | 45 ± 17 | 62 ± 31** |
| MaxQTc (ms1/2) | 408 ± 29 | 410 ± 31 | 419 ± 37* | 403 ± 20 | 437 ± 35** |
| MinQTc (ms1/2) | 363 ± 31 | 362 ± 24 | 355 ± 25 | 356 ± 22 | 375 ± 36 |
| QTc dispersion (ms1/2) | 45 ± 18 | 49 ± 17 | 64 ± 23* | 47 ± 16 | 62 ± 27** |
| T‐peak to T‐end (ms) | 173 ± 35 | 152 ± 48 | 175 ± 24 | 163 ± 29 | 168 ± 47 |
*P < 0.05 for comparison of group A; **P < 0.01 for comparison of group A.
RR = RR interval; MaxQT = maximum QT interval; MinQT = minimum QT interval; MaxQTc = corrected maximum QT interval; MinQTc = corrected minimum QT interval; QTc dispersion = corrected QT dispersion; T‐peak to T‐end = T‐peak to T‐end interval.
Figure 2.
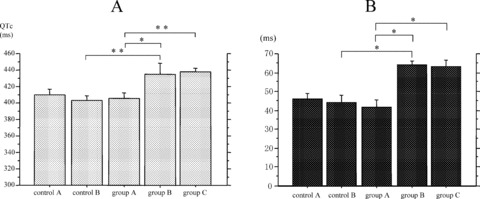
Maximum QTc interval (maxQTc) and QT dispersion in each group. (A) Max QTc of group B and group C was significantly longer than group A. (B) QT dispersion of group B and group C also increased than group A. *P < 0.05 for comparison to group A; **P < 0.01 for comparison to group A.
To make sure of the relationship between the QT variables and clinical outcome, we have followed mutation carriers prospectively. Fifty‐seven of 111 mutation carriers including 13 of group A, 6 of group B, and 38 of group C could be followed. Mean follow‐up periods were 6.9 years (1–15 years). There were four cases of SCDs and one case that was resuscitated from ventricular fibrillation in group C. ICD was implanted to the resuscitated patient after this episode. However, there was no cardiac death in group A and group B, although two nonlethal arrhythmias such as triplet PVC were detected by 24‐hour Holter monitoring in group B.
Interestingly, T‐peak to T‐end interval of the five subjects who had SCD or ventricular fibrillation were 188 ± 22 ms, which tended to be longer than those from the remaining subjects without SCD or ventricular fibrillation in the group C (165 ± 46 ms). It is important that LV function of five patients suffering from SCD or ventricular fibrillation was not impaired in comparison with the remaining patients.
DISCUSSION
The present study demonstrates that, in genotyped subjects, QT variables can be altered not only in subjects with LVH but also in those without LVH accompanying any ECG abnormalities such as abnormal Q wave, high voltage and inverted T wave. Under these conditions, lethal or nonlethal ventricular arrhythmia can occur in those subjects even without LVH.
In the present study QT variables did not change in mutation carriers lacking ECG abnormalities (group A), but were changed in those with ECG abnormalities even in absence of LVH (group B), although previous studies have revealed that HCM patients have longer maximum QTc interval and increased QT dispersion with respect to values in normal controls. 16 , 23 Jouven et al. 24 analyzed QT variables in mutation carriers in the beta‐myosin heavy‐chain or myosin‐binding protein‐C gene, and reported that QT dispersion in mutation carriers without either ECG abnormalities or wall hypertrophy (nonpenetrants) did not differ from that in controls, as observed in the present study. However, maximum QTc interval in non‐penetrants was increased over that in controls, although QTc interval was not prolonged in our group A subjects. This discrepancy may be due to differences in genes involved or in criteria for ECG abnormalities. Actually, in our group A subjects, the mutation of the beta‐myosin heavy‐chain gene or the myosin‐binding protein‐C gene was observed in only 12 out of 22. QT dispersion may be changed in association with appearance of any ECG abnormalities in mutation carriers even in the absence of prominent LVH.
QT dispersion is thought to reflect regional heterogeneity of ventricular repolarization, and it is thought that upper limit of 50 ms is highly specific. 25 Prolonged QT dispersion correlates with the incidence of ventricular tachyarrhythmias and sudden death in patients with HCM, 4 , 5 and usually precedes the appearance of LVH. 21 Before myocyte hypertrophy develops, electrophysiological activity may be changed in cardiomyopathic cells and may produce heterogeneities of ventricular depolarization and repolarization.
