

Abstract
Short QT syndrome (SQTS) is an inheritable primary electrical disease of the heart, discovered in 1999. It is characterized by an abnormally short QT interval (<300 ms) and a propensity to atrial fibrillation and sudden cardiac death (SCD). Like in the case of long QT syndrome there is more than one genetic mutation that can lead to a short QT interval in the ECG and so far two have been identified. Shortening of the effective refractory period combined with increased dispersion of repolarization is the likely substrate for reentry and life threatening tachyarrhythmias. Only 22 people have been classified as having SQTS: 15 from the actual measurement of a short QT interval in their ECG and 7 by history, all having died from SCD. It is very likely that several cases, especially among children, have been overlooked, since the shortness of the QT interval only becomes apparent at heart rates <80 beats/min. The best form of treatment is still not known, but prevention of atrial fibrillation has been accomplished by propafenone, and an implantable cardioverter defibrillator is recommended for prevention of SCD.
Keywords: short QT syndrome, sudden cardiac death, atrial fibrillation, sudden infant death syndrome
DEFINITION OF A SHORT QT INTERVAL
Even though there is some debate about the upper limit of normal for the QT interval, a corrected QT interval >450 ms in males and >460 ms in females is generally considered prolonged. Numbers for the lower limit of normal are seldom given. Based upon data from the study by Rautaharju et al. 1 we have previously suggested a short QT Interval to be <350 ms (two standard deviations below the mean predicted value) and an abnormally short QT interval to be <320 ms (<80% of mean predicted value), below which short QT syndrome (SQTS) should be strongly considered 2
In all published articles about SQTS to date, the diagnosis was based upon an ECG with a QT interval <300 ms at a normal heart rate. The QT interval is traditionally corrected for heart rate, but in patients with SQTS there are only minimal changes in the QT interval with a change in heart rate. 3 If the usual correction for heart rate is applied, a diagnosis of SQTS may be missed. Corrected by Bazett's formula, at a heart rate of 130–140 beats/min, the corrected QT interval in our patients would all be >350 ms. In order to make a diagnosis of SQTS, the heart rate has to be <130 beats/min, and preferably <100 beats/min. This is particularly important in children, whose heart rate is often fast, even at rest, and SQTS is a possible cause for some cases of sudden infant death syndrome (SIDS) 7 (concealed short QT syndrome).
SQTS can best be described as a new, inheritable, primary electrical disease characterized by an abnormally short QT interval and a propensity to atrial fibrillation and sudden cardiac death (SCD). It requires elimination of secondary short QT interval seen in hypercalcemia, hyperkalemia, hyperthermia, acidosis, and digitalis treatment. Also catecholamine and acetylcholine can occasionally shorten the QT interval. 4
DISCOVERY OF SQTS
In February 1999, a 17‐year‐old white female developed atrial fibrillation with fast ventricular response while undergoing laparoscopic cholecystectomy. Her ECG showed an unusually short QT interval (225 ms), which was not explained by hypercalcemia (Figs. 1 and 2) Further cardiac examination including Doppler echocardiography proved normal. She had no history of palpitations, syncope, or dizziness and her family history was negative for SCD. Her only sibling (a 21‐year‐old brother) and her 51‐year‐old mother also had short QT intervals in their ECGs (240 and 230 ms, respectively), while her father's ECG was normal. It was later discovered that her maternal grandfather (born in 1914 and an immigrant from Italy) had a long‐standing history of chronic atrial fibrillation and a similar short QT interval (245 ms). This family and a 37‐year‐old Spanish woman 5 who suddenly died shortly after an ECG revealed a QT interval of 266 ms, formed the basis for the first description of SQTS. 2
Figure 1.
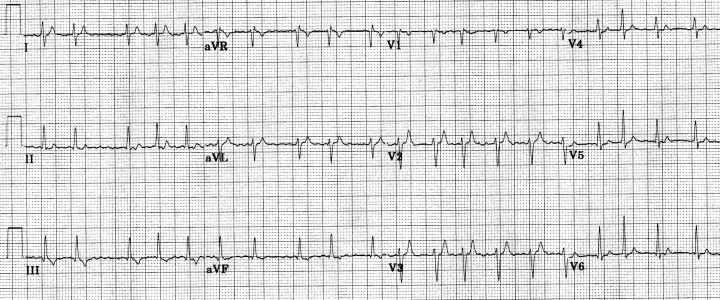
Twelve‐lead ECG from 17‐year‐old white female with atrial fibrillation (heart rate = 126 beats/min) in the setting of short QT interval.
