

Abstract
Background: Sudden cardiac death of a child is a devastating event for the family and an enormous challenge for the attending physician.
Methods and Results: We report a family with repeat events of sudden cardiac death and recurrent ventricular fibrillation in a teenage girl, where autopsy data and clinical investigations were inconclusive. The diagnosis of catecholaminergic polymorphic ventricular tachycardia (CPVT) was established only following finding a gene mutation in the cardiac ryanodine receptor.
Conclusions: Interpretation of autopsy data, provocation testing and genetic testing in victims of sudden death and family members are discussed to correctly identify the cause and properly manage asymptomatic carriers in such families.
Keywords: Molecular biology/genetics, Electrophysiology ‐ ventricular tachycardia, Sudden death
CASE PRESENTATION
A 39‐year‐old woman (IY, Fig. 1) presented in 2005 for a cardiological consultation because of family history of sudden death. Her 16‐year‐old brother suffered from recurrent syncope and eventually died suddenly during a soccer game. An autopsy reportedly showed hypertrophic cardiomyopathy (HCM) with subaortic stenosis. Remarkably, electrocardiogram (ECG) and echo performed following his syncope attacks were reported as normal. Several years later, her 8 year‐old son drowned in a public swimming pool. An autopsy was nondiagnostic but again suggested HCM.
Figure 1.
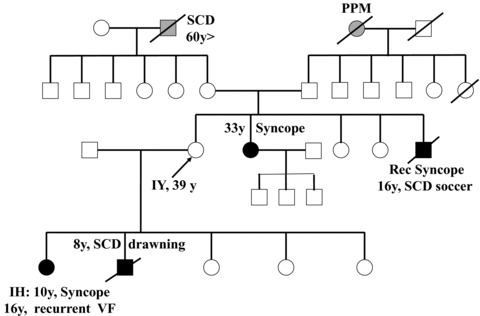
The pedigree. Solid symbols, affected individuals. Open symbols, not clinically affected. SCD = sudden cardiac death; PPM = permanent pacemaker; EP = electrophysiological.
Concomitantly, her daughter (designated IH) developed fainting episodes at age 10. The daughter underwent several echo Doppler, Holter recordings, EEG, tilt, and exercise testing all of which were normal. At age 16 an implanatable loop recorder was implanted. After 9 months while sitting on a sofa in a girlfriend's house, she felt dizzy, fainted, and developed convulsions. Physical exam shortly afterwards was reported to be unremarkable. The loop recorder tracing showed polymorphic ventricular tachycardia (VT), which terminated spontaneously, followed by 25‐second asystole and then sinus bradycardia. An implantable defibrillator (ICD) was implanted. In the following 2 years IH received 10 ICD shocks for ventricular fibrillation on various circumstances, without any remarkable trigger. An electrophysiological study performed at another hospital was essentially negative evoking a 5‐beat polymorphic VT during programmed stimulation using four premature stimuli. Empiric therapy with amiodarone or quiniduran was noneffective but low‐dose atenolol provided some relief. Because of family history compatible with malignant cardiomyopathy with minimal hypertrophy, a genetic testing of troponin T gene was performed and found to be negative.
IY's 33‐year‐old sister also experienced an episode of syncope. She had had a normal physical examination, ECG, echo Doppler, and MRI but because of the family history an ICD was recommended.
During physical examination, IY appeared younger than her age. Her physical examination, echo Doppler, and 24‐hour Holter recorder were normal. An ECG is shown in Figure 2. A maximal exercise test (9 minutes of Bruce protocol) was characterized by a flat blood pressure response to exercise and 0.5 mm horizontal ST depression in V2–V3. Both of her parents were healthy and doing well. The paternal grandmother and several of her siblings had a pacemaker in their sixties to seventies.
Figure 2.
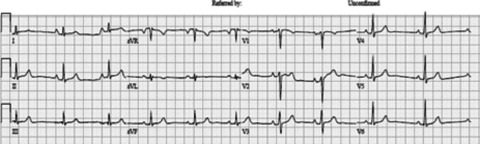
12‐Lead ECG of IY who is an obligate carrier.
Dr. Monserrat: Please let us know with what additional tests you would proceed in the case of IY, with a specific focus on possible further recommendations for genetic testing. In addition, please provide your thoughts regarding the management strategy you would recommend at this stage.