The relationship between QTc interval and left ventricular wall thickness was reported to be different between mutation genes, 24 and LVH in athletes was not associated with prolonged QTc interval. 26 This suggests that gene mutation itself may affect QTc interval independent of myocardial hypertrophy. Additionally, inhomogeneities of LV wall thickness also influence QT dispersion. 27 Therefore, in the advanced stages, morphological inhomogeneities such as asymmetrical septal hypertrophy as observed in the present study, in addition to electrical inhomogeneity at the myocyte level, may contribute to increasing QT dispersion.
Shimizu et al. reported that T‐peak to T‐end interval favorably reflect the transmural dispersion of repolarization because the end of repolarization of the epicardial cell corresponded to the peak of the T wave, whereas the end of repolarization of the M cell corresponded to the end of the T wave. 17 Indeed, we previously reported that prolonged T‐peak to T‐end interval was associated with SCD or malignant VT in the genotyped HCM subjects with troponin I gene mutation. 6 However, in the present study there was no difference in T‐peak to T‐end interval between studied groups. This may be explained by the fact that the number of the cases with SCD or malignant VTs was small and different kinds of genotyped patients were included for analysis. Even under these conditions, it is interesting that the five subjects with SCD or ventricular fibrillation during follow‐up period exhibited prolonged T‐peak to T‐end without statistical significance.
Clinical Implications and Limitation
The present results suggest that we must be aware of the potential for occurrence of lethal arrhythmias/sudden cardiac death in individuals carrying the disease‐causing gene mutation of HCM when any ECG abnormalities are observed. To test this hypothesis, the enrolled patients were followed up over an average of 6.9 years. Surprisingly, we identified five patients from group C (1.64%/year) and two patients from group B suffered from sudden cardiac death or ventricular arrhythmias during follow‐up periods. Previous study pointed out that the annual rate of sudden death or ICD discharge was 1.02% in clinical HCM patients with evident LVH. 28 The present data suggest that mutation carriers may suffer from serious cardiac events if they have increased maximal QTc interval and/or QT dispersion even without LVH.
In the present study, we specifically examined subjects with HCM‐related gene mutation carriers. However, we did not consider the effects of conventional medical treatment, although the use of amiodarone and/or ICD was avoided until the occurrence of cardiac event. Further study with medical intervention may demonstrate clinical significance of QT variables in the occurrence of lethal cardiac events in genotyped subjects with ECG abnormalities.
CONCLUSION
Maximal QTc interval and/or QT dispersion increased in HCM‐related gene mutation carriers with ECG abnormalities in the absence or presence of LVH. We suggest the possible occurrence of lethal arrhythmia in mutation carriers of HCM when ECG abnormalities including QT variables are observed before the appearance of LVH.
REFERENCES
- 1. Jarcho JA, McKenna W, Pare JA. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med 1989;321:1372–1378. [DOI] [PubMed] [Google Scholar]
- 2. Maron BJ, Olivotto I, Spirito P, et al Epidemiology of hypertrophic cardiomyopathy‐related death. Revisited in a large non‐referral‐based patient population. Circulation 2000;102:858–864. [DOI] [PubMed] [Google Scholar]
- 3. Maron BJ, Roberts WC, Edwards JE, et al Sudden death in patients with hypertrophic cardiomyopathy: Characterization of 26 patients with functional limitation. Am J Cardiol 1978;41:803–810. [DOI] [PubMed] [Google Scholar]
- 4. Buja G, Miorelli M, Turrini P, et al Comparison of QT dispersion in hypertrophic cardiomyopathy between patients with and without ventricular arrhythmias and sudden death. Am J Cardiol 1993;72:973–976. [DOI] [PubMed] [Google Scholar]
- 5. Yetman AT, Hamilton RM, Benson LN, et al Long‐term outcome and prognostic determinants in children with hypertrophic cardiomyopathy. J Am Coll Cardiol 1998;32:1943–1950. [DOI] [PubMed] [Google Scholar]
- 6. Shimizu M, Ino H, Okeie K, et al T‐peak to T‐end interval may be a better predictor of high‐risk patients with hypertrophic cardiomyopathy associated with a cardiac troponin I mutation than QT dispersion. Clin Cardiol 2002;25:335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kokado H, Shimizu M, Yoshio H, et al Clinical features of hypertrophic cardiomyopathy caused by a Lys183 deletion mutation in the cardiac troponin I gene. Circulation 2000;102:663–669. [DOI] [PubMed] [Google Scholar]