Figure 2.
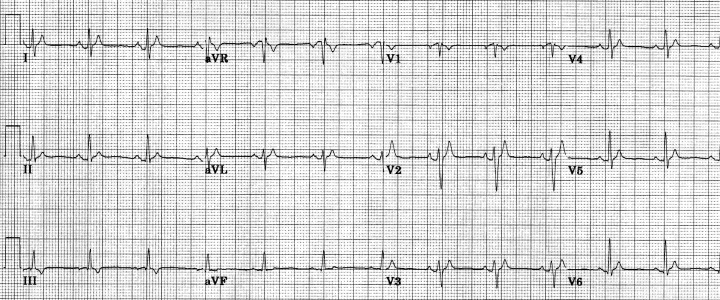
Twelve‐lead ECG from the same patient as in Figure 1, following restoration of normal sinus rhythm (heart rate = 76 beats/min, QT = 225 ms).
PATHOPHYSIOLOGY
Within the last year (2004), two different genetic mutations have been identified in families with SQTS. Brugada et al. 6 examined three families. In two of them, two different missense mutations were discovered, revealing the same amino acid change (N588K) in the S5‐P loop region of the cardiac I Kr channel HERG (KCNH2). These mutations dramatically increased I Kr, leading to heterogeneous abbreviation of action potential duration and refractoriness, reducing the affinity of the channel to I Kr‐blockers.
Bellocq et al. 7 examined a 70‐year‐old man presenting with idiopathic ventricular fibrillation and a QT interval of 290 ms. Analysis of candidate genes identified a g919c substitution in KCNQ1 encoding the K+ channel KvLQT1. It is of interest to note that mutations in the same two genes also can lead to a decrease in function of the affected potassium channels and QT prolongation as actually seen in the LQT2 and LQT1 syndromes. This finding suggests that SQTS is a genetically heterogeneous disease similar to other inherited arrhythmogenic disorders. In patients with SQTS, the effect of the genetic mutation is a gain in function in late potassium channels leading to an abbreviation of the action potential and refractoriness. Shortening of the effective refractory period (ERP) combined with increased dispersion of repolarization is the likely substrate for reentry, which in the atria can lead to atrial fibrillation and in the ventricles to ventricular fibrillation and SCD. So far no patient has been monitored during the onset of an episode of either atrial or ventricular fibrillation, and the circumstances, when such episodes are most likely to occur, are unknown. In patients with some forms of long QT syndrome, for instance, many episodes are related to physical or emotional stress but in others occurring during sleep, and the initiating trigger is usually a premature beat. Due to the very short refractory period in patients with SQTS, it is very likely that the trigger also would be a very early premature beat in these patients, and since the QT interval is most abnormal during slow heart rates, it is possible that episodes of tachyarrhythmia are most likely to occur when the heart rate is slow.
CURRENT POPULATION OF PUBLISHED CASES OF SHORT QT
A total of six families with at least one member having short QT interval in their ECGs have been identified, including two sporadic cases where SQTS was suspected. 5 , 7 In the other four families, attempts have been made to collect information about symptoms and ECG findings (Table 1).
Table 1.