Expert
We evaluate an asymptomatic woman with a strong family history of sudden death affecting both her siblings and descendants. Even though the autopsies of both deceased brother and son suggested HCM, these diagnoses seem unlikely and could be misleading. Electrocardiograms, echocardiography, and MRI were normal in all the evaluated relatives. HCM may cause sudden death with normal echocardiography, for example, in patients with some troponin T mutations, but even in those cases the ECG is usually abnormal and it is very rare the absence of both LV hypertrophy on echocardiography and ECG abnormalities in all the affected relatives. In any case we would suggest the reevaluation of the performed autopsies by an expert pathologist.
In this family the differential diagnosis should focus in causes of syncope with polymorphic VT, ventricular fibrillation, and sudden death without structural heart disease. The most likely etiologies are those related with long QT (LQT) and Brugada syndromes, and we could also consider catecholamine dependent polymorphic ventricular tachycardia (CPVT). Exercise‐related syncope and sudden death at a young age without structural heart disease are more suggestive of either LQT or CPVT. Swimming is a well‐recognized trigger for sudden death associated with LQT1 (mutations in KCNQ1), but may also happen with other etiologies. The absence of ECG abnormalities is compatible with a diagnosis of CPVT, but in this condition the exercise test usually uncovers the diagnosis with the development of ventricular arrhythmias during exercise. The prolonged asystole and bradycardia after the episode of polymorphic VT shown by the loop recorder in the patient's daughter suggest the presence of conduction abnormalities, which were also present in the family history. Conduction system disease is part of the phenotypic spectrum of mutations associated with LQT, especially in LQT3 (mutations in SCN5A). The beneficial effect of atenolol in the prevention of syncopal episodes in this patient also supports the diagnosis of LQT. Beta‐blockers are more effective in LQT1, but may also improve symptoms in other LQT types and in CPVT.
The ECG of our index does not show relevant abnormalities. ECG may be normal both in LQT and CPVT. Concealed LQT may be uncovered doing repeated ECGs, evaluating the ECGs of the relatives, and with the evaluation of the modifications of LQT during exercise, on Holter monitoring or with a provocation test with adrenaline, which is especially useful in LQT1. With all these considerations, our first option for the diagnosis would be LQT and we would recommend to perform repeated ECGs in the index patient, in her affected relatives, and in her parents, to reevaluate the LQT duration on exercise and to perform a provocation test with adrenaline. We would also recommend a genetic study including the main LQT‐related genes: KCNQ1, KCNH2, KCNE1, KCNE2, and SCN5A. If these studies are negative we should consider the possibility of CPVT (screening of the RYR2 and CASQ2 genes), Brugada syndrome (SCN5A gene may be negative and a flecainide or ajmaline test would be useful), or less likely a cardiomyopathy with mild structural abnormalities such as arrhytmogenic cardiomyopathy, dilated cardiomyopathy due to a lamin A/C mutation (associated with high incidence of sudden death and conduction system disease), or HCM without hypertrophy. While we pursue these studies, we would recommend to avoid intense exercise, drugs that could be associated with LQT prolongation and to correct any potential electrolyte imbalance.
CASE PRESENTATION
Because IY was an obligate carrier with a malignant family history, an ICD was implanted. We then obtained IY DNA and began genetic testing by sequencing the 18 exons of the cardiac ryanodine receptor gene, RYR2, which contain most of CPVT1 mutations. 1 , 2 A heterozygous substitution of guanine to adenine was found in exon 14 predicting a nonconservative amino acid change of arginine (R) to glutamine (Q) at position 420 (Fig. 3).
Figure 3.
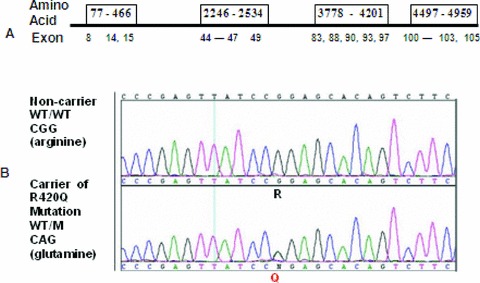
Ryr2 genotyping. (A) The strategy of targeting the “hot” exons containing most of CPVT1 mutations; (B) sequence trace of exon 14 in wild type (WT) compared to heterozygous mutation encoding the R420Q amino acid change.
Dr. Monserrat: Please let us know if you think the identified mutation is indeed the disease‐causing or if you would like to proceed with further genetic testing in our index patient and/or her first degree family members.