- 8. Fujino N, Shimizu M, Ino H, et al A novel mutation Lys273Glu in the cardiac troponin T gene shows high degree of penetrance and transition from hypertrophic to dilated cardiomyopathy. Am J Cardiol 2002;89:29–33. [DOI] [PubMed] [Google Scholar]
- 9. Shimizu M, Ino H, Okeie K, et al Septal wall thinning and systolic dysfunction in patients with hypertrophic cardiomyopathy caused by a cardiac troponin I gene mutation. Am Heart J 2002;143:690–695. [DOI] [PubMed] [Google Scholar]
- 10. Konno T, Shimizu M, Ino H, et al A novel missense mutation in the myosin binding protein‐C gene is responsible hypertrophic cardiomyopathy with left ventricular dysfunction and dilation in elderly patients. J Am Coll Cardiol 2003;41:781–786. [DOI] [PubMed] [Google Scholar]
- 11. Konno T, Shimizu M, Ino H, et al A novel mutation in the cardiac myosin‐binding protein‐C gene is responsible for hypertrophic cardiomyopathy with severe ventricular hypertrophy and sudden death. Clin Sci (Lond) 2006;110:125–131. [DOI] [PubMed] [Google Scholar]
- 12. Nishi H, Kimura A, Harada H, et al Possible gene dose effect of a mutant cardiac beta‐myosin heavy chain gene on the clinical expression of familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun 1994;200:549–556. [DOI] [PubMed] [Google Scholar]
- 13. Fujino N, Shimizu M, Ino H, et al Cardiac troponin T Arg92Trp mutation and progression from hypertrophic to dilated cardiomyopathy. Clin Cardiol 2001;24:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fujino N, Shimizu M, Ino H, et al A novel mutation Lys273Glu in the cardiac troponin T gene shows high degree of penetrance and transition from hypertrophic to dilated cardiomyopathy. Am J Cardiol 2002;89:29–33. [DOI] [PubMed] [Google Scholar]
- 15. Anan R, Shono H, Tei C. Novel cardiac beta‐myosin heavy chain gene missense mutations (R869C and R870C) that cause familial hypertrophic cardiomyopathy. Hum Mutat 2000;15:584. [DOI] [PubMed] [Google Scholar]
- 16. Dritsas A, Sbarouni E, Gilligan D, et al QT‐interval abnormalities in hypertrophic cardiomyopathy. Clin Cardiol 1992;15:739–742. [DOI] [PubMed] [Google Scholar]
- 17. Shimizu W, Antzelevitch C. Sodium channel block with Mexiletine is effective in reducing dispersion of repolarization and preventing Torsade de Pointes in LQT2 and LQT3 models of the long‐QT syndrome. Circulation 1997;96:2038–2047. [DOI] [PubMed] [Google Scholar]
- 18. Ryan MP, Cleland JF, French JA, et al The standard electrocardiogram as a screening test for hypertrophic cardiomyopathy. Am J Cardiol 1995;76:689–694. [DOI] [PubMed] [Google Scholar]
- 19. Romhilt DW, Estes EH. A point‐score system for the ECG diagnosis of left ventricular hypertrophy. Am Heart J 1968;75:752–758. [DOI] [PubMed] [Google Scholar]
- 20. Charron P, Dubourg O, Desnos M, et al Diagnostic value of electrocardiography and echocardiography for familial hypertrophic cardiomyopathy in a genotyped adult population. Circulation 1997;96:214–219. [DOI] [PubMed] [Google Scholar]
- 21. Shimizu M, Ino H, Yamaguchi M, et al Chronologic electrocardiographic changes in patients with hypertrophic cardiomyopathy associated with cardiac troponin I mutation. Am Heart J 2002;143:289–293. [DOI] [PubMed] [Google Scholar]
- 22. Charron P, Dubourg O, Desnos M, et al Clinical features and prognostic implications of familial hypertrophic cardiomyopathy related to the cardiac myosin‐binding protein C gene. Circulation 1998;97:2230–2236. [DOI] [PubMed] [Google Scholar]
- 23. Zaidi M, Robert A, Fesler R, et al Dispersion of ventricular repolarization in hypertrophic cardiomyopathy. J Electrocardiol 1996;29(Suppl.):89–94. [DOI] [PubMed] [Google Scholar]
- 24. Jouven X, Hagege A, Charron P, et al Relation between QT duration and maximal wall thickness in familial hypertrophic cardiomyopathy. Heart 2002;88:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Macfarlane PW, McLaughlin SC, Rodger JC. Influence of lead selection and population on automated measurement of QT dispersion. Circulation 1998;98:2160–2167. [DOI] [PubMed] [Google Scholar]
- 26. Mayet J, Kanagaratnam P, Shahi M, et al QT dispersion in athletic left ventricular hypertrophy. Am Heart J 1999;137:678–681. [DOI] [PubMed] [Google Scholar]
- 27. Sakata K, Shimizu M, Ino H, et al QT dispersion and left ventricular morphology in patients with hypertrophic cardiomyopathy. Heart 2003;89:882–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elliott PM, Gimeno JR, Thaman R, et al Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006;92:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]