Short QT Syndrome
| Reference | Age | Gender | Short QT Interval | SCD | Aborted SCD | Atrial Fibrillation | Inducible VF | ICD |
|---|---|---|---|---|---|---|---|---|
| Brugada 5 | 37 years | Femalea | Yes | Yes | ||||
| Gussak et al. 2 | 84 years | Male | Yes | Yes | ||||
| 51 years | Female | Yes | Yes | Yes | Yes | |||
| 21 years | Male | Yes | Yes | Yes | Yes | |||
| 17 years | Female | Yes | Yes | Yes | Yes | |||
| Gaita 4 | 35 years | Male | Yes | Yes | Yes | Yes | ||
| 31 years | Female | Yes | Yes | Yes | ||||
| 6 years | Male | Yes | Yes (8 months) | |||||
| 3 months | Male | Yes | ||||||
| 39 years | Male | Yes | ||||||
| 49 years | Female | Yes | ||||||
| unknown | Female | Yes | ||||||
| 39 years | Female | Yes | ||||||
| Gaita 4 | 67 years | Female | Yes | Yes | Yes | Yes | ||
| 15 years | Male | Yes | No | Yes | ||||
| 40 years | Female | Yes | ||||||
| 45 years | Female | Yes | ||||||
| 62 years | Female | Yes | Yes | Yes | ||||
| 26 years | Male | Yes | ||||||
| Brugada 6 | 51 years | Male | Yes | Yes | ||||
| 20 years | Male | Yes | ||||||
| Bellocq 7 | 70 years | Male | Yes | Yes | No | Yes | ||
| Summary | 3 months– | 11 Females | 15 | 9 | 3 | 7 | 6/8 | 8 |
| 87 years | 11 Males | |||||||
aThis patient is also described in reference 2.
Abbreviations as in text.
Out of 52 members from these families, 22 have been classified as SQTS patients: 15 from the actual measurement of short QT interval in their ECG and 7 by history, all having died from SCD. There are an equal number of males and females in the group, their ages ranging from 3 months to 84 years.
CLINICAL PRESENTATION
In 1977, Brugada 2 , 5 documented the case of a patient who died shortly after an ECG revealed a QT interval of 266 ms, but it was an article by Gaita et al. 4 that brought attention to the high incidence of SCD in members of families with short QT interval. In one Italian family, six members died suddenly. Of those six, one had documented short QT interval. Two living members also were known to have short QT interval.
In a family from Germany, three members died suddenly, one with documented short QT interval. Two others with short QT interval are alive.
At the present time, there are reports of six individuals with short QT interval who have either died suddenly or have been resuscitated with defibrillation following sudden collapse (Table 1). This places short QT interval as one of the high risks for SCD, similar to Brugada syndrome. In addition, an episode of aborted SCD in an 8‐month‐old baby with short QT interval suggests that it may be one of the possible explanations for SIDS.
In the first family with SQTS, the development of atrial fibrillation at a very early age led to the detection of short QT interval. 2 The youngest patient was only 17 years old when she presented with recurrent atrial fibrillation. Her brother was 25 years old when an asymptomatic episode of atrial fibrillation was detected by the implantable cardioverter defibrillator (ICD). Their mother at the age of 55 also had atrial fibrillation and their maternal grandfather had chronic atrial fibrillation for many years prior to his death at age 85. Three more patients with short QT intervals have been reported to have a history of atrial fibrillation (ages 35, 62, and 67 years), bringing the total number of reported cases to 7 out of 15 (46%).
Based upon our current knowledge, it would seem reasonable to consider SQTS in everyone with primary short QT interval <320 ms at a normal heart rate as well as individuals with no prolongation in QT interval with slowing of heart rate. Subjects with a family member with SCD or SQTS should have an ECG performed in order to rule out a short QT interval.
ELECTROPHYSIOLOGY STUDIES
Atrial and ventricular‐programmed electrical stimulation studies have been performed in eight patients with short QT interval. 3 , 4 , 7 In one patient, 3 the shortest coupling interval applied was 180 ms, with ventricular ERP shorter than that, and no arrhythmias were induced. In the remaining seven patients, ERP was determined in both atria and ventricles. In five of these patients, ERP was found to be between 120 and 160 ms, while atrial fibrillation was induced in two patients.
AV‐node ERP was determined in two patients and found to be 210 and 440 ms. Ventricular ERP was remarkably consistent in all seven patients (130–140 ms). Ventricular fibrillation was inducted in five patients and a monomorph VT (CI 150 ms) in one patient.