Expert
We consider that the R420Q is indeed a disease‐causing mutation that is sufficient to explain the syncopal episodes, the polymorphic VT, the ventricular fibrillation episodes, and the sudden deaths that occurred in the family. The Arginine 420, located in the N‐terminal cytosolic portion of the protein, is highly preserved during evolution. Two different mutations affecting this amino acid have been associated with the development of CPVT: R420W and R420Q.
The R420W mutation has been identified in at least eight families from different countries, associated with either unexplained sudden death or CPVT. Seventeen mutation carriers have been described (six with possible or definitive CPVT, one with a diagnosis of arrhythmogenic cardiomyopathy, four sudden deaths of unknown cause, and six healthy carriers). Six of the carriers had previous syncopal episodes. In these families there have also been described 11 sudden deaths of unknown cause in relatives without a genetic diagnosis. Most sudden deaths related to the R420W mutation occurred in young individuals (age 13–40) and were related to either physical or emotional stress. The mutation was not found in at least 820 controls reported in these studies. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9
The R420Q mutation has been previously described in three publications. Medeiros‐Domingo et al. reported in 2009 two index patients with exertional syncope due to CPVT associated with this mutation. 10
In 2009, Zorio et al. reported the R420Q mutation in eight patients with CPVT from a Spanish family with four previous premature sudden deaths. 1 The mutation showed clear cosegregation with the disease (they studied 25 relatives in three generations). van der Werf et al. described a female patient with this mutation 8 who had been diagnosed at age 15 years in the study of syncopal episodes. She was treated with bisoprolol and later also received flecainide, which did not abolish the exercise‐induced arrhythmias in this patient and she received a defibrillator. In summary, R420Q has been clearly associated with CPVT with a high incidence of stress‐/exercise‐induced syncope and sudden death in young carriers. Sinus bradycardia and increased atrial/nodal automatism have also been associated with this mutation and other RYR2 mutations. 11 , 12
Our suggestion in this case would be to evaluate the presence of the R420Q mutation in all the affected individuals and also in all their first‐degree relatives (cascade screening). This would allow us to evaluate the cosegregation of the mutation with the disease, and also to identify asymptomatic carriers that could be at risk of disease‐associated complications and/or to transmit the disease to their offspring. Appropriate genetic counseling should be provided before and after the test. Available data support the use of beta‐blockers in all the mutation carriers. Exercise test may be useful in the evaluation of the response to treatment, but it is important to remember that the absence of arrhythmias during the exercise test does not ensure the absence of complications. In any case, it is usually accepted that the persistence of inducible arrhythmias or symptoms in patients treated with beta‐blockers could justify the introduction of additional treatments, such as flecainide, surgical sympathectomy, or implantation of a defibrillator.
CASE PRESENTATION
Because Arginine 420 is highly preserved during evolution and because another CPVT1 mutation R420W has been reported within 4 the same residue, we considered R420Q to be disease causing. Extensive family genotyping (Fig. 4) showed that the mutation originated in IY's father and was shared by her affected daughter and sister. In addition six asymptomatic relatives aged 2–22 were diagnosed. The father of IY, designated III‐2, had RBBB and suffered from sinus bradycardia. Remarkably, the conduction system disease that included advanced atrio‐ventricular block and necessitated pacing in his relatives in their sixties and seventies was not associated with Ryr2 mutation carrier state.
Figure 4.
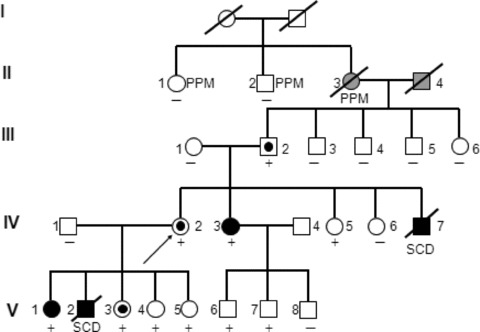
The pedigree after genotyping and provocation testing. Solid symbols, clinically affected individuals. Open symbols, not clinically affected. Targeted symbols, affected according to provocation testing without clinical symptoms. Ryr2 R420Q mutation carrier +, noncarrier –. SCD = sudden cardiac death; PPM = permanent pacemaker.
All genetically affected persons underwent provocation testing (Fig. 5). Epinephrine testing evoked polymorphic VT in IY (IV‐2) her sister IV‐3, her father III‐2, and her daughter V‐3. The carrier sister IH (designated IV‐5) had a 3‐beat monomorphic nonsustained VT. The affected IH (V‐1) had a negative test while on atenolol therapy. Symptom‐limited exercise test was positive (defined by multiple multifocal PVCs with or without couplets or triplets) in IV‐2, IV‐3, and III‐2).
Figure 5.
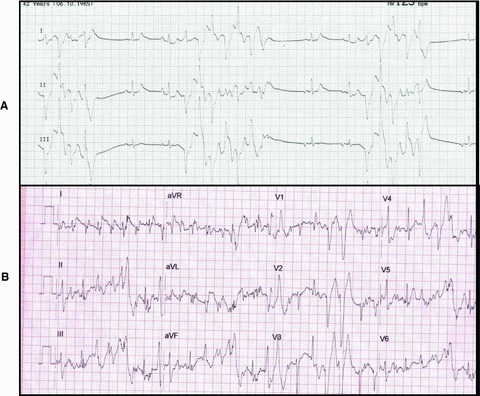
Examples of positive provocation tests in CPVT. (A) Epinephrine testing in IV‐2 showing recurrent events of bidirectional NSVT. (B) ECG trace during exercise testing in IV‐3 containing multifocal VPCS, a couplet, and a triplet of variable morphology.
All mutation carriers received beta‐blockers gradually increasing the dose. At the time of writing this report, no additional arrhythmic events were reported and no other family members underwent defibrillators implant. Remarkably, IY (IV‐2) experienced a series of inappropriate discharges due to device malfunction.
COMMENT
This story illustrates several commonly encountered problems in families inflicted by sudden unexpected death in young individuals. The presence of sudden death in two or more family members affected clearly suggests a genetic etiology. 1 An appropriately performed autopsy is often a clue for diagnosis. A negative autopsy (i.e., the absence of cardiomyopathy) strongly suggests a primary arrhythmic disorder. In our case autopsy diagnosis misled the clinicians by diverting attention to HCM. The normal wall thickness of formalin‐fixed left ventricle approaches 15 mm (range 10–20) due to heart arrest in contraction (rigor mortis) and an ensuing edema. Therefore an autopsy diagnosis of HCM shall be based on heart weight and requires histological confirmation by myofiber disarray involving at least 5–10% of muscle cross‐sectional area. 13 , 14 The discussant rightfully expressed skepticism of the postmortem diagnosis and suggested a review of autopsy specimens by an expert pathologist. This attitude may be in particular relevant for diagnosis of cardiomyopathy with minimal hypertrophy or arrhythmogenic right ventricular cardiomyopathy, especially in cases afflicted by a localized disease.
A second point deserving attention is the failure of initial exercise testing to provide a clue for stress‐induced arrhythmia. After having a genetic diagnosis, repeat testing of IV‐2, IV‐3 turned out to be positive, once performed in a dedicated setup. Regretfully, many of the stress tests carried out in an outpatient facility are prematurely terminated at submaximal heart rate or due to nondiagnostic findings. The value of provocation testing in investigating familial sudden death was documented by CASPER investigators. 15 We therefore recommend carrying out all studies related to familial sudden death in a specialized arrhythmia‐oriented facility.
An interesting issue encountered in this family was familial conduction system disease coinciding with CPVT. Patients with CPVT suffer from sinus bradycardia attributable to abnormal activity of autonomic nervous system or calcium clock. 16 Sinus bradycardia often complicates beta‐blocker therapy and requires a very careful dose up‐titration. Conduction disease necessitating pacemaker implantation in association with CPVT and dilated cardiomyopathy has been linked to Ryr2 exon 3 deletion, 17 but in our family R420Q mutation was not found in any of the pacemaker carriers.
The last topic already addressed by a discussing expert regards management of genetically affected individuals carrying a CPVT1 mutation. This issue is of particular importance because the first presentation of disease may be sudden cardiac death. Once clinical/genetic diagnosis is obtained most of the arrhythmia may be prevented by behavioral modification and prophylactic beta‐blocker therapy. Defibrillator implantation may usually be avoided. Instead, close follow‐up with repeat Holter telemetry and exercise testing to verify arrhythmia suppression become crucially important. 18
Acknowledgments
Acknowledgment: We are grateful to Ms. Gail Rezenfeld and Mr. Guy Katz for technical assistance.
These studies were supported by the Israel Science Foundation (ISF Grants 876/2005 and 763/10) and the Bircher–Baner award from 2007 by Tel Aviv University, Genetic analysis in families with sudden cardiac death.