THERAPY
Due to the low number of patients with SQTS, the experience with antiarrhythmic drugs is very limited. Gaita et al. 8 tested the effect of flecainide, sotalol, ibutilide, and hydroquinidine on the QT interval in seven patients with SQTS. Flecainide caused a slight prolongation of the QT interval, primarily due to the prolongation of the QRS complex, whereas sotalol and ibutilide had no effect on the QT interval. Hydroquinidine caused QT prolongation, which reached the normal range, varying from 263 ± 12 to 362 ± 25 ms. The ST segment, which was almost absent at basal recording, appeared, while the T wave, which was high and peaked, increased in duration and decreased in amplitude. The ventricular ERP was prolonged to more than 200 ms and ventricular fibrillation was no longer inducible after hydroquinidine administration. We have used propafenone in two patients with paroxysmal atrial fibrillation without recurrence of the arrhythmia. We did not see any prolongation of the QT interval in excess of the widening of the QRS complex. 3
ICDs have been implanted in eight patients with SQTS for primary and secondary prevention of SCD. We implanted ICDs (Guidant Vitality AVT A135) for primary prevention in three of our patients, 2 and during 1‐year follow‐up, no shocks were delivered and the overall treatment was without complications. Schimpf et al. 9 reported their experience with ICD implantation in five patients with SQTS (one Medtronic, Inc. and four St. Jude Medical, Inc.). Despite normal sensing behavior during intraoperative and postoperative device testing, three patients experienced inappropriate shock therapies for T wave oversensing 30 ± 26 days after implantation (one Medtronic Marquis VR 7230, one St. Jude Atlas VR V‐199 and one St. Jude Photon Micro VR‐194 ICD). Reprogramming of the decay delay (St. Jude) and/or the sensitivity (St. Jude, Medtronic) prevented further inappropriate discharges, but the high incidence of T wave oversensing by ICDs in patients with SQTS calls for special attention to device selection and options available for reprogramming postimplantation.
SUMMARY
Despite having only four families and two sporadic cases from which to draw data, and fewer than 5 years of research, SQTS has been described in great detail with regard to genetics, clinical manifestations, and electrophysiologic substrate. There is an equal distribution among males and females, and SCD can occur at any age. One of the remaining questions is how best to treat these patients. Currently, an ICD seems the only viable option. With the small number of patients available for clinical trials, it seems unlikely that medical treatment for prevention of SCD will be an acceptable option in the foreseeable future. Propafenone has shown promising results in the treatment of paroxysmal atrial fibrillation.
REFERENCES
- 1. Rautaharju PM, Zhou SH, Wong S, et al Sex differences in the evolution of the electrocardiographic QT interval with age. Can J Cardiol 1992;8: 690–695. [PubMed] [Google Scholar]
- 2. Gussak I, Brugada P, Brugada J, et al Idiopathic short QT interval: A new clinical syndrome? Cardiology 2000;94: 99–102. [DOI] [PubMed] [Google Scholar]
- 3. Bjerregaard P, Gussak I. Atrial fibrillation in the setting of familial short QT syndrome. Heart Rhythm 2004;1(1S):522. [Google Scholar]
- 4. Gaita F, Giustetto C, Bianchi F, et al Short QT syndrome. A familial cause of sudden death. Circulation 2003;108: 965–970. [DOI] [PubMed] [Google Scholar]
- 5. Brugada J. Ardiaca A. 1999, personal communication.
- 6. Brugada R, Hong K, Dumaine R, et al Sudden death associated with short‐QT syndrome linked to mutations in HERG. Circulation 2004;109: 30–35. [DOI] [PubMed] [Google Scholar]
- 7. Bellocq C, Van Ginneken ACG, Bezzina CR, et al Mutation in the KCNQ1 gene leading to the short QT‐Interval syndrome. Circulation 2004;109: 2394–2397. [DOI] [PubMed] [Google Scholar]
- 8. Gaita F, Giustetto C, Bianchi F, et al Short QT syndrome: Pharmacological treatment. J Am Coll Cardiol 2004;43: 1494–1499. [DOI] [PubMed] [Google Scholar]
- 9. Schimpf R, Wolpert C, Bianchi F, et al Congenital short QT syndrome and implantable cardioverter defibrillator treatment: Inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003;14: 1273–1277. [DOI] [PubMed] [Google Scholar]