REFERENCES
- 1. Tan HL, Hofman N, van Langen IM, et al Sudden unexplained death: Heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 2005;112(2):207–213. [DOI] [PubMed] [Google Scholar]
- 2. Tester DJ, Spoon DB, Valdivia HH, et al Targeted molecular autopsy of 49 medical examiner/coroner's cases. Mayo Clin Proc 2004;79(11):1380–1384. [DOI] [PubMed] [Google Scholar]
- 3. Bauce B, Rampazzo A, Basso C, et al Screening for ryanodine receptor type 2 mutations in families with effort‐induced polymorphic ventricular arrhythmias and sudden death: Early diagnosis of asymptomatic carriers. J Am Coll Cardiol 2002;40:341–349. [DOI] [PubMed] [Google Scholar]
- 4. Wehrens XH, Marks AR. Sudden unexplained death caused by cardiac ryanodine receptor (RyR2) mutations. Mayo Clin Proc 2004;79(11):1367–1371. [DOI] [PubMed] [Google Scholar]
- 5. Nishio H, Iwata M, Suzuki K. Postmortem molecular screening for cardiac ryanodine receptor type 2 mutations in sudden unexplained death: R420W mutated case with characteristics of status thymico‐lymphatics. Circ J 2006;70(11):1402–1406. [DOI] [PubMed] [Google Scholar]
- 6. Tiso N, Bauce B, Rampazzo A, et al Gene symbol: RYR2. Disease: Arrhythmogenic right ventricular cardiomyopathy type 2. Hum Genet 2004;114(4):405. [PubMed] [Google Scholar]
- 7. Sy RW, Gollob MH, Klein GJ, et al Arrhythmia characterization and long‐term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2011;8(6):864–871. [DOI] [PubMed] [Google Scholar]
- 8. van der Werf C, Kannankeril PJ, Sacher F, et al Flecainide therapy reduces exercise‐induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol 2011;57(22):2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steriotis AK, Nava A, Rampazzo A, et al Follow‐up with exercise test of effort‐induced ventricular arrhythmias linked to ryanodine receptor type 2 gene mutations. Am J Cardiol 2012;109(7):1015–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Medeiros‐Domingo A, Bhuiyan ZA, Tester DJ, et al The RYR2‐encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise‐induced long QT syndrome: A comprehensive open reading frame mutational analysis. J Am Coll Cardiol 2009;54(22):2065–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zorio GE, Cano PO, Navarro MJ, et al Asociación entre bradicardia sinusal y arritmias auriculares con el diagnóstico clínico de taquicardia ventricular polimórfica catecolaminérgica. Rev Esp Cardiol 2009; 62 Suppl 3:167 [http://www.revespcardiol.org/watermark/ctl_servlet?_f=10&pident_articulo=13142762&pident_usuario=0&pident_revista=25&fichero=25v62nSupl.3a13142762pdf001.pdf&ty=44&accion=L&origen=cardio&web=www.revespcardiol.org&lan=es]19232190 [Google Scholar]
- 12. Postma AV, Denjoy I, Kamblock J, et al Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet 2005;42:863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basso C, Burke M, Fornes P, et al Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch 2008;452:11–18. [DOI] [PubMed] [Google Scholar]
- 14. Davies MJ. The investigation of sudden death. Histopathology 1999; 34:93–98. [DOI] [PubMed] [Google Scholar]
- 15. Krahn AD, Healey JS, Chauhan V, et al Systematic assessment of patients with unexplained cardiac arrest. Cardiac Arrest Survivors with Preserved Ejection Fraction Registry (CASPER). Circulation 2009;120:278–285. [DOI] [PubMed] [Google Scholar]
- 16. Katz G, Arad M, Eldar M. Catecholaminergic polymorphic ventricular tachycardia: From bedside to bench and over. Curr Probl Cardiol 2009;34(1):1–44. [DOI] [PubMed] [Google Scholar]
- 17. Bhuiyan ZA, van den Berg MP, van Tintelen JP, et al Expanding spectrum of human RYR2‐related disease: New electrocardiographic, structural, and genetic features. Circulation 2007;116(14):1569–1576. [DOI] [PubMed] [Google Scholar]
- 18. Nof E, Belhassen B, Arad M, et al Postpacing abnormal repolarization in catecholaminergic polymorphic ventricular tachycardia associated with a mutation in the cardiac ryanodine receptor gene. Heart Rhythm 2011;8(10):1546–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]